Genomic Signatures of Reinforcement

 , , and
, , and 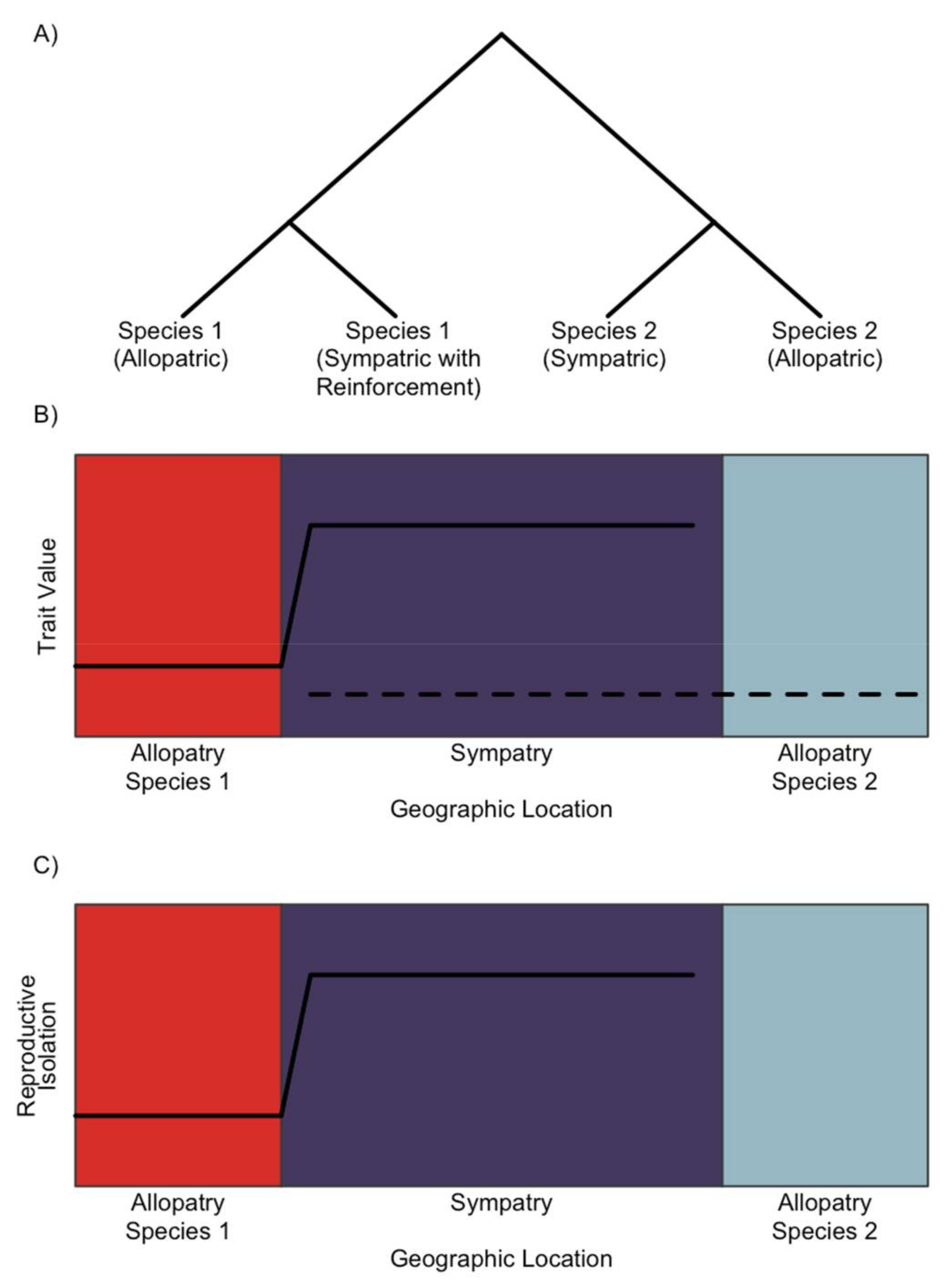{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
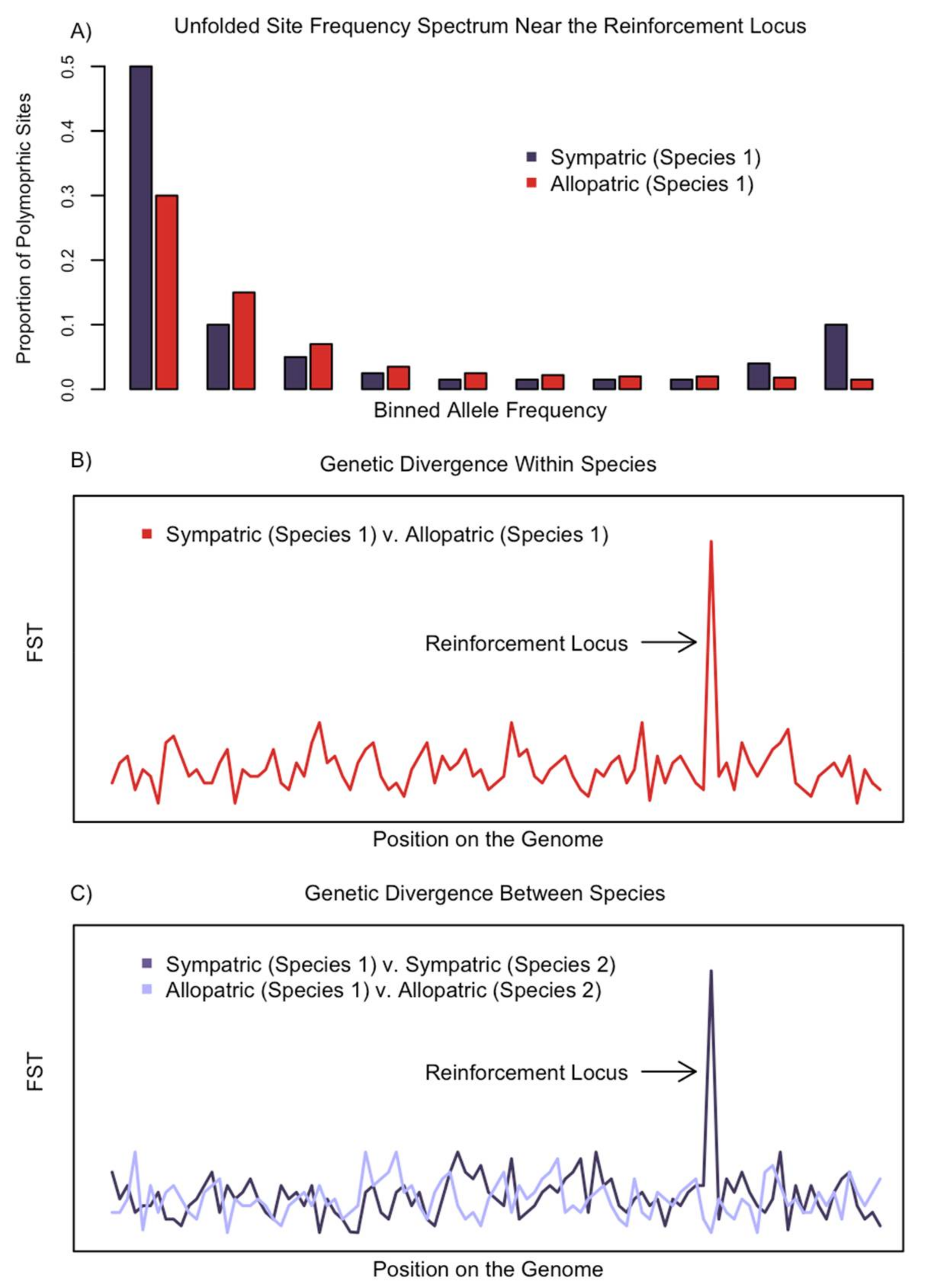
2. The Genomic Signature of Reinforcement
3. Alternative Hypotheses to the Signatures of Reinforcement
3.1. Ecological Character Displacement
3.2. Local Adaptation
3.3. Adaptive Introgression and the One-Allele Model
4. Complicating Factors to Identifying Genomic Signatures of Reinforcement
4.1. History of Mutation
4.2. Recombination
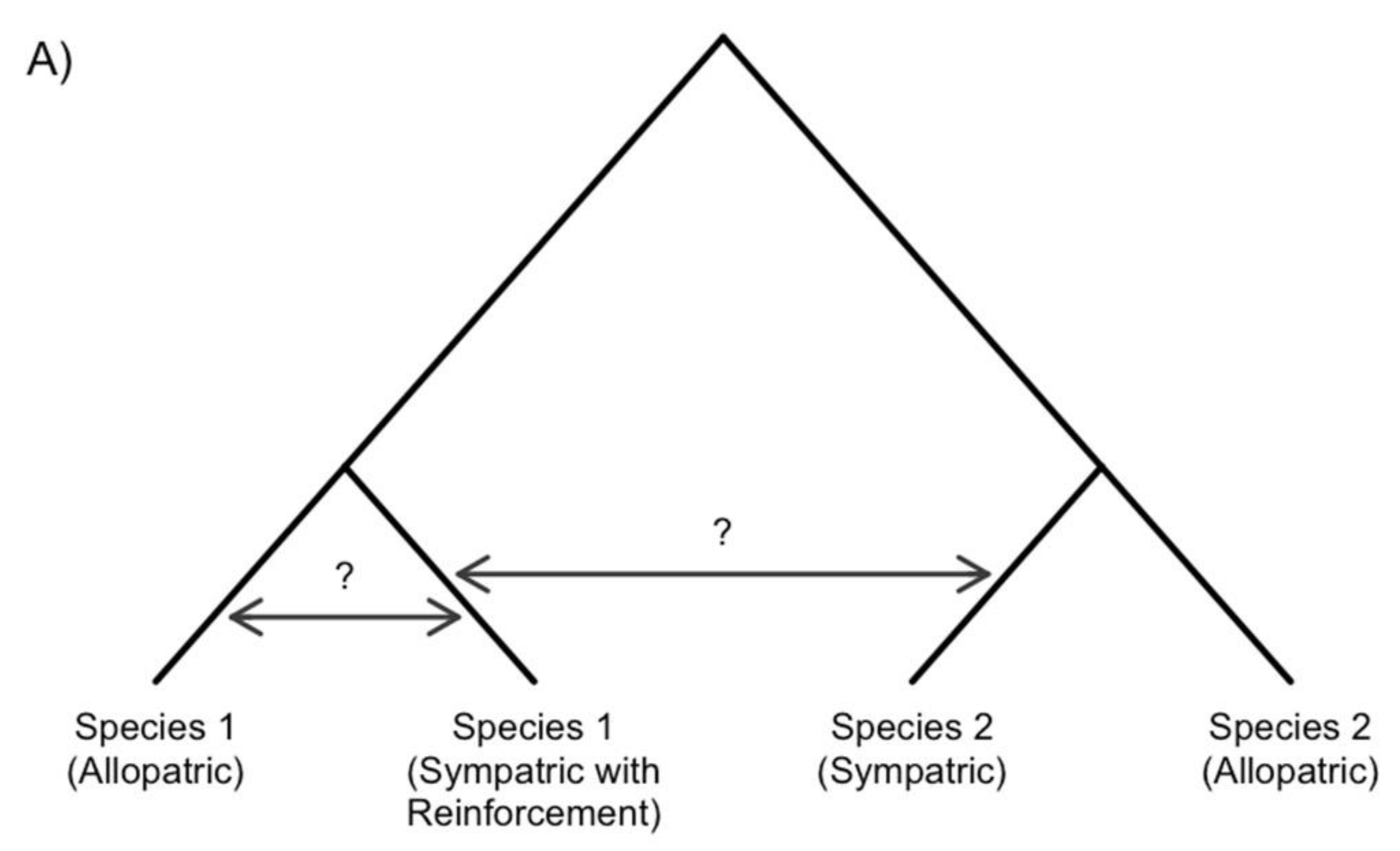
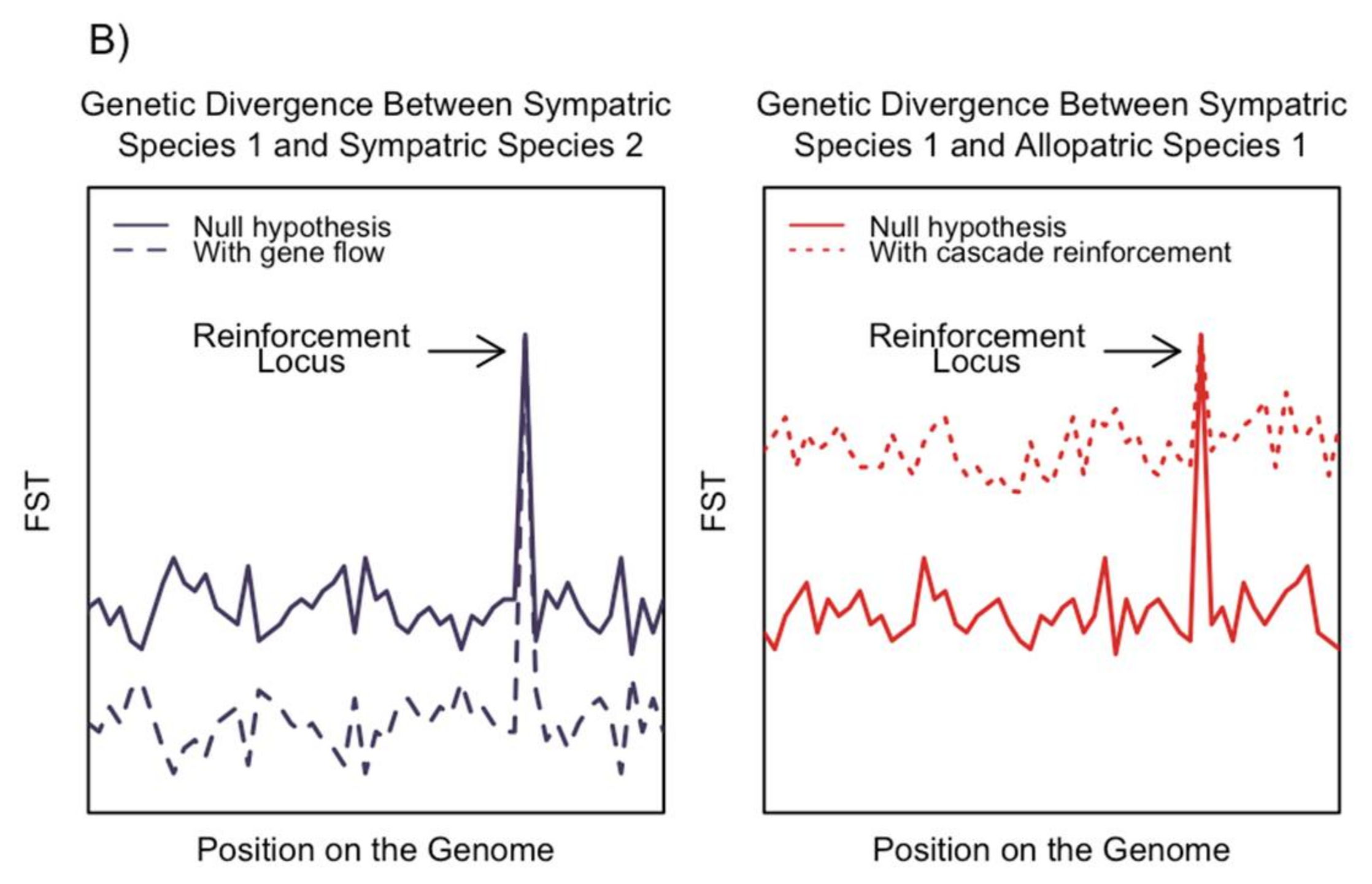
5. Gene Flow and the Evolution of Reinforcement
5.1. Gene Flow between Species
5.2. Gene Flow within Species
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Arnegard, M.E.; McGee, M.D.; Matthews, B.; Marchinko, K.B.; Conte, G.L.; Kabir, S.; Bedford, N.; Bergek, S.; Chan, Y.F.; Jones, F.C.; et al. Genetics of ecological divergence during speciation. Nature 2014, 511, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Marques, D.A.; Lucek, K.; Meier, J.I.; Mwaiko, S.; Wagner, C.E.; Excoffier, L.; Seehausen, O. Genomics of rapid incipient speciation in sympatric threespine stickleback. PLoS Genet. 2016, 12, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Roda, F.; Ambrose, L.; Walter, G.M.; Liu, H.L.; Schaul, A.; Lowe, A.; Pelser, P.B.; Prentis, P.; Rieseberg, L.H.; Ortiz-Barrientos, D. Genomic evidence for the parallel evolution of coastal forms in the Senecio lautus complex. Mol. Ecol. 2013, 22, 2941–2952. [Google Scholar] [CrossRef] [PubMed]
- Stankowski, S.; Sobel, J.M.; Streisfeld, M.A. Geographic cline analysis as a tool for studying genome-wide variation: A case study of pollinator-mediated divergence in a monkeyflower. Mol. Ecol. 2017, 26, 107–122. [Google Scholar] [CrossRef] [PubMed]
- Howard, D.J. Reinforcement: Origin, dynamics, and fate of an evolutionary hypothesis. In Hybrid Zones and the Evolutionary Process; Oxford University Press: Oxford, UK, 1993; pp. 46–69. [Google Scholar]
- Hopkins, R. Reinforcement in plants. New Phytol. 2013, 197, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Servedio, M.R.; Noor, M.A.F. The role of reinforcement in speciation: Theory and data. Annu. Rev. Ecol. Evol. Syst. 2003, 34, 339–364. [Google Scholar] [CrossRef]
- Butlin, R.K.; Ritchie, M.G. Pulling together or pulling apart: Hybridization in theory and practice. J. Evol. Biol. 2013, 26, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Dobzhansky, T. Genetic nature of species differences. Am. Nat. 1937, 71, 404–420. [Google Scholar]
- Dobzhansky, T. Genetics and the Origin of Species; Columbia University Press: New York, NY, USA, 1937; Volume 11. [Google Scholar]
- Dobzhansky, T. Speciation as a stage in evolutionary divergence. Am. Nat. 1940, 109, 235–238. [Google Scholar]
- Kay, K.M.; Schemske, D.W. Natural selection reinforces speciation in a radiation of neotropical rainforest plants. Evolution 2008, 62, 2628–2642. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, R.; Rausher, M.D. Identification of two genes causing reinforcement in the Texas wildflower Phlox drummondii. Nature 2011, 469, 411–414. [Google Scholar] [CrossRef] [PubMed]
- Fishman, L.; Wyatt, R. Pollinator-mediated competition, reproductive character displacement, and the evolution of selfing in Arenaria uniflora (Caryophyllaceae). Evolution 2018, 53, 1723–1733. [Google Scholar] [CrossRef] [PubMed]
- Rundle, H.D.; Schluter, D. Reinforcement of stickleback mate preferences: Sympatry breeds contempt. Evolution 1998, 52, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Gregorio, O.; Berdan, E.L.; Kozak, G.M.; Fuller, R.C. Reinforcement of male mate preferences in sympatric killifish species Lucania goodei and Lucania parva. Behav. Ecol. Sociobiol. 2012, 66, 1429–1436. [Google Scholar] [CrossRef]
- Moran, R.; Fuller, R. Male-driven reinforcement and cascade reinforcement in darters. bioRxiv 2017. [Google Scholar] [CrossRef]
- Rundle, H.D.; Nosil, P. Ecological speciation. Ecol. Lett. 2005, 8, 336–352. [Google Scholar] [CrossRef]
- Lukhtanov, V.A.; Kandul, N.P.; Plotkin, J.B.; Dantchenko, A.V.; Haig, D.; Pierce, N.E. Reinforcement of pre-zygotic isolation and karyotype evolution in Agrodiaetus butterflies. Nature 2005, 436, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Jaenike, J.; Dyer, K.A.; Cornish, C.; Minhas, M.S. Asymmetrical reinforcement and Wolbachia infection in Drosophila. PLoS Biol. 2006, 4. [Google Scholar] [CrossRef] [PubMed]
- Kronforst, M.R.; Young, L.G.; Gilbert, L.E. Reinforcement of mate preference among hybridizing Heliconius butterflies. J. Evol. Biol. 2007, 20, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Urbanelli, S.; Porretta, D. Evidence of reinforcement of premating isolation between two species of the genus Ochthebius (Coleoptera: Hydraenidae). Evolution 2008, 62, 1520–1527. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, H.C. Reproductive character displacement of female mate choice in the grey treefrog, Hyla chrysoscelis. Anim. Behav. 1994, 47, 959–969. [Google Scholar] [CrossRef]
- Pfennig, K.S. A test of alternative hypotheses for the evolution of reproductive isolation between spadefoot toads: Support for the reinforcement hypothesis. Evolution 2003, 57, 2842–2851. [Google Scholar] [CrossRef] [PubMed]
- Hoskin, C.J.; Higgie, M.; Mcdonald, K.R.; Moritz, C. Reinforcement drives rapid allopatric speciation. Nature 2005, 437, 1353. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, E.M.; Lemmon, A.R. Reinforcement in chorus frogs: Lifetime fitness estimates including intrinsic natural selection and sexual selection against hybrids. Evolution 2010, 64, 1748–1761. [Google Scholar] [CrossRef] [PubMed]
- Sætre, G.-P.; Moum, T.; Bureš, S.; Král, M.; Adamjan, M.; Moreno, J. A sexually selected character displacement in flycatchers reinforces premating isolation. Nature 1997, 387, 589. [Google Scholar] [CrossRef]
- Smadja, C.; Ganem, G. Asymmetrical reproductive character displacement in the house mouse. J. Evol. Biol. 2005, 18, 1485–1493. [Google Scholar] [CrossRef] [PubMed]
- Bímová, B.V.; MacHolán, M.; Baird, S.J.E.; Munclinger, P.; Dufková, P.; Laukaitis, C.M.; Karn, R.C.; Luzynski, K.; Tucker, P.K.; Piálek, J. Reinforcement selection acting on the European house mouse hybrid zone. Mol. Ecol. 2011, 20, 2403–2424. [Google Scholar] [CrossRef] [PubMed]
- Noor, M.A.F. Reinforcement and other consequences of sympatry. Heredity 1999, 83, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Servedio, M.R. The what and why of research on reinforcement. PLoS Biol. 2004, 2. [Google Scholar] [CrossRef] [PubMed]
- Ortíz-Barrientos, D.; Grealy, A.; Nosil, P. The genetics and ecology of reinforcement. Ann. N. Y. Acad. Sci. 2009, 1168, 156–182. [Google Scholar] [CrossRef] [PubMed]
- Butlin, R.K.; Smadja, C.M. Coupling, reinforcement, and speciation. Am. Nat. 2018, 191, 155–172. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R. Molecular signatures of natural selection. Annu. Rev. Genet. 2005, 39, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Oleksyk, T.K.; Smith, M.W.; O’Brien, S.J. Genome-wide scans for footprints of natural selection. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 185–205. [Google Scholar] [CrossRef] [PubMed]
- Cutter, A.D.; Payseur, B.A. Genomic signatures of selection at linked sites: Unifying the disparity among species. Nature 2013, 14, 262. [Google Scholar] [CrossRef] [PubMed]
- Vitti, J.J.; Grossman, S.R.; Sabeti, P.C. Detecting natural selection in genomic data. Annu. Rev. Genet. 2013, 47, 97–120. [Google Scholar] [CrossRef] [PubMed]
- Seehausen, O.; Butlin, R.K.; Keller, I.; Wagner, C.E.; Boughman, J.W.; Hohenlohe, P.A.; Peichel, C.L.; Sætre, G.-P. Genomics and the origin of species. Nat. Rev. Genet. 2014, 15, 176. [Google Scholar] [CrossRef] [PubMed]
- Ravinet, M.; Faria, R.; Butlin, R.K.; Galindo, J.; Bierne, N.; Rafajlović, M.; Noor, M.A.F.; Mehlig, B.; Westram, A.M. Interpreting the genomic landscape of speciation: A road map for finding barriers to gene flow. J. Evol. Biol. 2017, 30, 1450–1477. [Google Scholar] [CrossRef] [PubMed]
- Ortíz-Barrientos, D.; Reiland, J.; Hey, J.; Noor, M.A.F. Recombination and the divergence of hybridizing species. In Genetics of Mate Choice: From Sexual Selection to Sexual Isolation; Springer: Dordrecht, The Netherlands, 2002; Volume 116, pp. 167–178. [Google Scholar]
- Ortíz-Barrientos, D.; Counterman, B.A.; Noor, M.A.F. The genetics of speciation by reinforcement. PLoS Biol. 2004, 2, e416. [Google Scholar] [CrossRef] [PubMed]
- Sæther, S.A.; Sætre, G.-P.; Borge, T.; Wile, C.; Svedin, N.; Andersson, G.; Veen, T.; Haavie, J.; Servedio, M.R.; Bureš, S.; et al. Sex chromosome-linked species recognition and evolution of reproductive isolation in flycatchers. Science 2007, 318, 95. [Google Scholar] [CrossRef] [PubMed]
- Loire, E.; Tusso, S.; Caminade, P.; Severac, D.; Boursot, P.; Ganem, G.; Smadja, C.M. Do changes in gene expression contribute to sexual isolation and reinforcement in the house mouse? Mol. Ecol. 2017, 26, 5189–5202. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, R.; Levin, D.A.; Rausher, M.D. Molecular signatures of selection on reproductive character displacement of flower color in Phlox drummondii. Evolution 2012, 66, 469–485. [Google Scholar] [CrossRef] [PubMed]
- Smadja, C.M.; Loire, E.; Caminade, P.; Thoma, M.; Latour, Y.; Roux, C.; Thoss, M.; Penn, D.J. Seeking signatures of reinforcement at the genetic level: A hitchhiking mapping and candidate gene approach in the house mouse. Mol. Ecol. 2015, 24, 4222–4237. [Google Scholar] [CrossRef] [PubMed]
- Coyne, J.A.; Orr, H.A. Speciation; Sinauer: Sunderland, MA, USA, 2004. [Google Scholar]
- Brown, W.L.; Wilson, E.O. Character displacement. Syst. Biol. 1956, 5, 49–64. [Google Scholar] [CrossRef]
- Butlin, R. Speciation by reinforcement. Trends Ecol. Evol. 1987, 2, 8–13. [Google Scholar] [CrossRef]
- Noor, M.A.F. How often does sympatry affect sexual isolation in Drosophila? Am. Nat. 1997, 149, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, N.L.; Hudson, R.R.; Langley, C.H. The “hitchhiking effect” revisited. Genetics 1989, 123, 887–899. [Google Scholar] [PubMed]
- Barton, N.H. Genetic hitchhiking. Philos. Trans. R. Soc. B Biol. Sci. 2000, 355, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Pavlidis, P.; Alachiotis, N. A survey of methods and tools to detect recent and strong positive selection. J. Biol. Res. 2017, 24, 7. [Google Scholar] [CrossRef] [PubMed]
- Booker, T.R.; Jackson, B.C.; Keightley, P.D. Detecting positive selection in the genome. BMC Biol. 2017, 15, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pool, J.E.; Hellmann, I.; Jensen, J.D.J.; Nielsen, R. Population genetic inference from genomic sequence variation. Genome Res. 2010, 20, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Stephan, W.; Wiehe, T.H.E.; Lenz, M.W. The effect of strongly selected substitutions on neutral polymorphism: Analytical results based on diffusion theory. Theor. Popul. Biol. 1992, 41, 237–254. [Google Scholar] [CrossRef]
- Sabeti, P.C.; Reich, D.E.; Higgins, J.M.; Levine, H.Z.; Richter, D.J.; Schaffner, S.F.; Gabriel, S.B.; Platko, J.V.; Patterson, N.J.; McDonald, G.J.; et al. Detecting recent positive selection in the human genome from haplotype structure. Nature 2002, 419, 832. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Nielsen, R. Linkage disequilibrium as a signature of selective sweeps. Genetics 2004, 167, 1513–1524. [Google Scholar] [CrossRef] [PubMed]
- Jensen, J.D.; Thornton, K.R.; Bustamante, C.D.; Aquadro, C.F. On the utility of linkage disequilibrium as a statistic for identifying targets of positive selection in nonequilibrium populations. Genetics 2007, 176, 2371–2379. [Google Scholar] [CrossRef] [PubMed]
- Pfaffelhuber, P.; Lehnert, A.; Stephan, W. Linkage disequilibrium under genetic hitchhiking in finite populations. Genetics 2008, 179, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Fay, J.C.; Wu, C.I. Hitchhiking under positive Darwinian selection. Genetics 2000, 155, 1405–1413. [Google Scholar] [PubMed]
- Braverman, J.M.; Hudson, R.R.; Kaplan, N.L.; Langley, C.H.; Stephan, W. The hitchhiking effect on the site frequency spectrum of DNA polymorphisms. Genetics 1995, 140, 783–796. [Google Scholar] [PubMed]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [PubMed]
- Voight, B.F.; Kudaravalli, S.; Wen, X.; Pritchard, J.K. A map of recent positive selection in the human genome. PLoS Biol. 2006, 4, e72. [Google Scholar] [CrossRef]
- Degiorgio, M.; Huber, C.D.; Hubisz, M.J.; Hellmann, I.; Nielsen, R. SweepFinder2: Increased sensitivity, robustness and flexibility. Bioinformatics 2016, 32, 1895–1897. [Google Scholar] [CrossRef] [PubMed]
- Pavlidis, P.; Zivkovic, D.; Stamatakis, A.; Alachiotis, N. SweeD: Likelihood-based detection of selective sweeps in thousands of genomes. Mol. Biol. Evol. 2013, 30, 2224–2234. [Google Scholar] [CrossRef] [PubMed]
- Alachiotis, N.; Stamatakis, A.; Pavlidis, P. OmegaPlus: A scalable tool for rapid detection of selective sweeps in whole-genome datasets. Bioinformatics 2012, 28, 2274–2275. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, R.; Rausher, M.D. The cost of reinforcement: Selection on flower color in allopatric populations of Phlox drummondii. Am. Nat. 2014, 183, 693–710. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Comeault, A.A.; Venkat, A.; Matute, D.R. Correlated evolution of male and female reproductive traits drive a cascading effect of reinforcement in Drosophila yakuba. Proc. Biol. Sci. 2016, 283, 23–48. [Google Scholar] [CrossRef] [PubMed]
- Holsinger, K.E.; Weir, B.S. Genetics in geographically structured populations: Defining, estimating and interpreting FST. Nat. Rev. Genet. 2009, 10, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Meirmans, P.G.; Hedrick, P.W. Assessing population structure: FST and related measures. Mol. Ecol. 2011, 11, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Cruickshank, T.E.; Hahn, M.W. Reanalysis suggests that genomic islands of speciation are due to reduced diversity, not reduced gene flow. Mol. Ecol. 2014, 23, 3133–3157. [Google Scholar] [CrossRef] [PubMed]
- Stuart, Y.E.; Inkpen, S.A.; Hopkins, R.; Bolnick, D.I. Character displacement is a pattern: So, what causes it? Biol. J. Linn. Soc. 2017, 121, 711–715. [Google Scholar] [CrossRef]
- Hopkins, R.; Rausher, M.D. Pollinator-mediated selection on flower color allele drives reinforcement. Science 2012, 335, 1090–1092. [Google Scholar] [CrossRef] [PubMed]
- Kauer, M.O.; Dieringer, D.; Schlötterer, C. A microsatellite variability screen for positive selection associated with the “out of Africa” habitat expansion of Drosophila melanogaster. Genetics 2003, 165, 1137–1148. [Google Scholar] [PubMed]
- Schlötterer, C.; Dieringer, D. A novel test statistic for the identification of local selective sweeps based on microsatellite gene diversity. In Selective Sweep; Springer: New York, NY, USA, 2005; pp. 55–64. [Google Scholar]
- Pfennig, K.S.; Pfennig, D.W. Character displacement: Ecological and reproductive responses to a common evolutionary problem. Q. Rev. Biol. 2009, 84, 253–276. [Google Scholar] [CrossRef] [PubMed]
- Schluter, D.; Mcphail, J.D. Ecological character displacement and speciation in sticklebacks. Am. Nat. 2010, 140, 85–108. [Google Scholar] [CrossRef] [PubMed]
- Schluter, D. Ecological character displacement in adaptive radiation. Am. Nat. 2000, 156, S4–S16. [Google Scholar] [CrossRef]
- Losos, J.B. Ecological character displacement and the study of adaptation. Proc. Natl. Acad. Sci. USA 2000, 97, 5693–5695. [Google Scholar] [CrossRef] [PubMed]
- Otte, D.; Endler, J.A. Speciation and Its Consequences; Sinauer: Sunderland, MA, USA, 1989. [Google Scholar]
- Kawecki, T.J.; Ebert, D. Conceptual issues in local adaptation. Ecol. Lett. 2004, 7, 1225–1241. [Google Scholar] [CrossRef]
- Hereford, J. A quantitative survey of local adaptation and fitness trade-offs. Am. Nat. 2009, 173, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Tigano, A.; Friesen, V.L. Genomics of local adaptation with gene flow. Mol. Ecol. 2016, 25, 2144–2164. [Google Scholar] [CrossRef] [PubMed]
- Tiffin, P.; Ross-ibarra, J. Advances and limits of using population genetics to understand local adaptation. Trends Ecol. Evol. 2014, 29, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Savolainen, O.; Lascoux, M.; Merilä, J. Ecological genomics of local adaptation. Nat. Rev. Genet. 2013, 14, 807–820. [Google Scholar] [CrossRef] [PubMed]
- Coop, G.; Witonsky, D.; Di Rienzo, A.; Pritchard, J.K. Using environmental correlations to identify loci underlying local adaptation. Genetics 2010, 185, 1411–1423. [Google Scholar] [CrossRef] [PubMed]
- Joost, S.; Bonin, A.; Bruford, M.W.; Després, L.; Conord, C.; Erhardt, G.; Taberlet, P. A spatial analysis method (SAM) to detect candidate loci for selection: Towards a landscape genomics approach to adaptation. Mol. Ecol. 2007, 16, 3955–3969. [Google Scholar] [CrossRef] [PubMed]
- Poncet, B.N.; Herrmann, D.; Gugerli, F.; Taberlet, P.; Holderegger, R.; Gielly, L.; Rioux, D.; Thuiller, W.; Aubert, S.; Manel, S. Tracking genes of ecological relevance using a genome scan in two independent regional population samples of Arabis alpina. Mol. Ecol. 2010, 19, 2896–2907. [Google Scholar] [CrossRef] [PubMed]
- Günther, T.; Coop, G. Robust identification of local adaptation from allele frequencies. Genetics 2013, 195, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Frichot, E.; Schoville, S.D.; Bouchard, G.; François, O. Testing for associations between loci and environmental gradients using latent factor mixed models. Mol. Biol. Evol. 2013, 30, 1687–1699. [Google Scholar] [CrossRef] [PubMed]
- Servedio, M.R. The evolution of premating isolation: Local adaptation and natural and sexual selection against hybrids. Evolution 2004, 58, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, M. Reinforcement and divergence under assortative mating. Proc. R. Soc. Lond. B Biol. Sci. 2000, 267, 1649–1655. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.; Stebbins, G.L. Hybridization as an evolutionary stimulus. Evolution 1954, 8, 378–388. [Google Scholar] [CrossRef]
- Pease, J.B.; Haak, D.C.; Hahn, M.W.; Moyle, L.C. Phylogenomics reveals three sources of adaptive variation during a rapid radiation. PLoS Biol. 2016, 14, e1002379. [Google Scholar] [CrossRef] [PubMed]
- Geraldes, A.; Farzaneh, N.; Grassa, C.J.; Mckown, A.D.; Guy, R.D.; Mansfield, S.D.; Douglas, C.J.; Cronk, Q.C.B. Landscape genomics of Populus trichocarpa: The role of hybridization, limited gene flow, and natural selection in shaping patterns of population structure. Evolution 2014, 68, 3260–3280. [Google Scholar] [CrossRef] [PubMed]
- Turissini, D.A.; Matute, D.R. Fine scale mapping of genomic introgressions within the Drosophila yakuba clade. PLoS Genet. 2017, 13. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Skepticism towards Santa Rosalia, or why are there so few kinds of animals? Evolution 1981, 35, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Kopp, M.; Servedio, M.R.; Mendelson, T.C.; Safran, R.J.; Rodríguez, R.L.; Hauber, M.E.; Scordato, E.C.; Symes, L.B.; Balakrishnan, C.N.; Zonana, D.M.; et al. Mechanisms of assortative mating in speciation with gene flow: Connecting theory and empirical research. Am. Nat. 2017, 191, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Ortíz-Barrientos, D.; Noor, M.A.F. Evidence for a one-allele assortative mating locus. Science 2005, 310, 1467. [Google Scholar] [CrossRef] [PubMed]
- Hermisson, J.; Pennings, P.S. Soft sweeps and beyond: Understanding the patterns and probabilities of selection footprints under rapid adaptation. Methods Ecol. Evol. 2017, 8, 700–716. [Google Scholar] [CrossRef]
- Servedio, M.R. The role of linkage disequilibrium in the evolution of premating isolation. Heredity 2009, 102, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Templeton, A.R. Mechanisms of speciation—A population genetic approach. Ann. Rev. Eeol. Syst. 1981, 12, 23–48. [Google Scholar] [CrossRef]
- Liou, L.W.; Price, T.D. Speciation by reinforcement of premating isolation. Evolution 1994, 48, 1451–1459. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, M.; Servedio, M.R. The reinforcement of mating preferences on an island. Genetics 1999, 151, 865–884. [Google Scholar] [PubMed]
- Ortíz-Barrientos, D.; Engelstädter, J.; Rieseberg, L.H. Recombination rate evolution and the origin of species. Trends Ecol. Evol. 2016, 31, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Stapley, J.; Feulner, P.G.D.; Johnston, S.E.; Santure, A.W.; Smadja, C.M. Variation in recombination frequency and distribution across eukaryotes: Patterns and processes. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372. [Google Scholar] [CrossRef] [PubMed]
- Navarro, A.; Barton, N.H. Accumulating postzygotic isolation genes in parapatry: A new twist on chromosomal speciation. Evolution 2003, 57, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Noor, M.A.F.; Grams, K.L.; Bertucci, L.A.; Reiland, J. Chromosomal inversions and the reproductive isolation of species. Proc. Natl. Acad. Sci. USA 2001, 98, 12084–12088. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, A.R.; Kirkpatrick, M. Reinforcement and the genetics of hybrid incompatibilities. Genetics 2006, 173, 1145–1155. [Google Scholar] [CrossRef] [PubMed]
- Noor, M.A.F.; Bennett, S.M. Islands of speciation or mirages in the desert? Examining the role of restricted recombination in maintaining species. Heredity 2009, 103, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Corbett-detig, R.B.; Hartl, D.L. Population genomics of inversion polymorphisms in Drosophila melanogaster. PLoS Genet. 2012, 8, e1003056. [Google Scholar] [CrossRef] [PubMed]
- Mcgaugh, S.E.; Noor, M.A.F. Genomic impacts of chromosomal inversions in parapatric Drosophila species. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, R.F.; Rousset, F.; Kirkpatrick, M. Coalescent patterns for chromosomal inversions in divergent populations. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Bolnick, D.I.; Fitzpatrick, B.M. Sympatric speciation: Models and empirical evidence. Annu. Rev. Ecol. Evol. Syst. 2007, 38, 459–487. [Google Scholar] [CrossRef]
- Wu, C. The genic view of the process of speciation. J. Evol. Biol. 2001, 14, 851–865. [Google Scholar] [CrossRef]
- Servedio, M.R.; Kirkpatrick, M. The effects of gene flow on reinforcement. Evolution 1997, 51, 1764–1772. [Google Scholar] [CrossRef] [PubMed]
- Kisel, Y.; Barraclough, T.G. Speciation has a spatial scale that depends on levels of gene flow. Am. Nat. 2018, 175, 316–334. [Google Scholar] [CrossRef] [PubMed]
- Roda, F.; Hopkins, R. Correlated evolution of self and interspecific incompatibly across the range of a Texas wildflower. bioRxiv 2017. [Google Scholar] [CrossRef]
- Kulathinal, R.J.; Stevison, L.S.; Noor, M.A.F. The genomics of speciation in Drosophila: Diversity, divergence, and introgression estimated using low-coverage genome sequencing. PLoS Genet. 2009, 5, e1000550. [Google Scholar] [CrossRef] [PubMed]
- Roda, F.; Mendes, F.K.; Hahn, M.W.; Hopkins, R. Genomic evidence of gene flow during reinforcement in Texas Phlox. Mol. Ecol. 2017, 26, 2317–2330. [Google Scholar] [CrossRef] [PubMed]
- Wen, D.; Yu, Y.; Nakhleh, L. Bayesian inference of reticulate phylogenies under the multispecies network coalescent. PLoS Genet. 2016. [Google Scholar] [CrossRef] [PubMed]
- Payseur, B.A.; Rieseberg, L.H. A genomic perspective on hybridization and speciation. Mol. Ecol. 2016, 25, 2337–2360. [Google Scholar] [CrossRef] [PubMed]
- Abbott, R.J.; Barton, N.H.; Good, J. Genomics of hybridization and its evolutionary consequences. Mol. Ecol. 2016, 25, 2325–2332. [Google Scholar] [CrossRef] [PubMed]
- Gompert, Z.; Buerkle, C.A. Analyses of genetic ancestry enable key insights for molecular ecology. Mol. Ecol. 2013, 22, 5278–5294. [Google Scholar] [CrossRef] [PubMed]
- Hey, J. Recent advances in assessing gene flow between diverging populations and species. Curr. Opin. Genet. Dev. 2006, 16, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Green, R.E.; Krause, J.; Briggs, A.W.; Maricic, T.; Stenzel, U.; Kircher, M.; Patterson, N.; Li, H.; Zhai, W.; Fritz, M.H.; et al. A draft sequence of the Neandertal genome. Science 2010, 328, 710–722. [Google Scholar] [CrossRef] [PubMed]
- Reich, D.; Thangaraj, K.; Patterson, N.; Price, A.L.; Singh, L. Reconstructing Indian population history. Nature 2009, 461, 489. [Google Scholar] [CrossRef] [PubMed]
- Solís-Lemus, C.; Ané, C. Inferring phylogenetic networks with maximum pseudolikelihood under incomplete lineage sorting. PLoS Genet. 2016, 12, e1005896. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Dupanloup, I.; Huerta-Sánchez, E.; Sousa, V.C.; Foll, M. Robust demographic inference from genomic and SNP data. PLoS Genet. 2013, 9, e1003905. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Estoup, A.; Cornuet, J.M. Bayesian analysis of an admixture model with mutations and arbitrarily linked markers. Genetics 2005, 169, 1727–1738. [Google Scholar] [CrossRef] [PubMed]
- Beaumont, M.A.; Zhang, W.; Balding, D.J. Approximate Bayesian computation in population genetics. Genetics 2002, 162, 2025–2035. [Google Scholar] [PubMed]
- Gutenkunst, R.N.; Hernandez, R.D.; Williamson, S.H.; Bustamante, C.D. Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genet. 2009, 5, e1000695. [Google Scholar] [CrossRef] [PubMed]
- Nosil, P.; Funk, D.J.; Ortíz-Barrientos, D. Divergent selection and heterogeneous genomic divergence. Mol. Ecol. 2009, 18, 375–402. [Google Scholar] [CrossRef] [PubMed]
- Littlejohn, M.J. Reproductive isolation: A critical review. In Evolution and Speciation; Atchley, W.R., Woodruff, D.S., Eds.; Cambridge University Press: Cambridge, UK, 1981; p. 298334. [Google Scholar]
- Hoskin, C.J.; Higgie, M. Speciation via species interactions: The divergence of mating traits within species. Ecol. Lett. 2010, 13, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Nosil, P.; Crespi, B.J.; Sandoval, C.P. Reproductive isolation driven by the combined effects of ecological adaptation and reinforcement. Proc. R. Soc. Lond. B 2003, 270, 1911–1918. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, D.P.; Rundle, H.D.; Dyer, K.A. Patterns of reproductive isolation in the Drosophila subquinaria complex: Can reinforced premating isolation cascade to other species? Curr. Zool. 2017, 62, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Kozak, G.M.; Roland, G.; Rankhorn, C.; Falater, A.; Emma, L.; Fuller, R.C.; Kozak, G.M.; Roland, G.; Rankhorn, C.; Falater, A.; et al. Behavioral isolation due to cascade reinforcement in Lucania killifish. Am. Nat. 2015, 185, 491–506. [Google Scholar] [CrossRef] [PubMed]
- Dyer, K.A.; White, B.E.; Sztepanacz, J.L.; Bewick, E.R.; Rundle, H.D. Reproductive character displacement of epicuticular compounds and their contributon to mate choice in Drosophila subquinaria and Drosophila recens. Evolution 2014, 68, 1163–1175. [Google Scholar] [CrossRef] [PubMed]
- Higgie, M.; Blows, M.W. Also under sexual selection? Am. Nat. 2007, 170, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, E.M. Diversification of conspecific signals in sympatry: Geographic overlap drives multidimensional reproductive character displacement in frogs. Evolution 2009, 63, 1155–1170. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garner, A.G.; Goulet, B.E.; Farnitano, M.C.; Molina-Henao, Y.F.; Hopkins, R. Genomic Signatures of Reinforcement. Genes 2018, 9, 191. https://doi.org/10.3390/genes9040191
Garner AG, Goulet BE, Farnitano MC, Molina-Henao YF, Hopkins R. Genomic Signatures of Reinforcement. Genes. 2018; 9(4):191. https://doi.org/10.3390/genes9040191
Chicago/Turabian StyleGarner, Austin G., Benjamin E. Goulet, Matthew C. Farnitano, Y. Franchesco Molina-Henao, and Robin Hopkins. 2018. "Genomic Signatures of Reinforcement" Genes 9, no. 4: 191. https://doi.org/10.3390/genes9040191
APA StyleGarner, A. G., Goulet, B. E., Farnitano, M. C., Molina-Henao, Y. F., & Hopkins, R. (2018). Genomic Signatures of Reinforcement. Genes, 9(4), 191. https://doi.org/10.3390/genes9040191