Evolutionary Mechanisms of Varying Chromosome Numbers in the Radiation of Erebia Butterflies


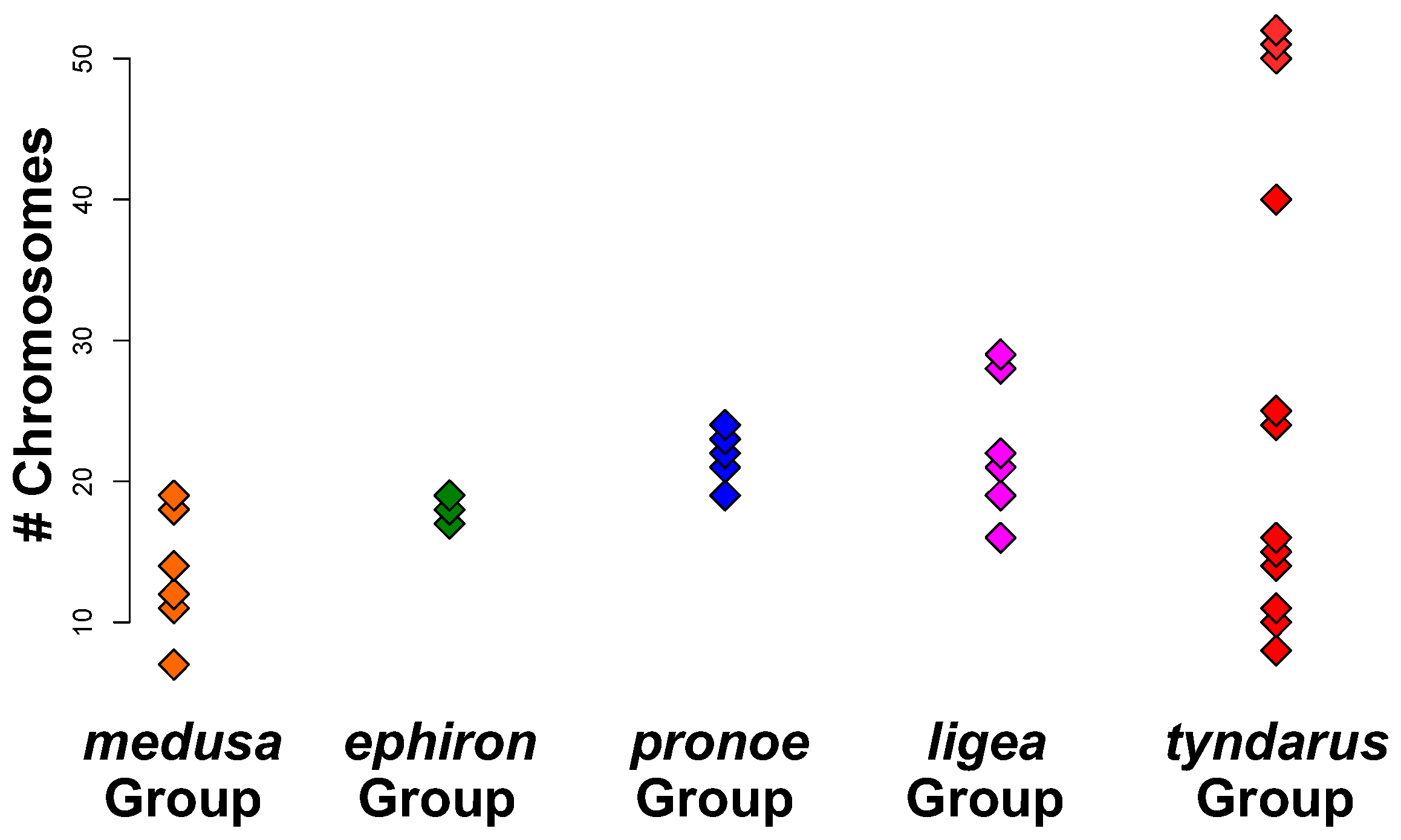{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Phylogenetic Analyses
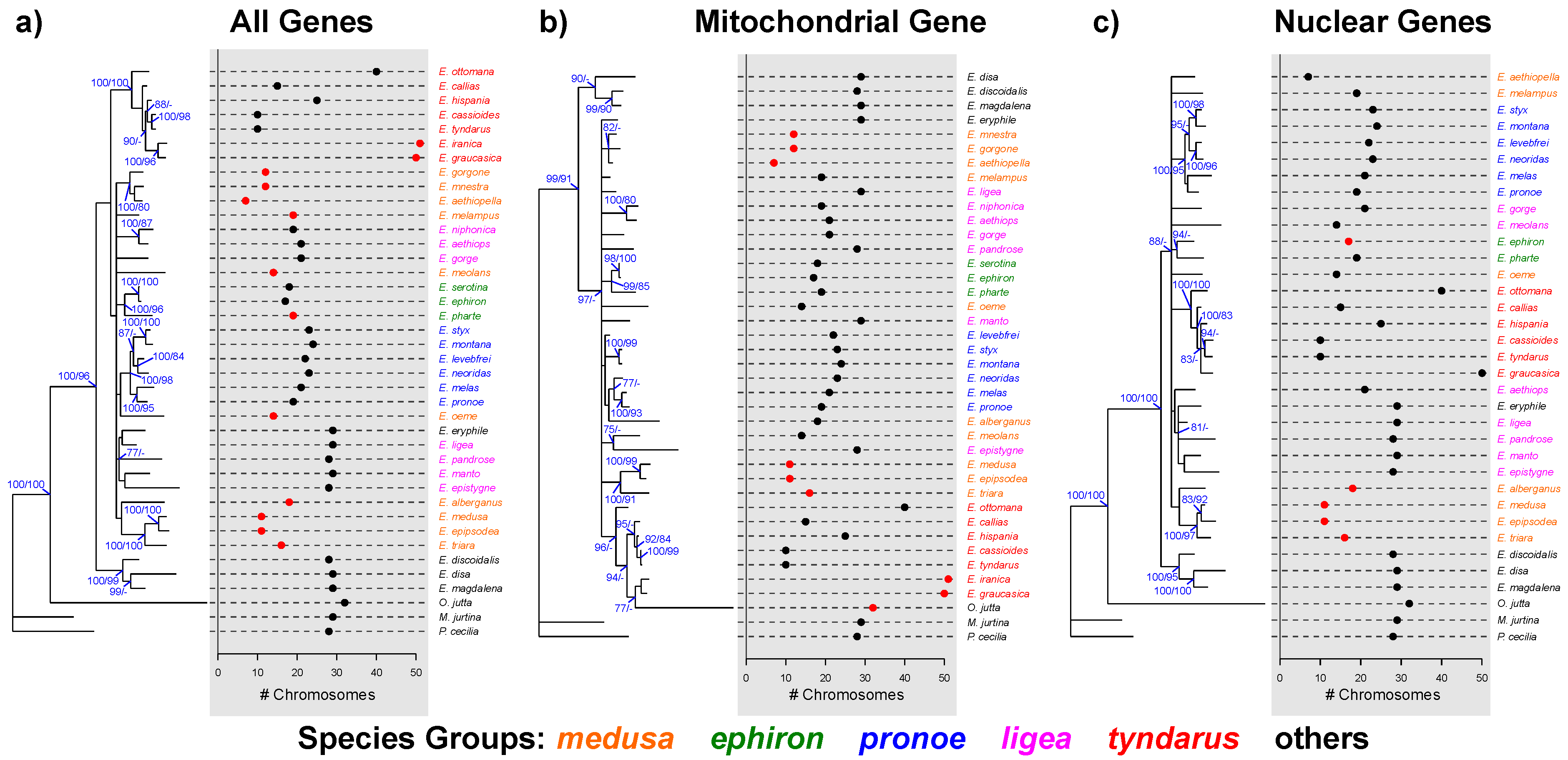
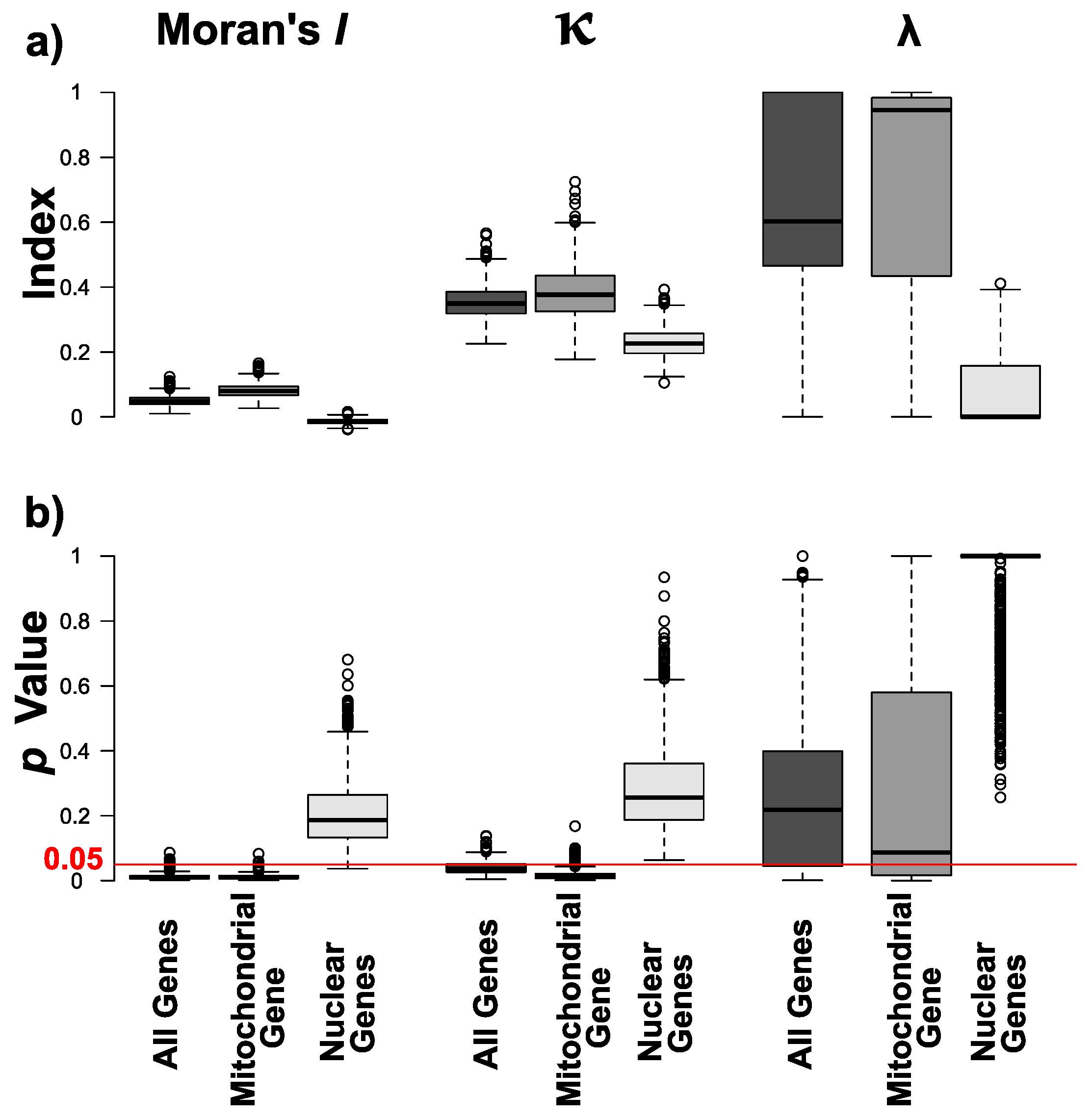
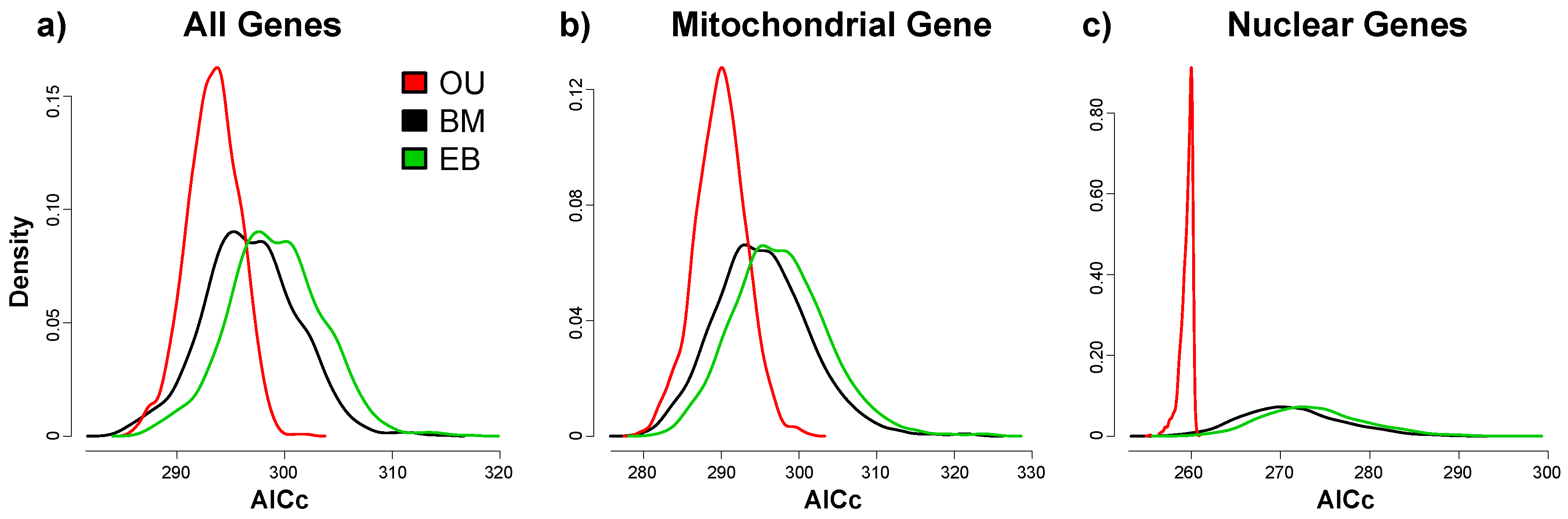
3. Results
3.1. Phylogenetic Reconstruction
3.2. Phylogenetic Signals
4. Discussion
Supplementary Materials
Acknowledgments
Conflict of interest
References
- Hendry, A.P. Ecological speciation! Or the lack thereof? Can. J. Fish. Aquat. Sci. 2009, 66, 1383–1398. [Google Scholar] [CrossRef]
- Nosil, P. Ecological Speciation; Oxford University Press: Oxford, UK, 2012. [Google Scholar]
- Seehausen, O.; Butlin, R.K.; Keller, I.; Wagner, C.E.; Boughman, J.W.; Hohenlohe, P.A.; Peichel, C.L.; Saetre, G.-P.; Bank, C.; Brännström, Å.; et al. Genomics and the origin of species. Nat. Rev. Genet. 2014, 15, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Nosil, P.; Harmon, L.J.; Seehausen, O. Ecological explanations for (incomplete) speciation. Trends Ecol. Evol. 2009, 24, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Butlin, R.K. Recombination and speciation. Mol. Ecol. 2005, 14, 2621–2635. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, M.; Barton, N.J. Chromosome inversions, local adaptation and speciation. Genetics 2006, 173, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Via, S. Natural selection in action during speciation. Proc. Natl. Acad. Sci. USA 2009, 106 (Suppl. 1), 9939–9946. [Google Scholar] [CrossRef] [PubMed]
- Kulmuni, J.; Westram, A.M. Intrinsic incompatibilities evolving as a by-product of divergent ecological selection: Considering them in empirical studies on divergence with gene flow. Mol. Ecol. 2017, 26, 3093–3103. [Google Scholar] [CrossRef] [PubMed]
- Martinez, P.A.; Zurano, J.P.; Amado, T.F.; Penone, C.; Betancur-R, R.; Bidau, C.J.; Jacobina, U.P. Chromosomal diversity in tropical reef fishes is related to body size and depth range. Mol. Phylogenet. Evol. 2015, 93, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Hooper, D.M.; Price, T.D. Chromosomal inversion differences correlate with range overlap in passerine birds. Nat. Ecol. Evol. 2017, 1, 1526–1534. [Google Scholar] [CrossRef] [PubMed]
- White, M.J.D. Modes of Speciation; W.H. Freeman: San Francisco, CA, USA, 1978. [Google Scholar]
- King, M. Species Evolution; Cambridge University Press: Cambridge, UK, 1995. [Google Scholar]
- Faria, R.; Navarro, A. Chromosomal speciation revisited: Rearranging theory with pieces of evidence. Trends Ecol. Evol. 2010, 25, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Dion-Côté, A.-M.; Barbash, D.A. Beyond speciation genes: An overview of genome stability in evolution and speciation. Curr. Opin. Genet. Dev. 2017, 47, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Cohuet, A.; Dia, I.; Simard, F.; Raymond, M.; Rousset, F.; Antonio-Nkondjio, C.; Awono-Ambene, P.H.; Wondji, C.S.; Fontenille, D. Gene flow between chromosomal forms of the malaria vector Anopheles funestus in Cameroon, Central Africa, and its relevance in malaria fighting. Genetics 2005, 169, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Polyakov, A.V.; White, T.A.; Jones, R.M.; Borodin, P.M.; Searle, J.B. Natural hybridization between extremely divergent chromosomal races of the common shrew (Sorex araneus, Soricidae, Soricomorpha): Hybrid zone in European Russia. J. Evol. Biol. 2011, 24, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Descimon, H.; Mallet, J. Bad Species. In Ecology of Butterflies in Europe; Settle, R., Shreeve, T., Konvicka, M., van Dyck, H., Eds.; Cambridge University Press: Cambridge, UK, 2009. [Google Scholar]
- Mallet, J.; Besansky, N.; Hahn, M.W. How reticulated are species? Bioessays 2016, 38, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Charron, G.; Leducq, J.-B.; Landry, C.R. Chromosomal variation segregates within incipient species and correlates with reproductive isolation. Mol. Ecol. 2014, 23, 4362–4372. [Google Scholar] [CrossRef] [PubMed]
- Potter, S.; Bragg, J.G.; Blom, M.P.K.; Deakin, J.E.; Kirkpatrick, M.; Eldridge, M.D.B.; Moritz, C. Chromosomal speciation in the genomics era: Disentangling phylogenetic evolution of rock-wallabies. Front. Genet. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Vershinina, A.O.; Lukhtanov, V.A. Evolutionary mechanisms of runaway chromosome number change in Agrodiaetus butterflies. Sci. Rep. 2017, 7, 8199. [Google Scholar] [CrossRef] [PubMed]
- Robinson, R. Lepidoptera Genetics; Pergamon Press: Oxford, UK, 1971. [Google Scholar]
- Lukhtanov, V.A.; Kandul, N.P.; Plotkin, J.B.; Dantchenko, A.V.; Haig, D.; Pierce, N.E. Reinforcement of pre-zygotic isolation and karyotype evolution in Agrodiaetus butterflies. Nature 2005, 436, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Talavera, G.; Lukhtanov, V.A.; Rieppel, L.; Pierce, N.E.; Vila, R. In the shadow of phylogenetic uncertainty: The recent diversification of Lysandra butterflies through chromosomal change. Mol. Phylogenet. Evol. 2013, 69, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Lorkovic, Z. Some peculiarities of spatially and sexually restricted gene exchange in the Erebia tyndarus group. Cold Spring Harb. Symp. Quant. Biol. 1958, 23, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Peña, C.; Witthauer, H.; Kleckova, I.; Fric, Z. Adaptive radiations in butterflies: Evolutionary history of the genus Erebia (Nymphalidae: Satyrinae). Biol. J. Linn. Soc. 2015, 116, 449–467. [Google Scholar] [CrossRef]
- Martin, J.F.; Gilles, A.; Lortscher, M.; Descimon, H. Phylogenetics and differentiation among the western taxa of the Erebia tyndarus group (Lepidoptera: Nymphalidae). Biol. J. Linn. Soc. 2002, 75, 319–332. [Google Scholar] [CrossRef]
- Albre, J.; Gers, C.; Legal, L. Molecular phylogeny of the Erebia tyndarus (Lepidoptera, Rhopalocera, Nymphalidae, Satyrinae) species group combining CoxII and ND5 mitochondrial genes: A case study of a recent radiation. Mol. Phylogenet. Evol. 2008, 47, 196–210. [Google Scholar] [CrossRef] [PubMed]
- Gratton, P.; Trucchi, E.; Trasatti, A.; Riccarducci, G.; Marta, S.; Allegrucci, G.; Cesaroni, D.; Sbordoni, V. Testing classical species properties with contemporary data: How “Bad Species” in the brassy ringlets (Erebia tyndarus complex, Lepidoptera) turned good. Syst. Biol. 2016, 65, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Lorkovic, Z. Die Speziationsstufen in der Erebia tyndarus Gruppe. Biol. Glas. 1957, 10, 61–110. [Google Scholar]
- Kleckova, I.; Konvicka, M.; Klecka, J. Thermoregulation and microhabitat use in mountain butterflies of the genus Erebia: Importance of fine-scale habitat heterogeneity. J. Therm. Biol. 2014, 41, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Maximum-likelihood estimation of evolutionary trees from continuous characters. Am. J. Hum. Genet. 1973, 25, 471–492. [Google Scholar] [PubMed]
- Harmon, L.J.; Losos, J.B.; Jonathan Davies, T.; Gillespie, R.G.; Gittleman, J.L.; Bryan Jennings, W.; Kozak, K.H.; McPeek, M.A.; Moreno-Roark, F.; Near, T.J.; et al. Early bursts of body size and shape evolution are rare in comparative data. Evolution 2010, 64, 2385–2396. [Google Scholar] [CrossRef] [PubMed]
- Cressler, C.E.; Butler, M.A.; King, A.A. Detecting adaptive evolution in phylogenetic comparative analysis using the Ornstein-Uhlenbeck model. Syst. Biol. 2015, 64, 953–968. [Google Scholar] [CrossRef] [PubMed]
- Albre, J.; Gers, C.; Legal, L. Taxonomic notes on the species of the Erebia tyndarus group (Lepidoptera, Nymphalidae, Satyrinae). Lépidoptères 2008, 17, 12–28. [Google Scholar]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Suchard, M.A.; Xie, D.; Drummond, A.J. Tracer v1.6. Available online: http://tree.bio.ed.ac.uk/software/tracer (accessed on 26 October 2017).
- Keck, F.; Rimet, F.; Bouchez, A.; Franc, A. Phylosignal: An R package to measure, test, and explore the phylogenetic signal. Ecol. Evol. 2016, 6, 2774–2780. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R Foundation for Statistical Computing; R 3.3.1; R Foundation for Statistical Computing: Vienna, Austria, 2016. [Google Scholar]
- Münkemüller, T.; Lavergne, S.; Bzeznik, B.; Dray, S.; Jombart, T.; Schiffers, K.; Thuiller, W. How to measure and test phylogenetic signal. Methods Ecol. Evol. 2012, 3, 743–756. [Google Scholar] [CrossRef]
- Diniz-Filho, J.A.F.; Santos, T.; Rangel, T.F.; Bini, L.M. A comparison of metrics for estimating phylogenetic signal under alternative evolutionary models. Genet. Mol. Biol. 2012, 35, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Gustavo, P.; Werner, G.; Caterina, P. SensiPhy: Sensitivity Analysis for Comparative Methods. Available online: https://github.com/paternogbc/sensiPhy (accessed on 6 December 2017).
- Clavel, J.; Escarguel, G.; Merceron, G. mv morph: An R package for fitting multivariate evolutionary models to morphometric data. Methods Ecol. Evol. 2015, 6, 1311–1319. [Google Scholar] [CrossRef]
- Sites, J.W., Jr.; Moritz, C. Chromosomal evolution and speciation revisited. Syst. Biol. 1987, 36, 153–174. [Google Scholar] [CrossRef]
- Kandul, N.P.; Lukhtanov, V.A.; Pierce, N.E. Karyotypic diversity and speciation in Agrodiaetus butterflies. Evolution 2007, 61, 546–559. [Google Scholar] [CrossRef] [PubMed]
- Toews, D.P.L.; Brelsford, A. The biogeography of mitochondrial and nuclear discordance in animals. Mol. Ecol. 2012, 21, 3907–3930. [Google Scholar] [CrossRef] [PubMed]
- Pardo-Diaz, C.; Salazar, C.; Baxter, S.W.; Merot, C.; Figueiredo-Ready, W.; Joron, M.; Mcmillan, W.O.; Jiggins, C.D. Adaptive introgression across species boundaries in Heliconius butterflies. PLoS Genet. 2012, 8, e1002752. [Google Scholar] [CrossRef] [PubMed]
- Narita, S.; Nomura, M.; Kato, Y.; Fukatsu, T. Genetic structure of sibling butterfly species affected by Wolbachia infection sweep: Evolutionary and biogeographical implications. Mol. Ecol. 2006, 15, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Gompert, Z.; Forister, M.L.; Fordyce, J.A.; Nice, C.C. Widespread mito-nuclear discordance with evidence for introgressive hybridization and selective sweeps in Lycaeides. Mol. Ecol. 2008, 17, 5231–5244. [Google Scholar] [CrossRef] [PubMed]
- Davies, T.J.; Kraft, N.J.B.; Salamin, N.; Wolkovich, E.M. Incompletely resolved phylogenetic trees inflate estimates of phylogenetic conservatism. Ecology 2012, 93, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Cooper, N.; Thomas, G.H.; Venditti, C.; Meade, A.; Freckleton, R.P. A cautionary note on the use of Ornstein Uhlenbeck models in macroevolutionary studies. Biol. J. Linn. Soc. 2016, 118, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Kandul, N.P.; Lukhtanov, V.A.; Dantchenko, A.V.; Coleman, J.W.S.; Sekercioglu, C.H.; Haig, D.; Pierce, N.E. Phylogeny of Agrodiaetus Hübner 1822 (Lepidoptera: Lycaenidae) inferred from mtDNA sequences of COI and COII and nuclear sequences of EF1α: Karyotype diversification and species radiation. Syst. Biol. 2004, 53, 278–298. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.A.; King, A.A. Phylogenetic comparative analysis: A modeling approach for adaptive evolution. Am. Nat. 2004, 164, 683–695. [Google Scholar] [CrossRef]
- Molina-Venegas, R.; Rodríguez, M.Á. Revisiting phylogenetic signal; strong or negligible impacts of polytomies and branch length information? BMC Evol. Biol. 2017, 17, 53. [Google Scholar] [CrossRef] [PubMed]
- Saura, A.; Von Schoultz, B.; Saura, A.O.; Brown, K.S.J. Chromosome evolution in neotropical butterflies. Hereditas 2013, 150, 26–37. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lucek, K. Evolutionary Mechanisms of Varying Chromosome Numbers in the Radiation of Erebia Butterflies. Genes 2018, 9, 166. https://doi.org/10.3390/genes9030166
Lucek K. Evolutionary Mechanisms of Varying Chromosome Numbers in the Radiation of Erebia Butterflies. Genes. 2018; 9(3):166. https://doi.org/10.3390/genes9030166
Chicago/Turabian StyleLucek, Kay. 2018. "Evolutionary Mechanisms of Varying Chromosome Numbers in the Radiation of Erebia Butterflies" Genes 9, no. 3: 166. https://doi.org/10.3390/genes9030166
APA StyleLucek, K. (2018). Evolutionary Mechanisms of Varying Chromosome Numbers in the Radiation of Erebia Butterflies. Genes, 9(3), 166. https://doi.org/10.3390/genes9030166