Hepatitis B Virus-Associated Hepatocellular Carcinoma and Hepatic Cancer Stem Cells

Abstract
1. Hepatitis
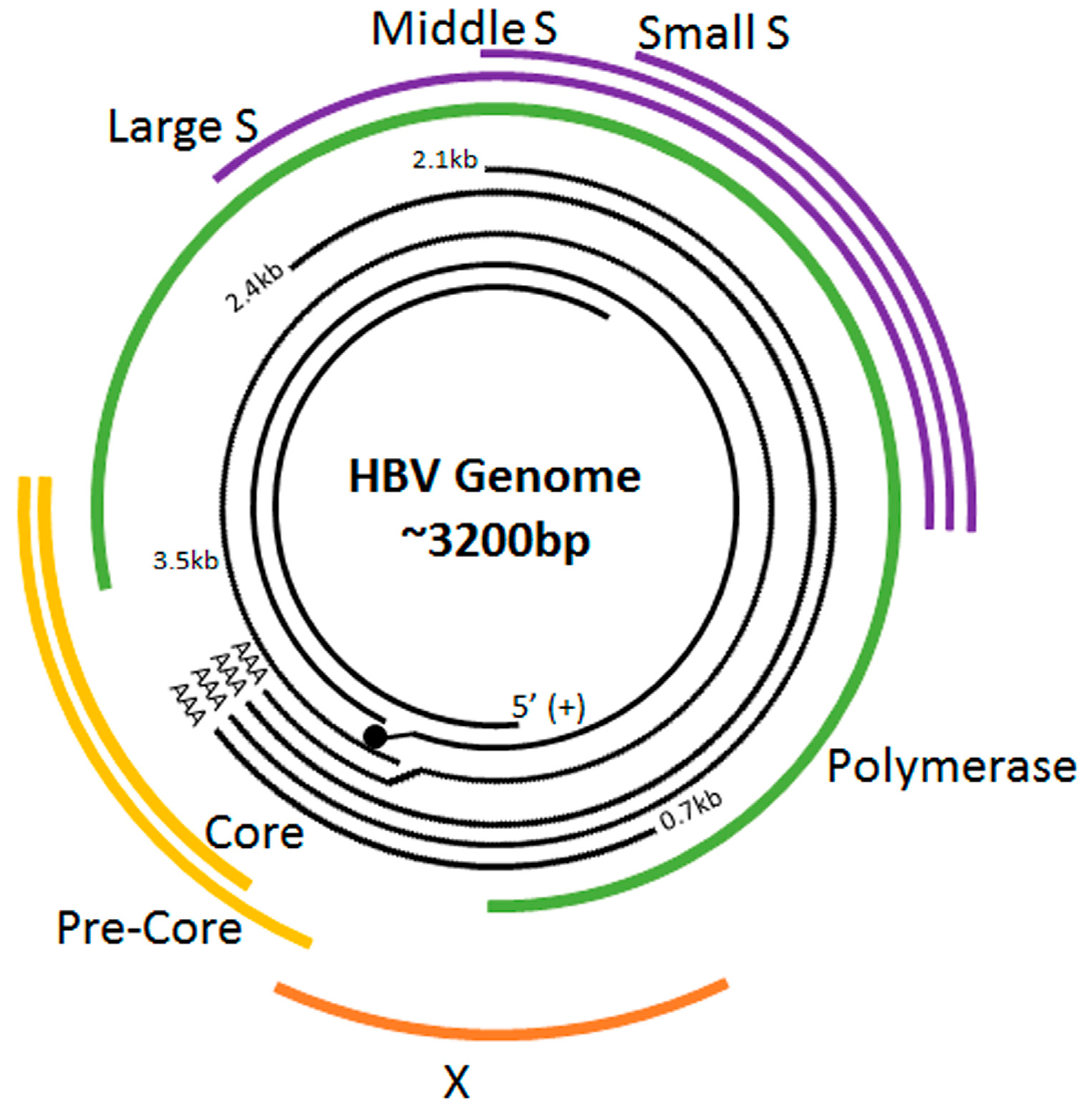
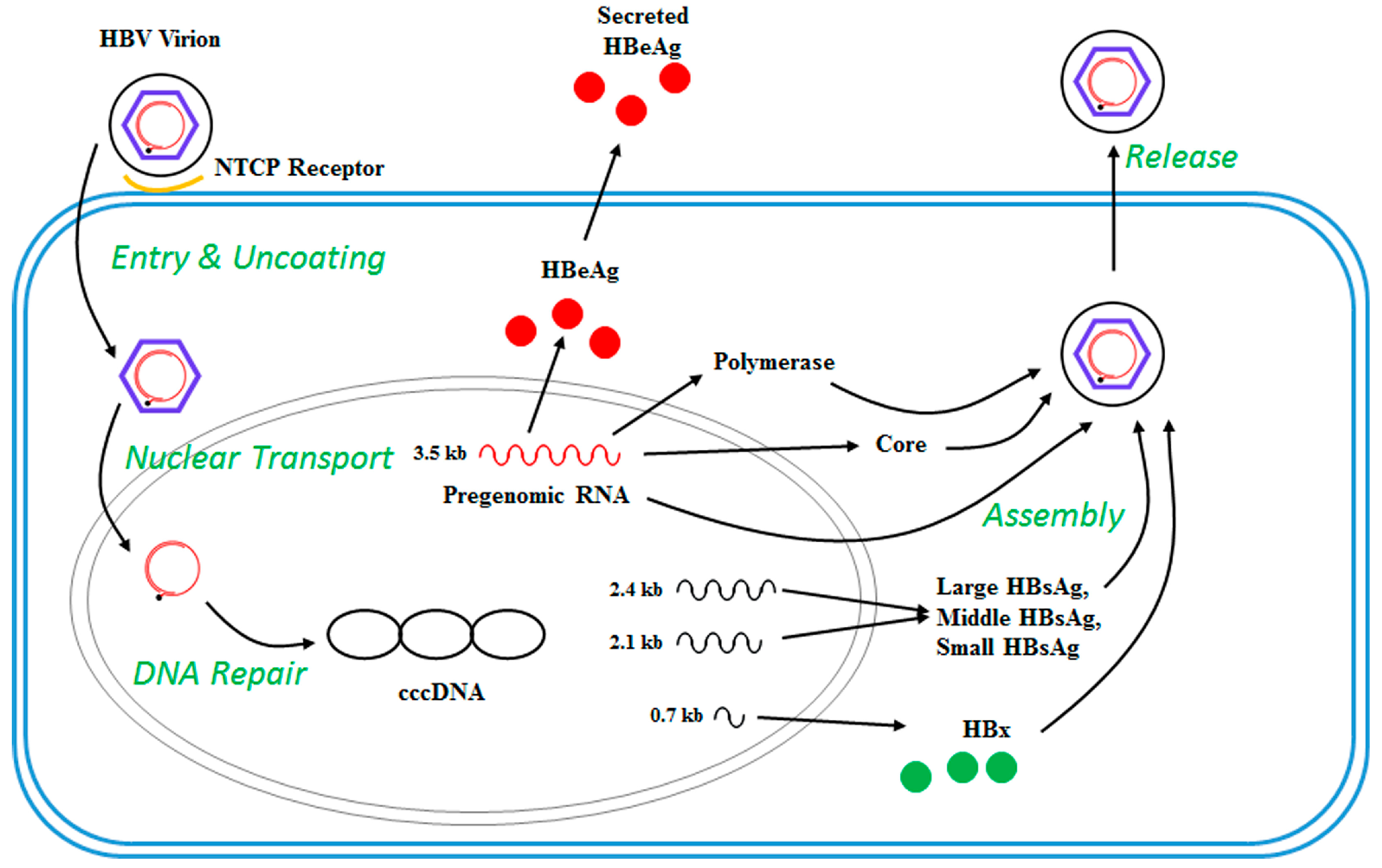
2. The HBV Life Cycle
3. Chronic Viral Hepatitis B and Hepatocellular Carcinoma (HCC)
4. Mutational Landscape of HBV-Related HCC
5. Epigenetic Mechanisms in HBV-Related HCC
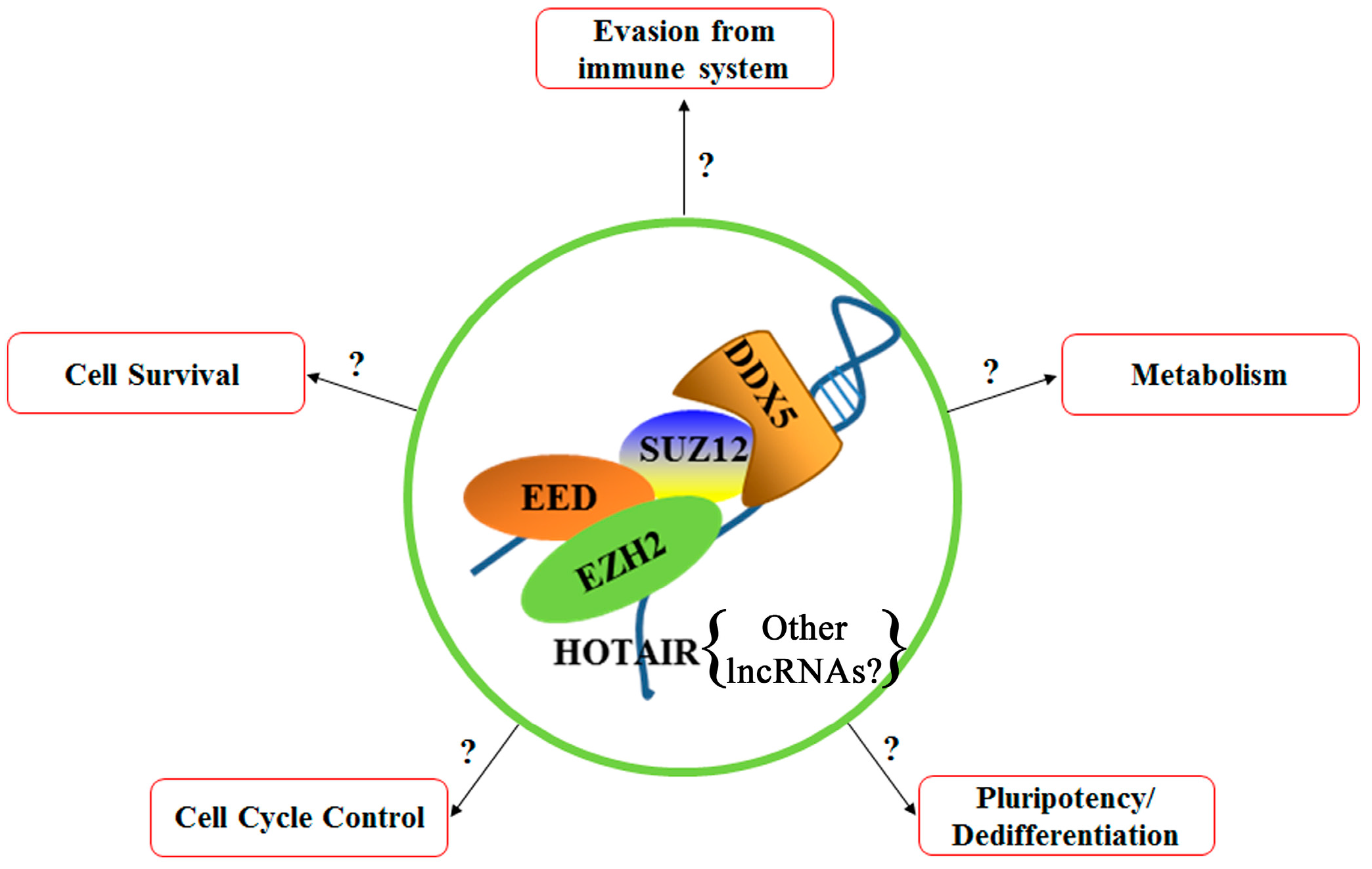
6. Polycomb Repressive Complex 2 (PRC2)
7. Epithelial Cell Adhesion Molecule (EpCAM) and Wnt Activation
8. P68 (DDX5)
9. Hox Transcript Antisense RNA (HOTAIR)
10. Summary and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lee, W.M.; Dienstag, J.L. Chapter 361: Toxic and Drug-Induced Hepatitis. In Harrison’s Principles of Internal Medicine; McGraw-Hill Education: New York, NY, USA, 2015; ISBN 0071373756. [Google Scholar]
- Mailliard, M.E.; Sorrell, M.F. Chapter 363: Alcoholic Liver Disease. In Harrison’s Principles of Internal Medicine; McGraw-Hill Education: New York, NY, USA, 2015; ISBN 0071373756. [Google Scholar]
- Friedman, L. Liver, biliary tract & pancreas disorders. In Current Medical Diagnosis and Treatment: 2015; McGraw-Hill Education: New York, NY, USA, 2015; pp. 658–663. ISBN 978-0-07-182486-6. [Google Scholar]
- Manns, M.P.; Czaja, A.J.; Gorham, J.D.; Krawitt, E.L.; Mieli-Vergani, G.; Vergani, D.; Vierling, J.M. Diagnosis and management of autoimmune hepatitis. Hepatology 2010, 51, 2193–2213. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H. Molecular mechanisms of hepatic ischemia-reperfusion injury and preconditioning. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 284, G15–G26. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Available online: http://www.who.int/mediacentre/factsheets/fs204/en/ (accessed on 29 January 2018).
- Harder, A.; Mehlhorn, H. Diseases Caused by Adult Parasites or Their Distinct Life Cycle Stages. In Comparative Hepatitis; Birkhäuser: Basel, Switzerland, 2008; pp. 161–216. ISBN 978-3-76-438557-6. [Google Scholar]
- Wisplinghoff, H.; Appleton, D.L. Bacterial Infections of the Liver. In Comparative Hepatitis; Birkhäuser: Basel, Switzerland, 2008; pp. 143–160. ISBN 978-3-76-438557-6. [Google Scholar]
- Zuckerman, A.J. Chapter 70 Hepatitis Viruses. In Medical Microbiology, 4th ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996; pp. 1–16. [Google Scholar]
- Robinson, W.S. The Genome of Hepatitis B Virus. Annu. Rev. Microbiol. 1977, 31, 357–377. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.M. Hepatitis B virus infection. N. Engl. J. Med. 1997, 337, 1733–1745. [Google Scholar] [CrossRef] [PubMed]
- Kao, J.H.; Chen, D.S. Overview of hepatitis B and C viruses. In Infectious Causes of Cancer Targets for Intervention; Humana Press: New York, NY, USA, 2000; pp. 313–330. ISBN 978-1-59259-024-7. [Google Scholar]
- Glebe, D.; Urban, S. Viral and cellular determinants involved in hepadnaviral entry. World J. Gastroenterol. 2007, 13, 22–38. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.; Nassal, M. Hepatitis B virus replication. World J. Gastroenterol. 2007, 13, 48–64. [Google Scholar] [CrossRef] [PubMed]
- Rehermann, B.; Nascimbeni, M. Immunology of hepatitis B virus and hepatitis C virus infection. Nat. Rev. Immunol. 2005, 5, 215–229. [Google Scholar] [CrossRef] [PubMed]
- GBD 2013 Mortality and Causes of Death Collaborators. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015, 385, 117–171. [Google Scholar] [CrossRef]
- Block, T.M.; Mehta, A.S.; Fimmel, C.J.; Jordan, R. Molecular viral oncology of hepatocellular carcinoma. Oncogene 2003, 22, 5093–5107. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.A.; Bonhoeffer, S.; Hill, A.M.; Boehme, R.; Thomas, H.C.; McDade, H. Viral dynamics in hepatitis B virus infection. Proc. Natl. Acad. Sci. USA 1996, 93, 4398–4402. [Google Scholar] [CrossRef] [PubMed]
- Kojima, T. Immune electron microscopic study of hepatitis B virus associated antigens in hepatocytes. Gastroenterol. Jpn. 1982, 17, 559–575. [Google Scholar] [PubMed]
- Kasai, H.; Sakamoto, M.; Yamaguchi, N.; Yokota, J. Increased Formation of Oxidative DNA Damage, 8-Hydroxydeoxyguanosine, in Human Livers with Chronic Hepatitis. Cancer Res. 1994, 54, 3171–3172. [Google Scholar]
- Galli, A.; Svegliati-Baroni, G.; Ceni, E.; Milani, S.; Ridolfi, F.; Salzano, R.; Tarocchi, M.; Grappone, C.; Pellegrini, G.; Benedetti, A.; et al. Oxidative stress stimulates proliferation and invasiveness of hepatic stellate cells via a MMP2-mediated mechanism. Hepatology 2005, 41, 1074–1084. [Google Scholar] [CrossRef] [PubMed]
- Tokino, T.; Tamura, H.; Hori, N.; Matsubara, K. Chromosome deletions associated with hepatitis B virus integration. Virology 1991, 185, 879–882. [Google Scholar] [CrossRef]
- Murakami, Y.; Saigo, K.; Takashima, H.; Minami, M.; Okanoue, T.; Bréchet, C.; Paterlini-Bréchot, P. Large scaled analysis of hepatitis B virus (HBV) DNA integration in HBV related hepatocellular carcinomas. Gut 2005, 54, 1162–1168. [Google Scholar] [CrossRef] [PubMed]
- Tarn, C.; Lee, S.; Hu, Y.; Ashendel, C.; Andrisani, O.M. Hepatitis B Virus X Protein Differentially Activates RAS-RAF-MAPK and JNK Pathways in X-transforming Versus Non-transforming AML12 Hepatocytes. J. Biol. Chem. 2001, 276, 34671–34680. [Google Scholar] [CrossRef] [PubMed]
- Nijhara, R.; Jana, S.S.; Goswami, S.K.; Rana, A.; Majumdar, S.S.; Kumar, V.; Sarkar, D.P. Sustained Activation of Mitogen-Activated Protein Kinases and Activator Protein 1 by the Hepatitis B Virus X Protein in Mouse Hepatocytes In Vivo. J. Virol. 2001, 75, 10348–10358. [Google Scholar] [CrossRef] [PubMed]
- Nijhara, R.; Jana, S.S.; Goswami, S.K.; Kumar, V.; Sarkar, D.P. An internal segment (residues 58-119) of the hepatitis B virus X protein is sufficient to activate MAP kinase pathways in mouse liver. FEBS Lett. 2001, 504, 59–64. [Google Scholar] [CrossRef]
- Maguire, H.F.; Hoeffler, J.P.; Siddiqui, A. HBV X protein alters the DNA binding specificity of CREB and ATF-2 by protein-protein interactions. Science 1991, 252, 842–844. [Google Scholar] [CrossRef] [PubMed]
- Lucito, R.; Schneider, R. Hepatitis B virus X protein activates transcription factor NF-kappa B without a requirement for protein kinase C. J. Virol. 1992, 66, 983–991. [Google Scholar] [PubMed]
- Terradillos, O.; Billet, O.; Renard, C.-A.; Levy, R.; Molina, T.; Briand, P.; Buendia, M.A. The hepatitis B virus X gene potentiates c-myc-induced liver oncogenesis in transgenic mice. Oncogene 1997, 14, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Madden, C.R.; Finegold, M.J.; Slagle, B.L. Hepatitis B virus X protein acts as a tumor promoter in development of diethylnitrosamine-induced preneoplastic lesions. J. Virol. 2001, 75, 3851–3858. [Google Scholar] [CrossRef] [PubMed]
- Rakotomalala, L.; Studach, L.; Wang, W.H.; Gregori, G.; Hullinger, R.L.; Andrisani, O. Hepatitis B virus X protein increases the Cdt1-to-geminin ratio inducing DNA re-replication and polyploidy. J. Biol. Chem. 2008, 283, 28729–28740. [Google Scholar] [CrossRef] [PubMed]
- Van Vugt, M.A.T.M.; Brás, A.; Medema, R.H. Polo-like kinase-1 controls recovery from a G2 DNA damage-induced arrest in mammalian cells. Mol. Cell 2004, 15, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Studach, L.L.; Rakotomalala, L.; Wang, W.-H.; Hullinger, R.L.; Cairo, S.; Buendia, M.-A.; Andrisani, O.M. Polo-like kinase 1 inhibition suppresses hepatitis B virus X protein-induced transformation in an in vitro model of liver cancer progression. Hepatology 2009, 50, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Ahodantin, J.; Bou-Nader, M.; Cordier, C.; Soussan, P.; Desdouets, C.; Kremsdorf, D. Alteration of hepatocytes polyploidization during liver disease in HBx transgenic mice. In Proceedings of the 2017 International Meeting on the Molecular Biology of Hepatitis B Viruses, Washington DC, USA, 3–7 September 2017. O-90. [Google Scholar]
- Totoki, Y.; Tatsuno, K.; Covington, K.R.; Ueda, H.; Creighton, C.J.; Kato, M.; Tsuji, S.; Donehower, L.A.; Slagle, B.L.; Nakamura, H.; et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat. Genet. 2014, 46, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- Guichard, C.; Amaddeo, G.; Imbeaud, S.; Ladeiro, Y.; Pelletier, L.; Maad, I.B.; Calderaro, J.; Bioulac-Sage, P.; Letexier, M.; Degos, F.; et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 2012, 44, 694–698. [Google Scholar] [CrossRef] [PubMed]
- Schulze, K.; Imbeaud, S.; Letouzé, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Deng, Q.; Wang, Q.; Li, K.Y.; Dai, J.H.; Li, N.; Zhu, Z.D.; Zhou, B.; Liu, X.Y.; Liu, R.F.; et al. Exome sequencing of hepatitis B virus-associated hepatocellular carcinoma. Nat. Genet. 2012, 44, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.A.; Roberts, L.R. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cancer Genome Atlas Res. Netw. Cell 2017, 169, 1327–1341. [Google Scholar] [CrossRef]
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.C.; Llovet, J.M. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology 2015, 149, 1226–1239. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, E.S.; Gopal, P.; Singal, A.G. The changing landscape of hepatocellular carcinoma: Etiology, genetics, and therapy. Am. J. Pathol. 2014, 184, 574–583. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xing, Z.; Mani, S.K.K.; Bancel, B.; Durantel, D.; Zoulim, F.; Tran, E.J.; Merle, P.; Andrisani, O. RNA helicase DEAD box protein 5 regulates Polycomb repressive complex 2/Hox transcript antisense intergenic RNA function in hepatitis B virus infection and hepatocarcinogenesis. Hepatology 2016, 64, 1033–1048. [Google Scholar] [CrossRef] [PubMed]
- Seeger, C.; Mason, W.S. Molecular biology of hepatitis B virus infection. Virology 2015, 479–480, 672–686. [Google Scholar] [CrossRef] [PubMed]
- Pollicino, T.; Belloni, L.; Raffa, G.; Pediconi, N.; Squadrito, G.; Raimondo, G.; Levrero, M. Hepatitis B virus replication is regulated by the acetylation status of hepatitis B virus cccDNA-bound H3 and H4 histones. Gastroenterology 2006, 130, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Vivekanandan, P.; Thomas, D.; Torbenson, M. Hepatitis B viral DNA is methylated in liver tissues. J. Viral Hepat. 2008, 15, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.; Maruvada, P.; Srivastava, S. Epigenetics and Cancer. Crit. Rev. Clin. Lab. Sci. 2004, 41, 585–607. [Google Scholar] [CrossRef] [PubMed]
- Song, M.A.; Tiirikainen, M.; Kwee, S.; Okimoto, G.; Yu, H.; Wong, L.L. Elucidating the Landscape of Aberrant DNA Methylation in Hepatocellular Carcinoma. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Sceusi, E.L.; Loose, D.S.; Wray, C.J. Clinical implications of DNA methylation in hepatocellular carcinoma. HPB 2011, 13, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.S.; Shim, Y.H.; Hwa, J.Y.; Lee, S.K.; Ro, J.Y.; Kim, J.S.; Yu, E. Expression of DNA methyltransferases in multistep hepatocarcinogenesis. Hum. Pathol. 2003, 34, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Shi, J.; Budhu, A.; Yu, Z.; Forgues, M.; Roessler, S.; Ambs, S.; Chen, Y.; Meltzer, P.S.; Croce, C.M.; et al. MicroRNA expression, survival, and response to interferon in liver cancer. N. Engl. J. Med. 2009, 361, 1437–1447. [Google Scholar] [CrossRef] [PubMed]
- Burchard, J.; Zhang, C.; Liu, A.M.; Poon, R.T.P.; Lee, N.P.Y.; Wong, K.F.; Sham, P.C.; Lam, B.Y.; Ferguson, M.D.; Tokiwa, G.; et al. MicroRNA-122 as a regulator of mitochondrial metabolic gene network in hepatocellular carcinoma. Mol. Syst. Biol. 2010, 6. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Yasuda, T.; Saigo, K.; Urashima, T.; Toyoda, H.; Okanoue, T.; Shimotohno, K. Comprehensive analysis of microRNA expression patterns in hepatocellular carcinoma and non-tumorous tissues. Oncogene 2006, 25, 2537–2545. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.M.; Yang, Z.; Zhou, L.; Zhang, F.; Xie, H.Y.; Feng, X.W.; Wu, J.; Zheng, S. Sen Identification of histone deacetylase 3 as a biomarker for tumor recurrence following liver transplantation in HBV-associated hepatocellular carcinoma. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.K.K.; Zhang, H.; Diab, A.; Pascuzzi, P.E.; Lefrançois, L.; Fares, N.; Bancel, B.; Merle, P.; Andrisani, O. EpCAM-regulated intramembrane proteolysis induces a cancer stem cell-like gene signature in hepatitis B virus-infected hepatocytes. J. Hepatol. 2016, 65, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.H.; Studach, L.L.; Andrisani, O.M. Proteins ZNF198 and SUZ12 are down-regulated in hepatitis B virus (HBV) X protein-mediated hepatocyte transformation and in HBV replication. Hepatology 2011, 53, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Diab, A.; Fan, H.; Mani, S.K.K.; Hullinger, R.; Merle, P.; Andrisani, O. PLK1 and HOTAIR accelerate proteasomal degradation of SUZ12 and ZNF198 during hepatitis B virus-induced liver carcinogenesis. Cancer Res. 2015, 75, 2363–2374. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, A.; Totoki, Y.; Abe, T.; Boroevich, K.A.; Hosoda, F.; Nguyen, H.H.; Aoki, M.; Hosono, N.; Kubo, M.; Miya, F.; et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat. Genet. 2012, 44, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhao, H.; Zhang, X.; Wood, L.D.; Anders, R.A.; Choti, M.A.; Pawlik, T.M.; Daniel, H.D.; Kannangai, R.; Offerhaus, G.J.A.; et al. Inactivating mutations of the chromatin remodeling gene ARID2 in hepatocellular carcinoma. Nat. Genet. 2011, 43, 828–829. [Google Scholar] [CrossRef] [PubMed]
- Theunissen, T.W.; Jaenisch, R. Mechanisms of gene regulation in human embryos and pluripotent stem cells. Development 2017, 144, 4496–4509. [Google Scholar] [CrossRef] [PubMed]
- Paksa, A.; Rajagopal, J. The epigenetic basis of cellular plasticity. Curr. Opin. Cell Biol. 2017, 49, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Toh, T.B.; Lim, J.J.; Chow, E.K.-H. Epigenetics in cancer stem cells. Mol. Cancer 2017. [Google Scholar] [CrossRef] [PubMed]
- Mancini, R.; Noto, A.; Pisanu, M.E.; De Vitis, C.; Maugeri-Saccà, M.; Ciliberto, G. Metabolic features of cancer stem cells: the emerging role of lipid metabolism. Oncogene 2018. [Google Scholar] [CrossRef] [PubMed]
- Safa, A.R. Resistance to Cell Death and Its Modulation in Cancer Stem Cells. Crit. Rev. Oncog. 2016, 21, 203–219. [Google Scholar] [CrossRef] [PubMed]
- Gorkin, D.U.; Leung, D.; Ren, B. The 3D genome in transcriptional regulation and pluripotency. Cell Stem Cell 2014, 14, 762–775. [Google Scholar] [CrossRef] [PubMed]
- Stadhouders, R.; Vidal, E.; Serra, F.; Di Stefano, B.; Le Dily, F.; Quilez, J.; Gomez, A.; Collombet, S.; Berenguer, C.; Cuartero, Y.; et al. Transcription factors orchestrate dynamic interplay between genome topology and gene regulation during cell reprogramming. Nat. Genet. 2018, 50, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-P.; Liu, C.-R.; Lee, C.-N.; Chan, T.-S.; Liu, H.E. Targeting c-Myc as a novel approach for hepatocellular carcinoma. World J. Hepatol. 2010, 2, 16. [Google Scholar] [CrossRef] [PubMed]
- Cliff, T.S.; Wu, T.; Boward, B.R.; Yin, A.; Yin, H.; Glushka, J.N.; Prestegaard, J.H.; Dalton, S. MYC Controls Human Pluripotent Stem Cell Fate Decisions through Regulation of Metabolic Flux. Cell Stem Cell 2017, 21, 502–516.e9. [Google Scholar] [CrossRef] [PubMed]
- Klarmann, G.J.; Decker, A.; Farrar, W.L. Epigenetic gene silencing in the Wnt pathway in breast cancer. Epigenetics 2008, 3, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.H.; Choi, Y.L.; Hua, X.Y.; Shin, Y.K.; Song, Y.J.; Youn, S.J.; Yun, H.Y.; Park, S.M.; Kim, W.J.; Kim, H.J.; et al. Increased expression of sonic hedgehog and altered methylation of its promoter region in gastric cancer and its related lesions. Mod. Pathol. 2006. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Vu, T.T.; Datta, P.K. STRAP mediates the stemness of human colorectal cancer cells by epigenetic regulation of Notch pathway. Cancer Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Kelnar, K.; Liu, B.; Chen, X.; Calhoun-Davis, T.; Li, H.; Patrawala, L.; Yan, H.; Jeter, C.; Honorio, S.; et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat. Med. 2011, 17, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.M.; Ng, A.V.; Lam, S.; Hung, J.Y. Side population in human lung cancer cell lines and tumors is enriched with stem-like cancer cells. Cancer Res. 2007, 67, 4827–4833. [Google Scholar] [CrossRef] [PubMed]
- Arzumanyan, A.; Friedman, T.; Ng, I.O.L.; Clayton, M.M.; Lian, Z.; Feitelson, M.A. Does the Hepatitis B Antigen HBx Promote the Appearance of Liver Cancer Stem Cells? Cancer Res. 2011, 71, 3701–3708. [Google Scholar] [CrossRef] [PubMed]
- Widschwendter, M.; Fiegl, H.; Egle, D.; Mueller-Holzner, E.; Spizzo, G.; Marth, C.; Weisenberger, D.J.; Campan, M.; Young, J.; Jacobs, I.; et al. Epigenetic stem cell signature in cancer. Nat. Genet. 2007, 39, 157–158. [Google Scholar] [CrossRef] [PubMed]
- Holczbauer, Á.; Factor, V.M.; Andersen, J.B.; Marquardt, J.U.; Kleiner, D.E.; Raggi, C.; Kitade, M.; Seo, D.; Akita, H.; Durkin, M.E.; et al. Modeling pathogenesis of primary liver cancer in lineage-specific mouse cell types. Gastroenterology 2013, 145, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Tschaharganeh, D.F.; Xue, W.; Calvisi, D.F.; Evert, M.; Michurina, T.V.; Dow, L.E.; Banito, A.; Katz, S.F.; Kastenhuber, E.R.; Weissmueller, S.; Huang, C.-H.; et al. p53-Dependent Nestin Regulation Links Tumor Suppression to Cellular Plasticity in Liver Cancer. Cell 2014, 158, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Fitamant, J.; Kottakis, F.; Benhamouche, S.; Tian, H.S.; Chuvin, N.; Parachoniak, C.A.; Nagle, J.M.; Perera, R.M.; Lapouge, M.; Deshpande, V.; et al. YAP Inhibition Restores Hepatocyte Differentiation in Advanced HCC, Leading to Tumor Regression. Cell Rep. 2015, 10, 1692–1707. [Google Scholar] [CrossRef] [PubMed]
- Tarlow, B.D.; Pelz, C.; Naugler, W.E.; Wakefield, L.; Wilson, E.M.; Finegold, M.J.; Grompe, M. Bipotential adult liver progenitors are derived from chronically injured mature hepatocytes. Cell Stem Cell 2014, 15, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Jörs, S.; Jeliazkova, P.; Ringelhan, M.; Thalhammer, J.; Dürl, S.; Ferrer, J.; Sander, M.; Heikenwalder, M.; Schmid, R.M.; Siveke, J.T.; et al. Lineage fate of ductular reactions in liver injury and carcinogenesis. J. Clin. Invest. 2015, 125, 2445–2457. [Google Scholar] [CrossRef] [PubMed]
- Quasdorff, M.; Hösel, M.; Odenthal, M.; Zedler, U.; Bohne, F.; Gripon, P.; Dienes, H.P.; Drebber, U.; Stippel, D.; Goeser, T.; et al. A concerted action of HNF4α and HNF1α links hepatitis B virus replication to hepatocyte differentiation. Cell. Microbiol. 2008, 10, 1478–1490. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Dao Thi, V.L.; Huang, Y.; Billerbeck, E.; Saha, D.; Hoffmann, H.-H.; Wang, Y.; Silva, L.A.V.; Sarbanes, S.; Sun, T.; et al. Intrinsic Immunity Shapes Viral Resistance of Stem Cells. Cell 2018, 172, 423–438.e25. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.P.; Chen, P.H.; Yu, C.Y.; Chuang, C.Y.; Stone, L.; Hsiao, W.C.; Li, C.L.; Tsai, S.C.; Chen, K.Y.; Chen, H.F.; et al. Epithelial cell adhesion molecule (EpCAM) complex proteins promote transcription factor-mediated pluripotency reprogramming. J. Biol. Chem. 2011, 286, 33520–33532. [Google Scholar] [CrossRef] [PubMed]
- Studach, L.L.; Menne, S.; Cairo, S.; Buendia, M.A.; Hullinger, R.L.; Lefrançois, L.; Merle, P.; Andrisani, O.M. Subset of Suz12/PRC2 target genes is activated during hepatitis B virus replication and liver carcinogenesis associated with HBV X protein. Hepatology 2012, 56, 1240–1251. [Google Scholar] [CrossRef] [PubMed]
- Booth, C.A.G.; Barkas, N.; Neo, W.H.; Boukarabila, H.; Soilleux, E.J.; Giotopoulos, G.; Farnoud, N.; Giustacchini, A.; Ashley, N.; Carrelha, J.; et al. Ezh2 and Runx1 Mutations Collaborate to Initiate Lympho-Myeloid Leukemia in Early Thymic Progenitors. Cancer Cell 2018, 33, 274–291.e8. [Google Scholar] [CrossRef] [PubMed]
- Czermin, B.; Melfi, R.; McCabe, D.; Seitz, V.; Imhof, A.; Pirrotta, V. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell 2002, 111, 185–196. [Google Scholar] [CrossRef]
- Shen, X.; Liu, Y.; Hsu, Y.J.; Fujiwara, Y.; Kim, J.; Mao, X.; Yuan, G.C.; Orkin, S.H. EZH1 Mediates Methylation on Histone H3 Lysine 27 and Complements EZH2 in Maintaining Stem Cell Identity and Executing Pluripotency. Mol. Cell 2008, 32, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Pasini, D.; Bracken, A.P.; Jensen, M.R.; Denchi, E.L.; Helin, K. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J. 2004, 23, 4061–4071. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, S.J.; Yee, D.; Magnuson, T. Polycomb Repressive Complex 2 Is Dispensable for Maintenance of Embryonic Stem Cell Pluripotency. Stem Cells 2008, 26, 1496–1505. [Google Scholar] [CrossRef] [PubMed]
- Faust, C.; Lawson, K.A.; Schork, N.J.; Thiel, B.; Magnuson, T. The Polycomb-group gene eed is required for normal morphogenetic movements during gastrulation in the mouse embryo. Development 1998, 125, 4495–4506. [Google Scholar] [PubMed]
- O’Carroll, D.; Erhardt, S.; Pagani, M.; Barton, S.C.; Surani, M.A.; Jenuwein, T. The Polycomb-Group Gene Ezh2 Is Required for Early Mouse Development. Mol. Cell. Biol. 2001, 21, 4330–4336. [Google Scholar] [CrossRef] [PubMed]
- Brookes, E.; De Santiago, I.; Hebenstreit, D.; Morris, K.J.; Carroll, T.; Xie, S.Q.; Stock, J.K.; Heidemann, M.; Eick, D.; Nozaki, N.; et al. Polycomb associates genome-wide with a specific RNA polymerase II variant, and regulates metabolic genes in ESCs. Cell Stem Cell 2012, 10, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Wu, Z.J.; Groner, A.C.; He, H.H.; Cai, C.; Lis, R.T.; Wu, X.; Stack, E.C.; Loda, M.; Liu, T.; et al. EZH2 Oncogenic Activity in Castration-Resistant Prostate Cancer Cells Is Polycomb-Independent. Science 2012, 338, 1465–1469. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.Z.; Yan, Y.; Wang, X.X.; Jiang, Y.; Xu, H.E. EZH2: biology, disease, and structure-based drug discovery. Acta Pharmacol. Sin. 2014, 35, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Kim, M.; Woo, D.H.; Shin, Y.; Shin, J.; Chang, N.; Oh, Y.T.; Kim, H.; Rheey, J.; Nakano, I.; et al. Phosphorylation of EZH2 Activates STAT3 Signaling via STAT3 Methylation and Promotes Tumorigenicity of Glioblastoma Stem-like Cells. Cancer Cell 2013, 23, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Au, S.L.K.; Wong, C.C.L.; Lee, J.M.F.; Fan, D.N.Y.; Tsang, F.H.; Ng, I.O.L.; Wong, C.M. Enhancer of zeste homolog 2 epigenetically silences multiple tumor suppressor microRNAs to promote liver cancer metastasis. Hepatology 2012, 56, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.S.L.; Lau, S.S.; Chen, Y.; Kondo, Y.; Li, M.S.; Feng, H.; Ching, A.K.; Cheung, K.F.; Wong, H.K.; Tong, J.H.; et al. EZH2-mediated concordant repression of Wnt antagonists promotes β-catenin-dependent hepatocarcinogenesis. Cancer Res. 2011, 71, 4028–4039. [Google Scholar] [CrossRef] [PubMed]
- Studach, L.; Wang, W.H.; Weber, G.; Tang, J.; Hullinger, R.L.; Malbrue, R.; Liu, X.; Andrisani, O. Polo-like kinase 1 activated by the hepatitis B virus X protein attenuates both the DNA damage checkpoint and DNA repair resulting in partial polyploidy. J. Biol. Chem. 2010, 285, 30282–30293. [Google Scholar] [CrossRef] [PubMed]
- Diab, A.M.; Foca, A.; Fusil, F.; Lahlali, T.; Jalaguier, P.; Amirache, F.; N’Guyen, L.; Isorce, N.; Cosset, F.-L.; Zoulim, F.; et al. Polo-like-kinase 1 is a proviral host-factor for hepatitis B virus replication. Hepatology 2017, 66, 1750–1765. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Pasini, D.; Bracken, A.P.; Hansen, J.B.; Capillo, M.; Helin, K. The polycomb group protein Suz12 is required for embryonic stem cell differentiation. Mol. Cell. Biol. 2007, 27, 3769–3779. [Google Scholar] [CrossRef] [PubMed]
- Litvinov, S.V.; Bakker, H.A.M.; Gourevitch, M.M.; Velders, M.P.; Warnaar, S.O. Evidence for a role of the epithelial glycoprotein 40 (ep-CAM) in epithelial cell-cell adhesion. Cell Commun. Adhes. 1994, 2, 417–428. [Google Scholar] [CrossRef]
- Gires, O. Context-dependent adaption of EpCAM expression in early systemic esophageal cancer. Oncogene 2014, 33, 4904–4915. [Google Scholar] [CrossRef]
- Maetzel, D.; Denzel, S.; Mack, B.; Canis, M.; Went, P.; Benk, M.; Kieu, C.; Papior, P.; Baeuerle, P.A.; et al. Nuclear signalling by tumour-associated antigen EpCAM. Nat. Cell Biol. 2009, 11, 162–171. [Google Scholar] [CrossRef] [PubMed]
- González, B.; Denzel, S.; Mack, B.; Conrad, M.; Gires, O. EpCAM is involved in maintenance of the murine embryonic stem cell phenotype. Stem Cells 2009, 27, 1782–1791. [Google Scholar] [CrossRef] [PubMed]
- Balzar, M.; Winter, M.J.; de Boer, C.J.; Litvinov, S. V The biology of the 17-1A antigen (Ep-CAM). J. Mol. Med. 1999, 77, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Munz, M.; Baeuerle, P.A.; Gires, O. The Emerging Role of EpCAM in Cancer and Stem Cell Signaling. Cancer Res. 2009, 69, 5627–5629. [Google Scholar] [CrossRef] [PubMed]
- Went, P.T.; Lugli, A.; Meier, S.; Bundi, M.; Mirlacher, M.; Sauter, G.; Dirnhofer, S. Frequent EpCam Protein Expression in Human Carcinomas. Hum. Pathol. 2004, 35, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Theise, N.; Chua, M.; Reid, L.M. The stem cell niche of human livers: Symmetry between development and regeneration. Hepatology 2008, 48, 1598–1607. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Ji, J.; Budhu, A.; Forgues, M.; Yang, W.; Wang, H.Y.; Jia, H.; Ye, Q.; Qin, L.X.; Wauthier, E.; et al. EpCAM-Positive Hepatocellular Carcinoma Cells Are Tumor-Initiating Cells With Stem/Progenitor Cell Features. Gastroenterology 2009, 136, 1012–1024. [Google Scholar] [CrossRef] [PubMed]
- Strnad, J.; Hamilton, A.E.; Beavers, L.S.; Gamboa, G.C.; Apelgren, L.D.; Taber, L.D.; Sportsman, J.R.; Bumol, T.F.; Sharp, J.D.; Gadski, R.A. Molecular cloning and characterization of a human adenocarcinoma/epithelial cell surface antigen complementary DNA. Cancer Res. 1989, 49, 314–317. [Google Scholar] [PubMed]
- Chaves-Pérez, A.; MacK, B.; Maetzel, D.; Kremling, H.; Eggert, C.; Harréus, U.; Gires, O. EpCAM regulates cell cycle progression via control of cyclin D1 expression. Oncogene 2013, 32, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Oishi, N.; Yamashita, T.; Kaneko, S. Molecular biology of liver cancer stem cells. Liver Cancer 2014, 3, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Fuller-Pace, F. V RNA helicases: modulators of RNA structure. Trends Cell Biol. 1994, 4, 271–274. [Google Scholar] [CrossRef]
- Iggo, R.D.; Lane, D.P. Nuclear protein p68 is an RNA-dependent ATPase. EMBO J. 1989, 8, 1827–1831. [Google Scholar] [PubMed]
- Schmid, S.R.; Linder, P. D-E-A-D protein family of putative RNA helicases. Mol. Microbiol. 1992, 6, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Endoh, H.; Maruyama, K.; Masuhiro, Y.; Kobayashi, Y.; Goto, M.; Tai, H.; Yanagisawa, J.; Metzger, D.; Hashimoto, S.; Kato, S. Purification and identification of p68 RNA helicase acting as a transcriptional coactivator specific for the activation function 1 of human estrogen receptor alpha. Mol. Cell. Biol. 1999, 19, 5363–5372. [Google Scholar] [CrossRef] [PubMed]
- Clark, E.L.; Coulson, A.; Dalgliesh, C.; Rajan, P.; Nicol, S.M.; Fleming, S.; Heer, R.; Gaughan, L.; Leung, H.Y.; Elliott, D.J.; et al. The RNA helicase p68 is a novel androgen receptor coactivator involved in splicing and is overexpressed in prostate cancer. Cancer Res. 2008, 68, 7938–7946. [Google Scholar] [CrossRef] [PubMed]
- Caretti, G.; Schiltz, R.L.; Dilworth, F.J.; Di Padova, M.; Zhao, P.; Ogryzko, V.; Fuller-Pace, F.V.; Hoffman, E.P.; Tapscott, S.J.; Sartorelli, V. The RNA Helicases p68/p72 and the Noncoding RNA SRA Are Coregulators of MyoD and Skeletal Muscle Differentiation. Dev. Cell 2006, 11, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Jensen, E.D.; Niu, L.; Caretti, G.; Nicol, S.M.; Teplyuk, N.; Stein, G.S.; Sartorelli, V.; Van Wijnen, A.J.; Fuller-Pace, F.V.; Westendorf, J.J. p68 (Ddx5) interacts with Runx2 and regulates osteoblast differentiation. J. Cell. Biochem. 2008, 103, 1438–1451. [Google Scholar] [CrossRef] [PubMed]
- Bates, G.J.; Nicol, S.M.; Wilson, B.J.; Jacobs, A.-M.F.; Bourdon, J.-C.; Wardrop, J.; Gregory, D.J.; Lane, D.P.; Perkins, N.D.; Fuller-Pace, F.V. The DEAD box protein p68: A novel transcriptional coactivator of the p53 tumour suppressor. EMBO J. 2005, 24, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Singh, C.; Haines, G.K.; Talamonti, M.S.; Radosevich, J.A. Expression of p68 in human colon cancer. Tumor Biol. 1995, 16, 281–289. [Google Scholar] [CrossRef]
- Haines, G.K.; Cajulis, R.; Hayden, R.; Duda, R.; Talamonti, M.; Radosevich, J.A. Expression of the double-stranded RNA-dependent protein kinase (p68) in human breast tissues. Tumor Biol. 1996, 17, 5–12. [Google Scholar] [CrossRef]
- Lin, S.; Tian, L.; Shen, H.; Gu, Y.; Li, J.L.; Chen, Z.; Sun, X.; James You, M.; Wu, L. DDX5 is a positive regulator of oncogenic NOTCH1 signaling in T cell acute lymphoblastic leukemia. Oncogene 2013, 32, 4845–4853. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Jiao, Z.; Li, R.; Yue, H.; Chen, L. p68 RNA helicase promotes glioma cell proliferation in vitro and in vivo via direct regulation of NF-kappaB transcription factor p50. Neuro. Oncol. 2012, 14, 1116–1124. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, N.; Ojima, H.; Shirakihara, T.; Shimizu, H.; Kokubu, A.; Urushidate, T.; Totoki, Y.; Kosuge, T.; Miyagawa, S.; Shibata, T. Downregulation of the microRNA biogenesis components and its association with poor prognosis in hepatocellular carcinoma. Cancer Sci. 2013, 104, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Andrisani, O.; Cui, Z. The oncogenic microRNA clusters miR17~92 and miR106b~25 drive HBV related HCC progression by downregulating DDX5. In Proceedings of the 2017 International Meeting on the Molecular Biology of Hepatitis B Viruses, Washington DC, USA, 3–7 September 2017. O-189. [Google Scholar]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; Chang, H.Y. Functional Demarcation of Active and Silent Chromatin Domains in Human HOX Loci by Noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Rivea Morales, D.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar] [CrossRef] [PubMed]
- Chiyomaru, T.; Fukuhara, S.; Saini, S.; Majid, S.; Deng, G.; Shahryari, V.; Chang, I.; Tanaka, Y.; Enokida, H.; Nakagawa, M.; et al. Long non-coding RNA hotair is targeted and regulated by MIR-141 in human cancer cells. J. Biol. Chem. 2014, 289, 12550–12565. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Xie, S.; Li, Q.; Ma, J.; Wang, G. Large Intervening Non-Coding RNA HOTAIR is Associated with Hepatocellular Carcinoma Progression. J. Int. Med. Res. 2011, 39, 2119–2128. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhou, L.; Wu, L.-M.; Lai, M.-C.; Xie, H.-Y.; Zhang, F.; Zheng, S.-S. Overexpression of Long Non-coding RNA HOTAIR Predicts Tumor Recurrence in Hepatocellular Carcinoma Patients Following Liver Transplantation. Ann. Surg. Oncol. 2011, 18, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Klingenberg, M.; Matsuda, A.; Diederichs, S.; Patel, T. Non-coding RNA in hepatocellular carcinoma: Mechanisms, biomarkers and therapeutic targets. J. Hepatol. 2017, 67, 603–618. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Abdelmohsen, K.; Kim, J.; Yang, X.; Martindale, J.L.; Tominaga-Yamanaka, K.; White, E.J.; Orjalo, A.V.; Rinn, J.L.; Kreft, S.G.; et al. Scaffold function of long non-coding RNA HOTAIR in protein ubiquitination. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Gene | Protein |
|---|---|
| P | Reverse transcriptase/DNA polymerase (Pol) |
| X | HBx protein |
| C | Capsid protein/Core antigen (HBcAg) |
| S | Surface/Envelope antigen (HBsAg) |
| Epigenetic Modification | Epigenetic Regulators |
|---|---|
| Global hypomethylation, promoter hypermethylation of tumor suppressor and anti-proliferative genes | DNA methyltransferases DNMT1, DNMT3A and DNMT3B over-expressed[48,49] |
| miRNA and Lnc RNA mis-expression | Downregulated: miR-26[50], miR-195, miR-199a, miR-200a, miR-125a, miR-122[51] Upregulated: miR-224, miR-106b~25, miR-17~92, miR-18 [52] |
| Histone modifications | Histone deacetylases: overexpression of HDAC1, HDAC2 and HDAC3 [53]. Overexpression of EZH2 [54] and proteasomal degradation of SUZ12 [55,56], both core subunits of PRC2. |
| Nucleosome re-structuring | Mutations in subunits of SWI/SNF (ARID1A, ARID1B) [57], poly-bromo and BRG1-associated (PBAF) remodeling complex (ARID2) [58] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mani, S.K.K.; Andrisani, O. Hepatitis B Virus-Associated Hepatocellular Carcinoma and Hepatic Cancer Stem Cells. Genes 2018, 9, 137. https://doi.org/10.3390/genes9030137
Mani SKK, Andrisani O. Hepatitis B Virus-Associated Hepatocellular Carcinoma and Hepatic Cancer Stem Cells. Genes. 2018; 9(3):137. https://doi.org/10.3390/genes9030137
Chicago/Turabian StyleMani, Saravana Kumar Kailasam, and Ourania Andrisani. 2018. "Hepatitis B Virus-Associated Hepatocellular Carcinoma and Hepatic Cancer Stem Cells" Genes 9, no. 3: 137. https://doi.org/10.3390/genes9030137
APA StyleMani, S. K. K., & Andrisani, O. (2018). Hepatitis B Virus-Associated Hepatocellular Carcinoma and Hepatic Cancer Stem Cells. Genes, 9(3), 137. https://doi.org/10.3390/genes9030137