Genome-Wide Transcriptome Analysis Reveals the Comprehensive Response of Two Susceptible Poplar Sections to Marssonina brunnea Infection


Abstract
1. Introduction
2. Materials and Methods
2.1. Host and Pathogen
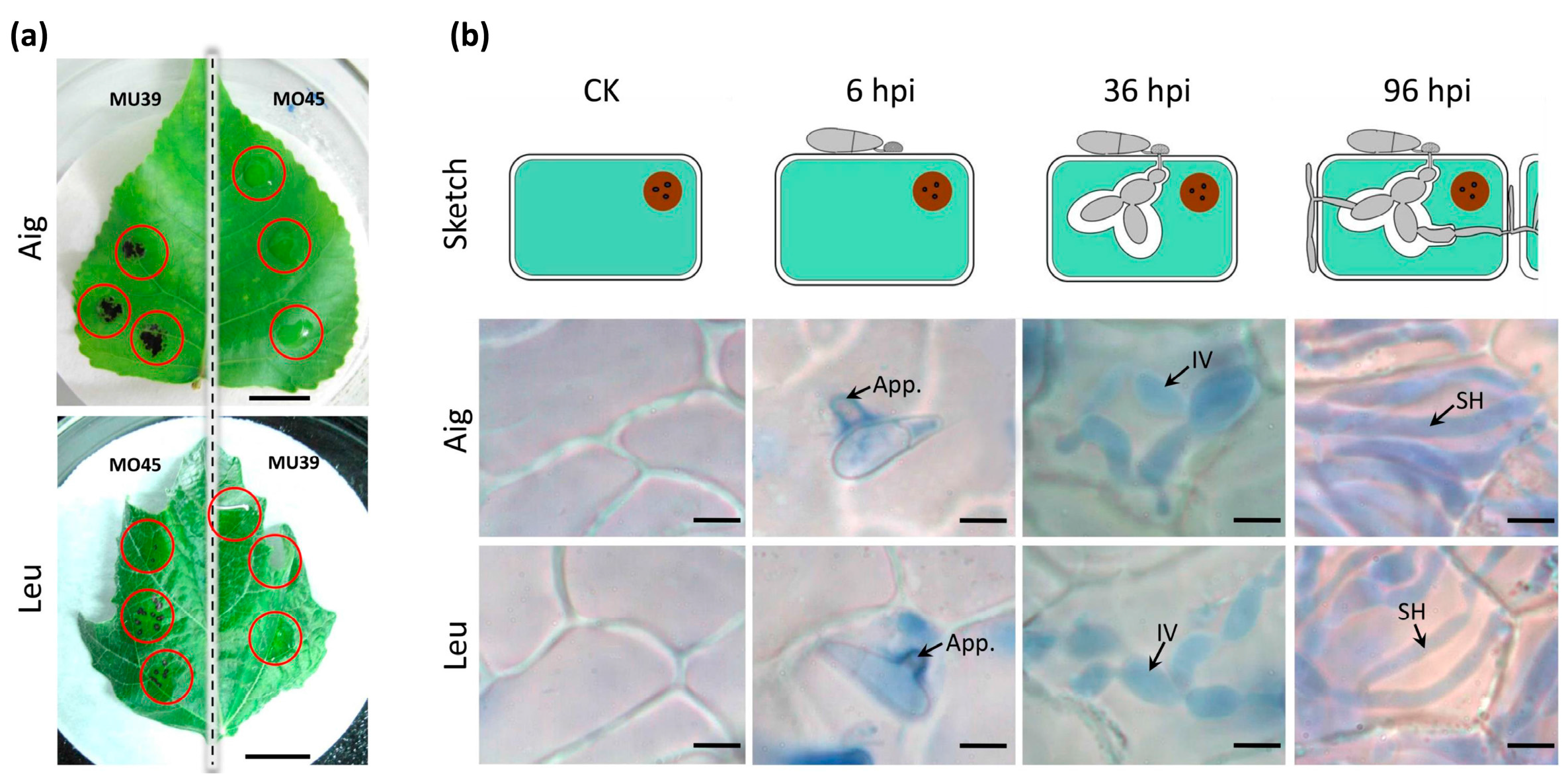
2.2. Microscopy
2.3. Inoculation and Sample Collection
2.4. RNA Isolation and Sequencing
2.5. Reads Mapping
2.6. Differentially Expressed Genes Analysis and Gene Function Annotation
2.7. Quantitative Reverse-Transcription PCR
3. Results
3.1. The Disease Development of Leaf Spot Disease of Poplar
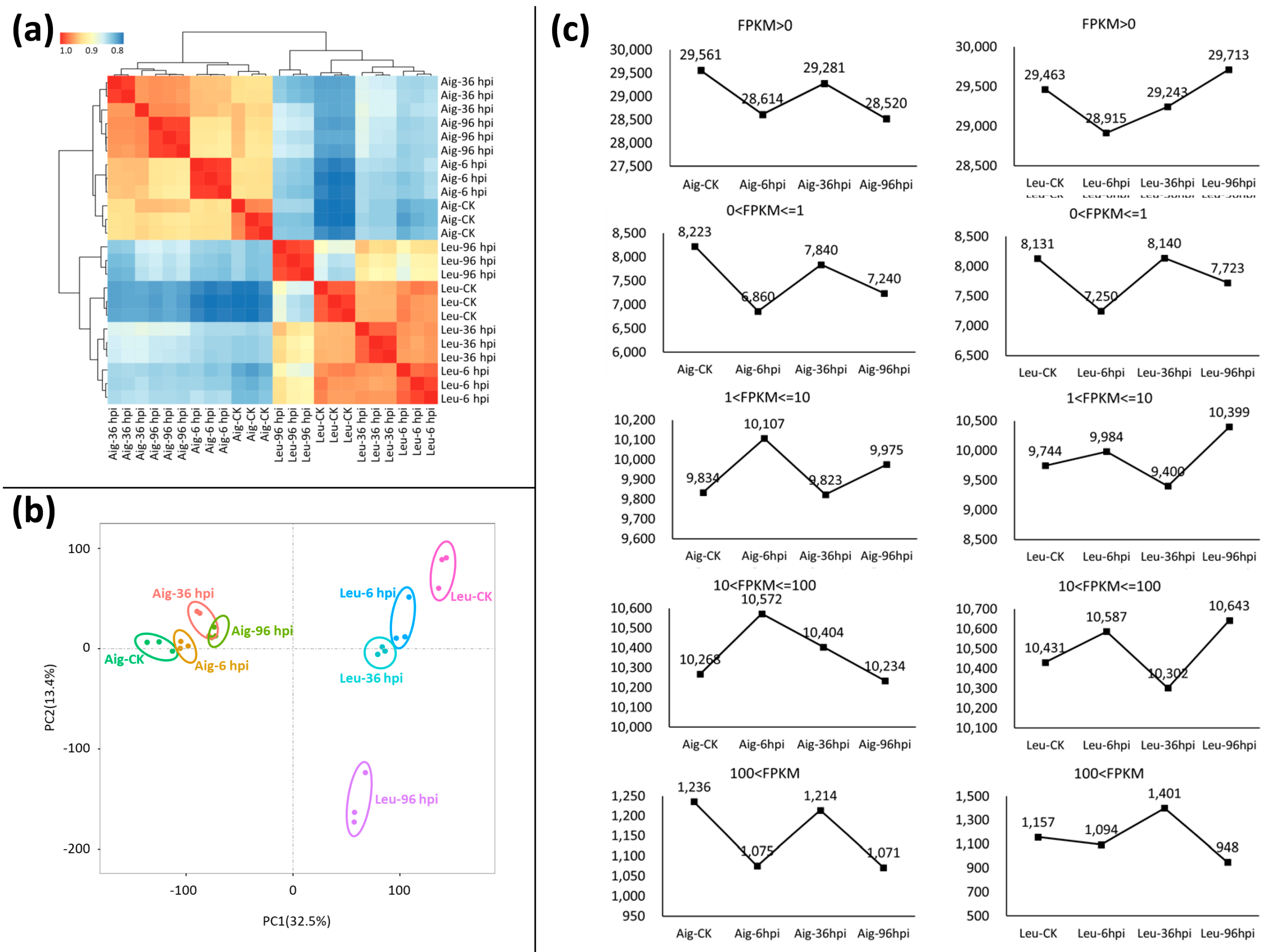
3.2. Summary of RNA Sequencing and Assembly
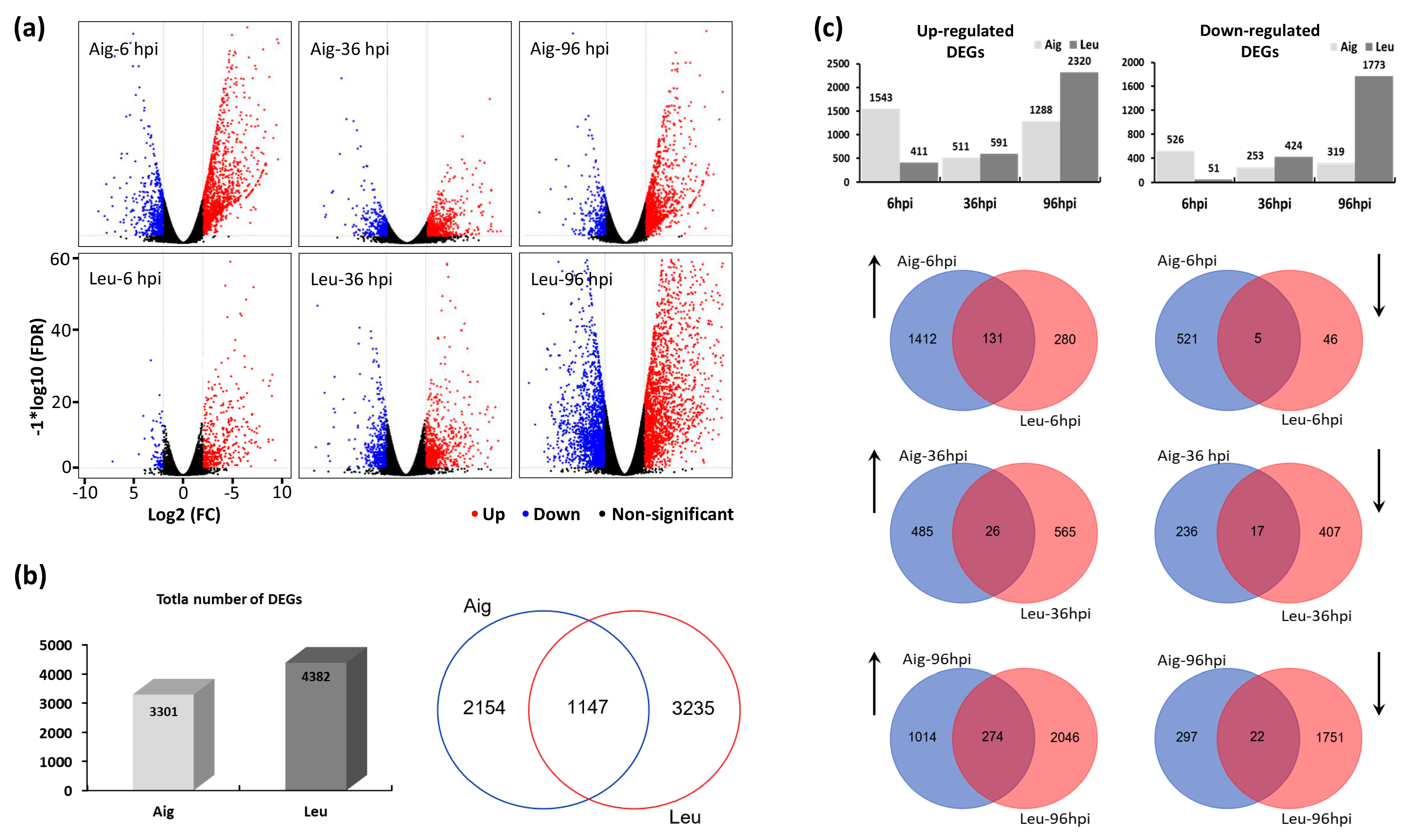
3.3. Analysis of Differentially Expressed Genes
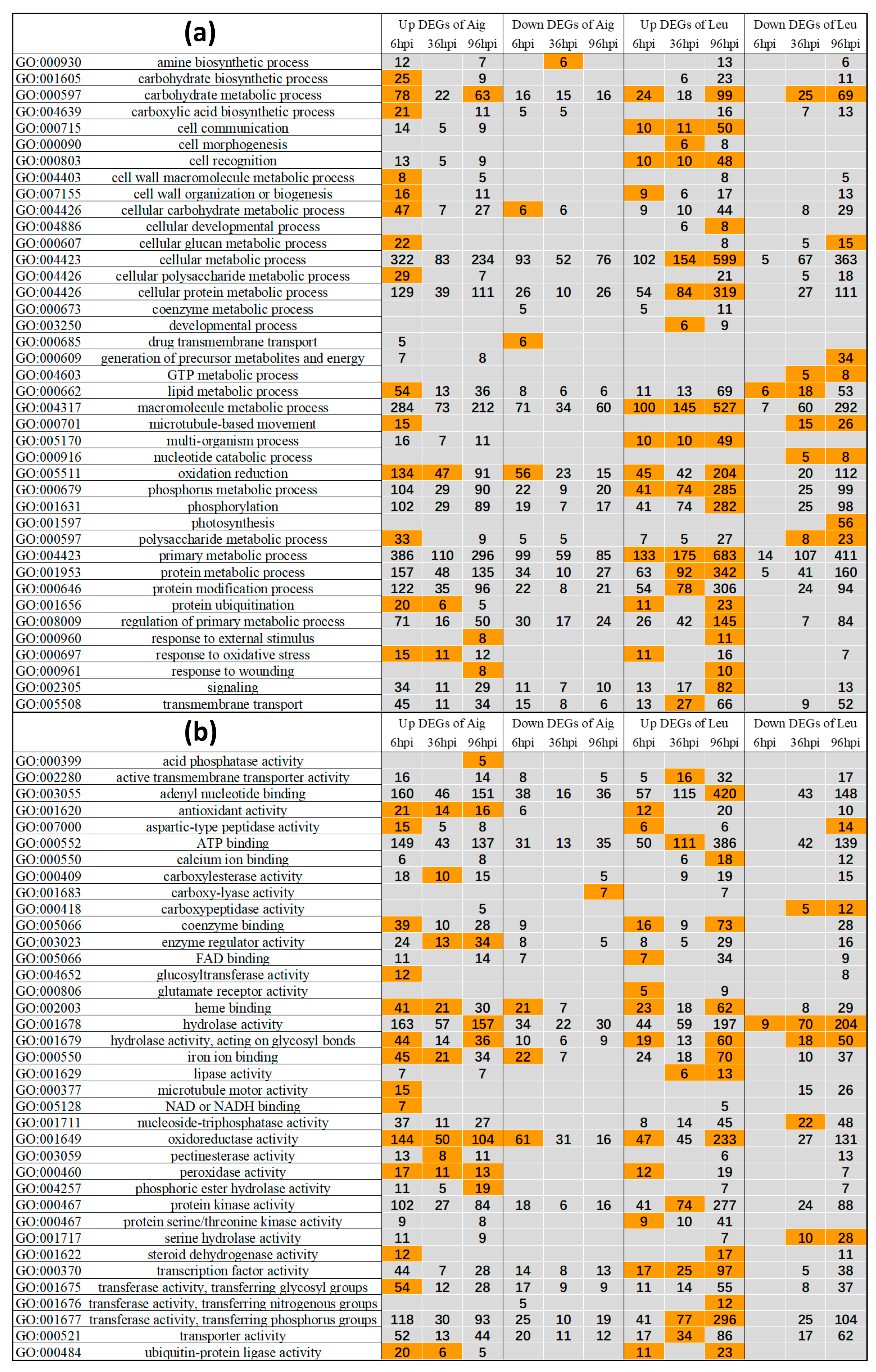
3.3.1. GO Enrichment Analysis of Differentially Expressed Genes
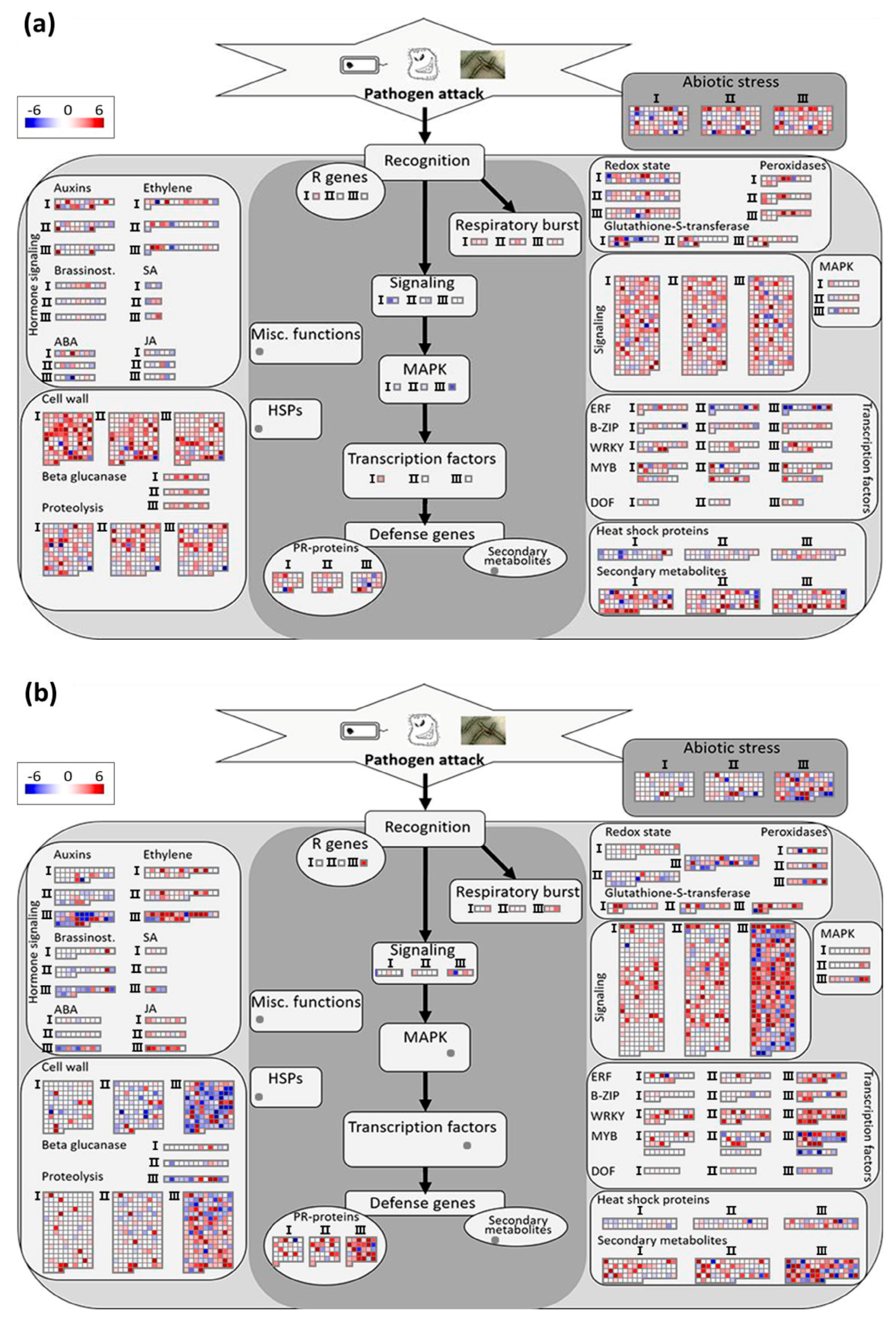
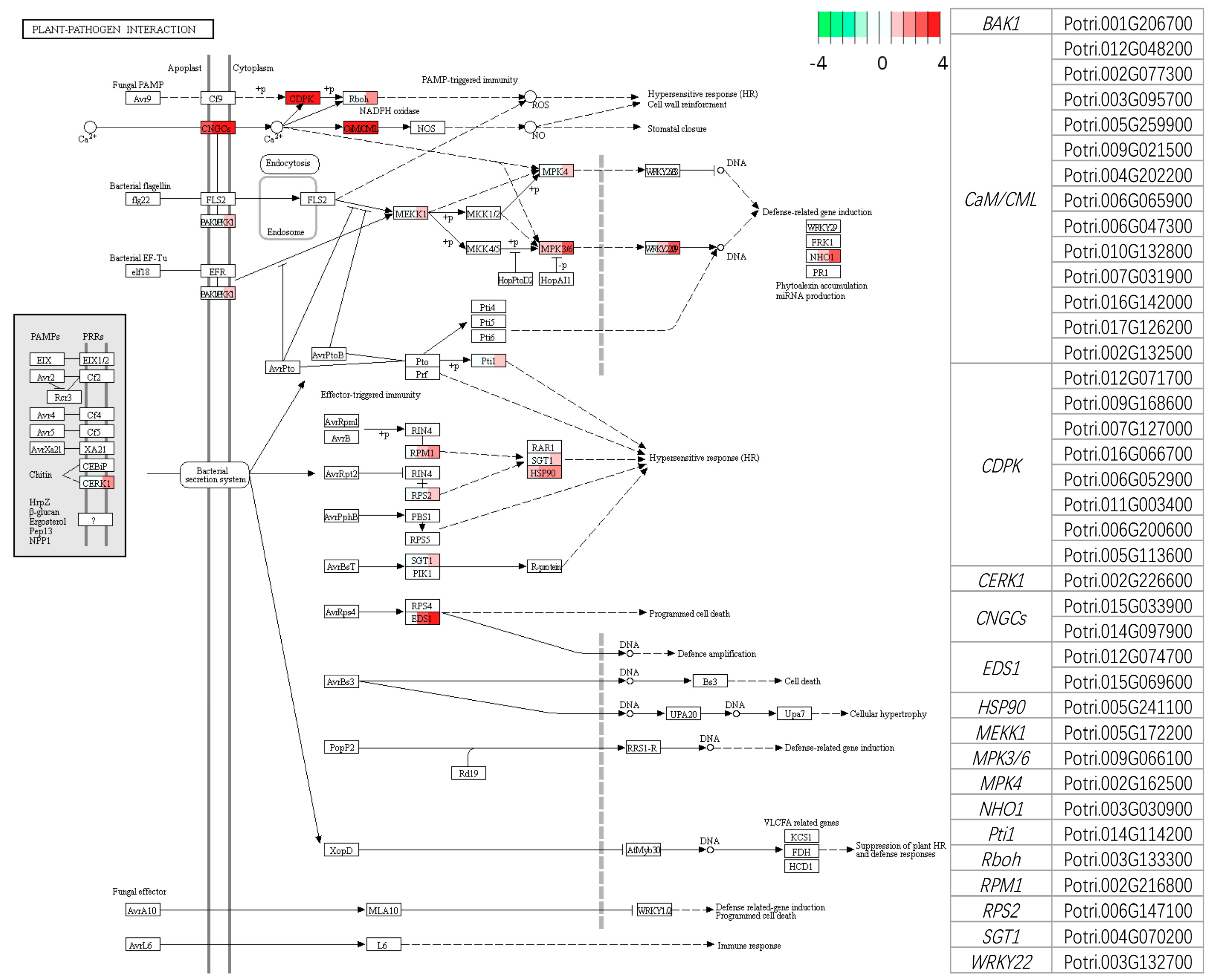
3.3.2. Analysis of Biotic Stress Response Differentially Expressed Genes
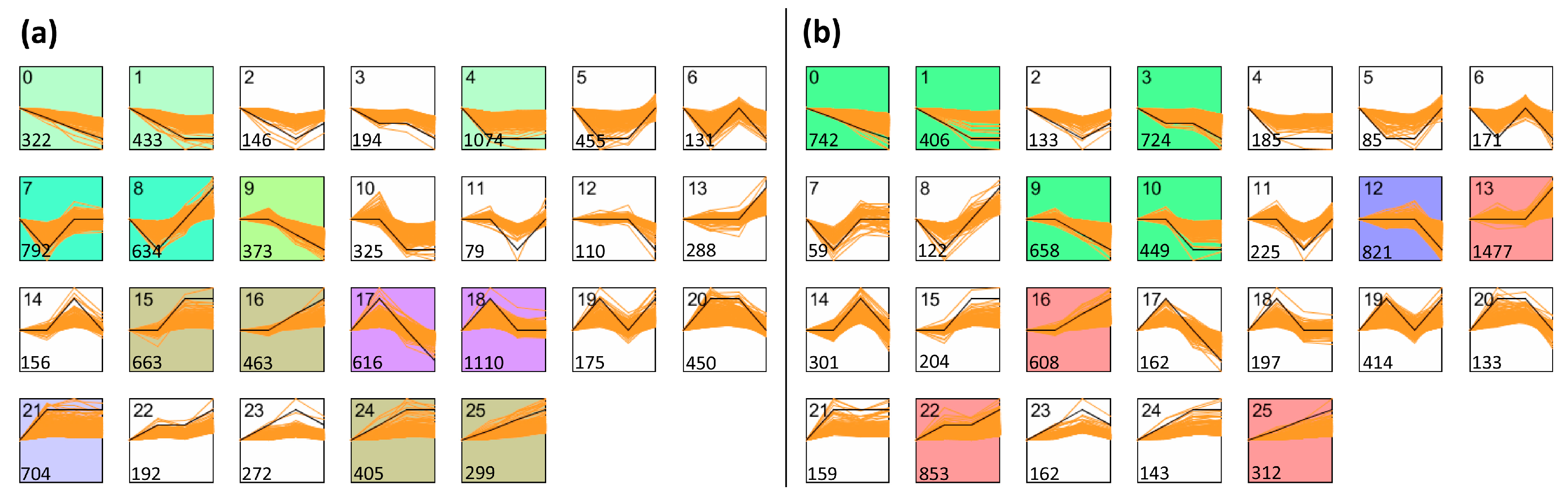
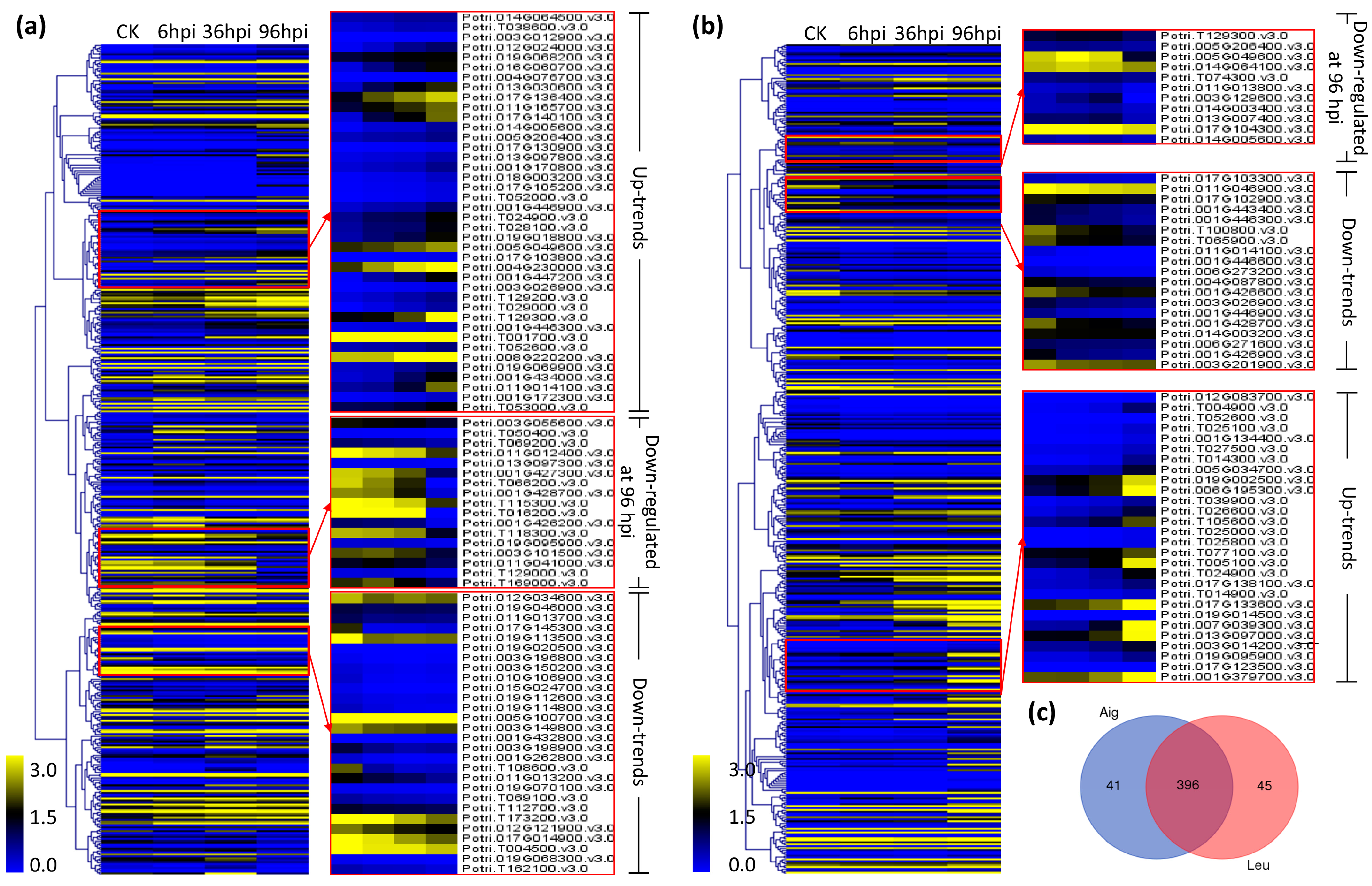
3.4. Analysis of Co-Expression
3.5. Expression Analysis of Genes Encoding Disease Resistant Proteins, Receptor-Like Kinases, Chitinases, and Defensins
4. Discussion
4.1. Recognition and Signaling
4.2. Transcriptional Regulation
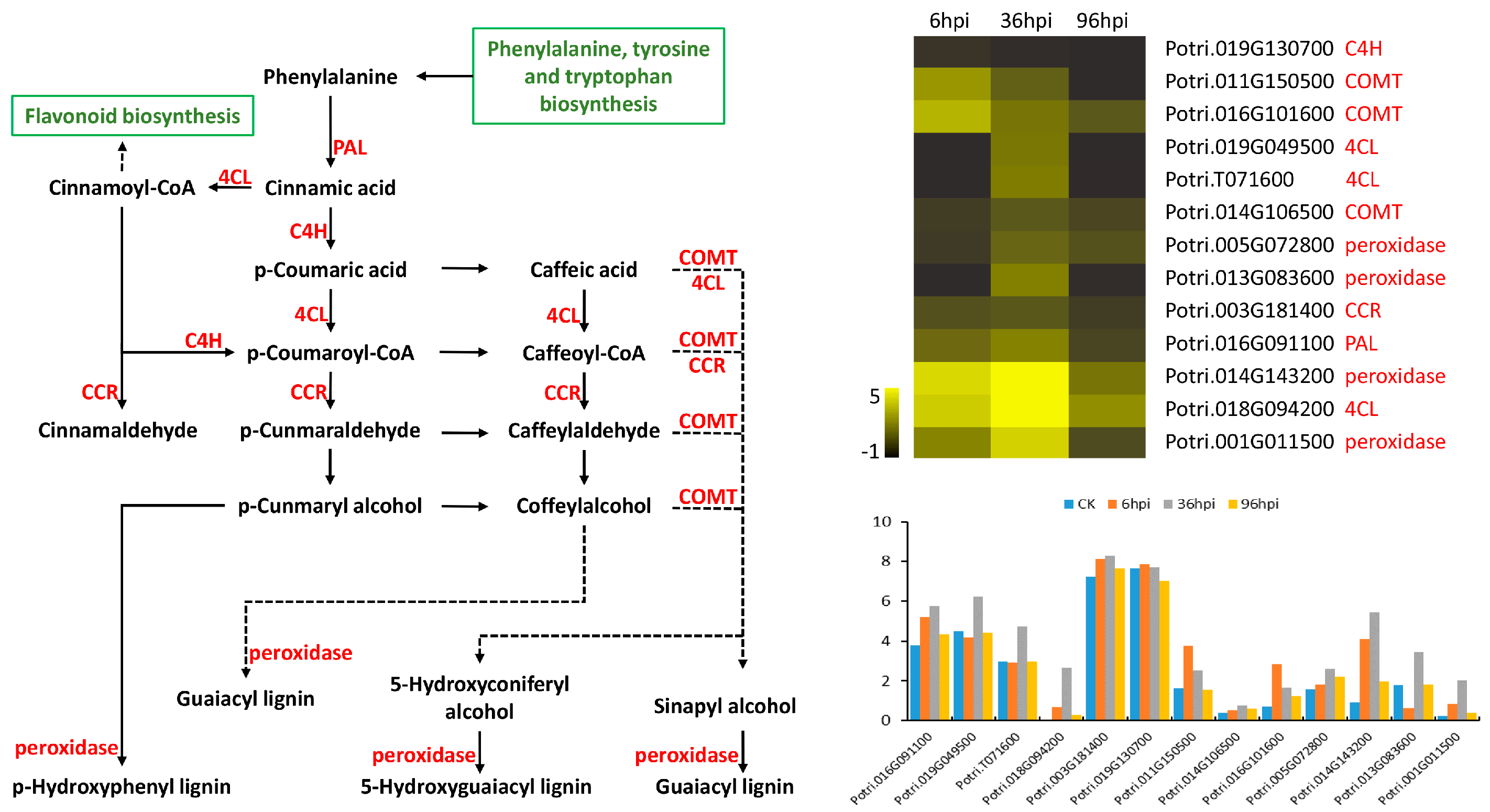
4.3. Primary and Secondary Metabolic Responses
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Boyd, L.A.; Ridout, C.; O’Sullivan, D.M.; Leach, J.E.; Leung, H. Plant-pathogen interactions: Disease resistance in modern agriculture. Trends Genet. 2013, 29, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Petit-Houdenot, Y.; Fudal, I. Complex interactions between fungal avirulence genes and their corresponding plant resistance genes and consequences for disease resistance management. Front. Plant Sci. 2017, 8, 1072. [Google Scholar] [CrossRef] [PubMed]
- Pavan, S.; Jacobsen, E.; Visser, R.G.F.; Bai, Y. Loss of susceptibility as a novel breeding strategy for durable and broad-spectrum resistance. Mol. Breed. 2010, 25. [Google Scholar] [CrossRef] [PubMed]
- Tuskan, G.A.; DiFazio, S.; Jansson, S.; Bohlmann, J.; Grigoriev, I.; Hellsten, U.; Putnam, N.; Ralph, S.; Rombauts, S.; Salamov, A.; et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 2006, 313, 1596–1604. [Google Scholar] [CrossRef] [PubMed]
- Eckenwalder, J.E. Biology of Populus and Its Implications for Management and Conservation; NRC Research Press: Ottawa, ON, Canada, 1996; pp. 7–32. [Google Scholar]
- Cellerino, G.P. Review of Fungal Diseases in Poplar; Food and Agriculture Organization of the United Nations: Rome, Italy, 1999; pp. 48–53. [Google Scholar]
- Wu, J.H.; Li, Z.; Zhou, Z.B.; Xue, Y.; Tao, W.Q.; Tian, C.M. Forecast model of poplar black spot caused by Marssonina brunnea. For. Pest Dis. 2012, 31, 5–8. [Google Scholar]
- Xi, B.; Bloomberg, M.; Watt, M.S.; Wang, Y.; Jia, L. Modeling growth response to soil water availability simulated by HYDRUS for a mature triploid Populus tomentosa plantation located on the North China Plain. Agric. Water Manag. 2016, 176, 243–254. [Google Scholar] [CrossRef]
- Spiers, A.G. Comparative studies of host specificity and symptoms exhibited by poplars infected with Marssonina brunnea, Marssonina castagnei and Marssonina populi. For. Pathol. 1984, 14, 202–218. [Google Scholar] [CrossRef]
- Li, C.D. Two specialized forms of Marssonina populi (Lib.) magn. J. Nanjing For. Univ. 1984, 8, 10–17. [Google Scholar]
- Beare, J.A.; Archer, S.A.; Bell, J.N.B. Marssonina leafspot disease of poplar under elevated ozone: Pre-fumigated host and in vitro studies. Environ. Pollut. 1999, 105, 409–417. [Google Scholar] [CrossRef]
- Erickson, J.E.; Stanosz, G.R.; Kruger, E.L. Photosynthetic consequences of Marssonina leaf spot differ between two poplar hybrids. New Phytol. 2004, 161, 577–583. [Google Scholar] [CrossRef]
- Cheng, Q.; Cao, Y.; Jiang, C.; Xu, L.; Wang, M.; Zhang, S.; Huang, M. Identifying secreted proteins of Marssonina brunnea by degenerate PCR. Proteomics 2010, 10, 2406–2417. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Cao, Y.; Pan, H.; Wang, M.; Huang, M. Isolation and characterization of two genes encoding polygalacturonase-inhibiting protein from Populus deltoides. J. Genet. Genom. 2008, 35, 631–638. [Google Scholar] [CrossRef]
- Cheng, Q.; Zhang, B.; Zhuge, Q.; Zeng, Y.; Wang, M.; Huang, M. Expression profiles of two novel lipoxygenase genes in Populus deltoides. Plant Sci. 2006, 170, 1027–1035. [Google Scholar] [CrossRef]
- Jiang, Y.; Duan, Y.; Yin, J.; Ye, S.; Zhu, J.; Zhang, F.; Lu, W.; Fan, D.; Luo, K. Genome-wide identification and characterization of the Populus WRKY transcription factor family and analysis of their expression in response to biotic and abiotic stresses. J. Exp. Bot. 2014, 65, 6629–6644. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, X.; Chen, Y.; Wang, Q.; Wang, M.; Huang, M. Function and chromosomal localization of differentially expressed genes induced by Marssonina brunnea f. sp. Multigermtubi in Populus deltoides. J. Genet. Genom. 2007, 34, 641–648. [Google Scholar] [CrossRef]
- He, W.; Yang, W. Host rang and distribution about three pathogens causing poplar black spot disease in parts of China. Sci. Silvae Sin. 1991, 27, 560–564. [Google Scholar]
- Han, Z.M.; Li, C.D. Comparative studies of isolates of Marssonina brunnea in China. Sci. Silvae Sin. 1998, 34, 59–65. [Google Scholar]
- Chen, C.; Yao, Y.; Zhang, L.; Xu, M.; Jiang, J.; Dou, T.; Lin, W.; Zhao, G.; Huang, M.; Zhou, Y. A comprehensive analysis of the transcriptomes of Marssonina brunnea and infected poplar leaves to capture vital events in host-pathogen interactions. PLoS ONE 2015, 10, e0134246. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.F.; He, W.; Yan, D.H. Histopathologic characterization of the process of Marssonina brunnea infection in poplar leaves. Can. J. For. Res. 2017. under review. [Google Scholar]
- Spiers, A.G. An agar leaf disc technique for screening poplars for resistance to Marssonina. Plant Dis. Rep. 1978, 62, 144–147. [Google Scholar]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- The Team of Bioinformatics and Evolutionary Genomics. Draw Veen Diagrams. Available online: http://bioinformatics.Psb.Ugent.Be/webtools/venn/ (accessed on 22 October 2017).
- Saeed, A.I.; Sharov, V.; White, J.; Li, J.; Liang, W.; Bhagabati, N.; Braisted, J.; Klapa, M.; Currier, T.; Thiagarajan, M.; et al. TM4: A free, open-source system for microarray data management and analysis. Biotechniques 2003, 34, 374–378. [Google Scholar] [PubMed]
- Ernst, J.; Bar-Joseph, Z. STEM: A tool for the analysis of short time series gene expression data. BMC Bioinform. 2006, 7, 191. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Zhou, X.; Ling, Y.; Zhang, Z.; Su, Z. agriGO: A GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010, 38, W64–W70. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2014; Available online: http://www.R-project.org/ (accessed on 22 August 2017).
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. ClusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Brouwer, C. Pathview: An R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 2013, 29, 1830–1831. [Google Scholar] [CrossRef] [PubMed]
- Thimm, O.; Bläsing, O.; Gibon, Y.; Nagel, A.; Meyer, S.; Krüger, P.; Selbig, J.; Müller, L.A.; Rhee, S.Y.; Stitt, M. MAPMAN: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004, 37, 914–939. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.L.; Chen, J.; Tian, Q.; Wang, S.; Xia, X.; Yin, W. Identification and validation of reference genes for Populus euphratica gene expression analysis during abiotic stresses by quantitative real-time PCR. Physiol. Plant. 2014, 152, 529–545. [Google Scholar] [CrossRef] [PubMed]
- Shiu, S.H.; Bleecker, A.B. Plant receptor-like kinase gene family: Diversity, function, and signaling. Sci. STKE 2001, 2001, re22. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Wang, Z.; Wang, J.; Wang, Y.; Wang, N.; Wang, Z.; Xu, M.; Su, X.; Wang, M.; Zhang, S.; et al. A quantitative model of transcriptional differentiation driving host-pathogen interactions. Brief. Bioinform. 2013, 14, 713–723. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhu, S.; Dai, Y.M.; Zhang, X.Y.; Ye, J.R.; Wang, M.X.; Huang, M.R. Untangling the transcriptome from fungus-infected plant tissues. Gene 2013, 519, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Ames, R.M. Using network extracted ontologies to identify novel genes with roles in appressorium development in the rice blast fungus Magnaporthe oryzae. Microorganisms 2017, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Westermann, A.J.; Gorski, S.A.; Vogel, J. Dual RNA-seq of pathogen and host. Nat. Rev. Microbiol. 2012, 10, 618. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, M.C.; Dagdas, Y.F.; Gupta, Y.K.; Mentlak, T.A.; Yi, M.; Martinez-Rocha, A.L.; Saitoh, H.; Terauchi, R.; Talbot, N.J.; Valent, B. Two distinct secretion systems facilitate tissue invasion by the rice blast fungus Magnaporthe oryzae. Nat. Commun. 2013, 4, 1996. [Google Scholar] [CrossRef] [PubMed]
- Wharton, P.S.; Julian, A.M.; O’Connell, R.J. Ultrastructure of the infection of Sorghum bicolor by Colletotrichum sublineolum. Phytopathology 2001, 91, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Oliveira-Garcia, E.; Deising, H.B. Infection structure–specific expression of β-1,3-glucan synthase is essential for pathogenicity of Colletotrichum graminicola and evasion of β-glucan–triggered immunity in maize. Plant Cell 2013, 25, 2356–2378. [Google Scholar] [CrossRef] [PubMed]
- Vargas, W.A.; Martín, J.M.S.; Rech, G.E.; Rivera, L.P.; Benito, E.P.; Díaz-Mínguez, J.M.; Thon, M.R.; Sukno, S.A. Plant defense mechanisms are activated during biotrophic and necrotrophic development of Colletotricum graminicola in maize. Plant Physiol. 2012, 158, 1342–1358. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, R.J.; Thon, M.R.; Hacquard, S.; Amyotte, S.G.; Kleemann, J.; Torres, M.F.; Damm, U.; Buiate, E.A.; Epstein, L.; Alkan, N.; et al. Lifestyle transitions in plant pathogenic Colletotrichum fungi deciphered by genome and transcriptome analyses. Nat. Genet. 2012, 44, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, M.C.; Valent, B. Filamentous plant pathogen effectors in action. Nat. Rev. Microbiol. 2013, 11, 800. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.D.G.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Glazebrook, J. Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annu. Rev. Phytopathol. 2005, 43, 205–227. [Google Scholar] [CrossRef] [PubMed]
- Mohr, T.J.; Mammarella, N.D.; Hoff, T.; Woffenden, B.J.; Jelesko, J.G.; McDowell, J.M. The Arabidopsis downy mildew resistance gene RPP8 is induced by pathogens and salicylic acid and is regulated by W box cis elements. Mol. Plant Microbe Interact. 2010, 23, 1303–1315. [Google Scholar] [CrossRef] [PubMed]
- Pedley, K.F.; Martin, G.B. Role of mitogen-activated protein kinases in plant immunity. Curr. Opin. Plant Biol. 2005, 8, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Su, H.; Han, L.; Wang, C.; Sun, Y.; Liu, F. Differential expression profiles of poplar map kinase kinases in response to abiotic stresses and plant hormones, and overexpression of Ptmkk4 improves the drought tolerance of poplar. Gene 2014, 545, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Hamel, L.P.; Miles, G.P.; Samuel, M.A.; Ellis, B.E.; Seguin, A.; Beaudoin, N. Activation of stress-responsive mitogen-activated protein kinase pathways in hybrid poplar (Populus trichocarpa × Populus deltoides). Tree Physiol. 2005, 25, 277–288. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yoo, S.D.; Sheen, J. MAPK signaling in plant hormone ethylene signal transduction. Plant Signal. Behav. 2008, 3, 848–849. [Google Scholar] [CrossRef] [PubMed]
- Van Loon, L.C.; Geraats, B.P.; Linthorst, H.J. Ethylene as a modulator of disease resistance in plants. Trends Plant Sci. 2006, 11, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Boyle, B.; Levee, V.; Hamel, L.P.; Nicole, M.C.; Seguin, A. Molecular and histochemical characterisation of two distinct poplar Melampsora leaf rust pathosystems. Plant Biol. 2010, 12, 364–376. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Du, L.; Poovaiah, B.W. Calcium signaling and biotic defense responses in plants. Plant Signal. Behav. 2014, 9, e973818. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Cox, L.K., Jr.; He, P. Functions of calcium-dependent protein kinases in plant innate immunity. Plants 2014, 3, 160–176. [Google Scholar] [CrossRef] [PubMed]
- Ranty, B.; Aldon, D.; Galaud, J.-P. Plant calmodulins and calmodulin-related proteins: Multifaceted relays to decode calcium signals. Plant Signal. Behav. 2006, 1, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Heo, W.D.; Sang, H.L.; Min, C.K.; Kim, J.C.; Chung, W.S.; Chun, H.J.; Lee, K.J.; Chan, Y.P.; Park, H.C.; Ji, Y.C. Involvement of specific calmodulin isoforms in salicylic acid-independent activation of plant disease resistance responses. Proc. Natl. Acad. Sci. USA 1999, 96, 766–771. [Google Scholar] [CrossRef] [PubMed]
- Takabatake, R.; Karita, E.; Seo, S.; Mitsuhara, I.; Kuchitsu, K.; Ohashi, Y. Pathogen-induced calmodulin isoforms in basal resistance against bacterial and fungal pathogens in tobacco. Plant Cell Physiol. 2007, 48, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Pennington, B.O.; Hua, J. Multiple R-like genes are negatively regulated by BON1 and BON3 in Arabidopsis. Mol. Plant Microbe Interact. 2009, 22, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Spitz, F.; Furlong, E.E.M. Transcription factors: From enhancer binding to developmental control. Nat. Rev. Genet. 2012, 13, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Foley, R.C.; Onate-Sanchez, L. Transcription factors in plant defense and stress responses. Curr. Opin. Plant Biol. 2002, 5, 430–436. [Google Scholar] [CrossRef]
- Grennan, A.K. Ethylene response factors in jasmonate signaling and defense response. Plant Physiol. 2008, 146, 1457–1458. [Google Scholar] [CrossRef] [PubMed]
- Jakoby, M.; Weisshaar, B.; Droge-Laser, W.; Vicente-Carbajosa, J.; Tiedemann, J.; Kroj, T.; Parcy, F. bZIP transcription factors in Arabidopsis. Trends Plant Sci. 2002, 7, 106–111. [Google Scholar] [CrossRef]
- Pandey, S.P.; Somssich, I.E. The role of WRKY transcription factors in plant immunity. Plant Physiol. 2009, 150, 1648–1655. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, L.; Zhou, X.; Zhou, M.; Lu, Y.; Ma, L.; Ma, H.; Zhang, Z. Transgenic wheat expressing Thinopyrum intermedium MYB transcription factor TiMYB2R-1 shows enhanced resistance to the take-all disease. J. Exp. Bot. 2013, 64, 2243–2253. [Google Scholar] [CrossRef] [PubMed]
- Ambawat, S.; Sharma, P.; Yadav, N.R.; Yadav, R.C. MYB transcription factor genes as regulators for plant responses: An overview. Physiol. Mol. Biol. Plants 2013, 19, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Alazem, M.; Lin, N.S. Roles of plant hormones in the regulation of host-virus interactions. Mol. Plant Pathol. 2015, 16, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Eckardt, N.A. Specificity and cross-talk in plant signal transduction. Plant Cell 2002, 14, S9–S14. [Google Scholar] [PubMed]
- Govrin, E.M.; Levine, A. The hypersensitive response facilitates plant infection by the necrotrophic pathogen Botrytis cinerea. Curr. Biol. 2000, 10, 751–757. [Google Scholar] [CrossRef]
- Horbach, R.; Navarro-Quesada, A.R.; Knogge, W.; Deising, H.B. When and how to kill a plant cell: Infection strategies of plant pathogenic fungi. J. Plant Physiol. 2011, 168, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.; Sinha, A.K.; Roitsch, T. Plant physiology meets phytopathology: Plant primary metabolism and plant-pathogen interactions. J. Exp. Bot. 2007, 58, 4019–4026. [Google Scholar] [CrossRef] [PubMed]
- Rojas, C.M.; Senthil-Kumar, M.; Tzin, V.; Mysore, K.S. Regulation of primary plant metabolism during plant-pathogen interactions and its contribution to plant defense. Front. Plant Sci. 2014, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Swarbrick, P.J.; Schulze-Lefert, P.; Scholes, J.D. Metabolic consequences of susceptibility and resistance (race-specific and broad-spectrum) in barley leaves challenged with powdery mildew. Plant Cell Environ. 2006, 29, 1061–1076. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Lian, L.; He, W.; Zhu, Y.; Cai, Q.; Xie, H.; Zhang, J. Genome-wide profiling of changes in gene expression in response to infection of the japonica rice variety Yunyin by Magnaporthe oryzae. Mol. Breed. 2014, 34, 1965–1974. [Google Scholar] [CrossRef]
- Kaplan, F.; Guy, C.L. β-amylase induction and the protective role of maltose during temperature shock. Plant Physiol. 2004, 135, 1674–1684. [Google Scholar] [CrossRef] [PubMed]
- Pusztahelyi, T.; Holb, I.J.; Pócsi, I. Secondary metabolites in fungus-plant interactions. Front. Plant Sci. 2015, 6, 573. [Google Scholar] [CrossRef] [PubMed]
- Vogt, T. Phenylpropanoid biosynthesis. Mol. Plant 2010, 3, 2–20. [Google Scholar] [CrossRef] [PubMed]
- War, A.R.; Paulraj, M.G.; War, M.Y.; Ignacimuthu, S. Role of salicylic acid in induction of plant defense system in chickpea (Cicer arietinum L.). Plant Signal. Behav. 2011, 6, 1787–1792. [Google Scholar] [CrossRef] [PubMed]
- Douglas, C.J. Phenylpropanoid metabolism and lignin biosynthesis: From weeds to trees. Trends Plant Sci. 1996, 1, 171–178. [Google Scholar] [CrossRef]
- Jin, M.; Zhang, X.; Zhao, M.; Deng, M.; Du, Y.; Zhou, Y.; Wang, S.; Tohge, T.; Fernie, A.R.; Willmitzer, L.; et al. Integrated genomics-based mapping reveals the genetics underlying maize flavonoid biosynthesis. BMC Plant Biol. 2017, 17, 17. [Google Scholar] [CrossRef] [PubMed]
- Underwood, W. The plant cell wall: A dynamic barrier against pathogen invasion. Front. Plant Sci. 2012, 3, 85. [Google Scholar] [CrossRef] [PubMed]
- Lionetti, V.; Raiola, A.; Camardella, L.; Giovane, A.; Obel, N.; Pauly, M.; Favaron, F.; Cervone, F.; Bellincampi, D. Overexpression of pectin methylesterase inhibitors in Arabidopsis restricts fungal infection by Botrytis cinerea. Plant Physiol. 2007, 143, 1871–1880. [Google Scholar] [CrossRef] [PubMed]
- Mierziak, J.; Kostyn, K.; Kulma, A. Flavonoids as important molecules of plant interactions with the environment. Molecules 2014, 19, 16240–16265. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Replicate 1 | Replicate 2 | Replicate 3 | |||
|---|---|---|---|---|---|---|
| Total Reads a | Mapped Reads b | Total Reads | Mapped Reads | Total Reads | Mapped Reads | |
| Aig-CK | 39,795,332 | 29,021,681 (72.93%) | 42,248,380 | 30,635,622 (72.51%) | 37,445,738 | 27,135,653 (72.47%) |
| Aig-6hpi | 109,470,584 | 79,720,175 (72.82%) | 66,456,714 | 47,641,251 (71.69%) | 74,871,338 | 54,150,365 (72.32%) |
| Aig-36hpi | 59,014,678 | 42,284,488 (71.65%) | 75,926,010 | 55,006,529 (72.45%) | 73,458,950 | 54,031,383 (73.55%) |
| Aig-96hpi | 62,112,734 | 42,113,564 (67.80%) | 66,706,786 | 44,826,241 (67.20%) | 88,780,204 | 61,825,740 (69.64%) |
| Leu-CK | 39,274,080 | 21,132,514 (53.81%) | 44,843,154 | 24,443,778 (54.51%) | 61,385,552 | 33,190,001 (54.07%) |
| Leu-6hpi | 82,348,652 | 43,884,193 (53.29%) | 71,055,446 | 36,835,745 (51.84%) | 88,607,696 | 46,844,883 (52.87%) |
| Leu-36hpi | 89,169,026 | 51,010,245 (57.21%) | 74,696,580 | 44,364,679 (59.39%) | 73,203,752 | 42,947,392 (58.67%) |
| Leu-96hpi | 69,355,868 | 35,080,780 (50.58%) | 92,556,954 | 47,437,815 (51.25%) | 85,561,166 | 44,456,042 (51.96%) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Tian, L.; Yan, D.-H.; He, W. Genome-Wide Transcriptome Analysis Reveals the Comprehensive Response of Two Susceptible Poplar Sections to Marssonina brunnea Infection. Genes 2018, 9, 154. https://doi.org/10.3390/genes9030154
Zhang Y, Tian L, Yan D-H, He W. Genome-Wide Transcriptome Analysis Reveals the Comprehensive Response of Two Susceptible Poplar Sections to Marssonina brunnea Infection. Genes. 2018; 9(3):154. https://doi.org/10.3390/genes9030154
Chicago/Turabian StyleZhang, Yanfeng, Longyan Tian, Dong-Hui Yan, and Wei He. 2018. "Genome-Wide Transcriptome Analysis Reveals the Comprehensive Response of Two Susceptible Poplar Sections to Marssonina brunnea Infection" Genes 9, no. 3: 154. https://doi.org/10.3390/genes9030154
APA StyleZhang, Y., Tian, L., Yan, D.-H., & He, W. (2018). Genome-Wide Transcriptome Analysis Reveals the Comprehensive Response of Two Susceptible Poplar Sections to Marssonina brunnea Infection. Genes, 9(3), 154. https://doi.org/10.3390/genes9030154