Biological Sexing of a 4000-Year-Old Egyptian Mummy Head to Assess the Potential of Nuclear DNA Recovery from the Most Damaged and Limited Forensic Specimens

 , , , , , ,
, , , , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Tooth Extraction
2.2. Strategy for Analyses
2.3. Sample Preparation
2.4. Bioinformatics
2.4.1. Post-Capture Mitochondrial DNA Sequence Analysis
2.4.2. Shotgun Sequence Analysis for Biological Sex Determination
3. Results
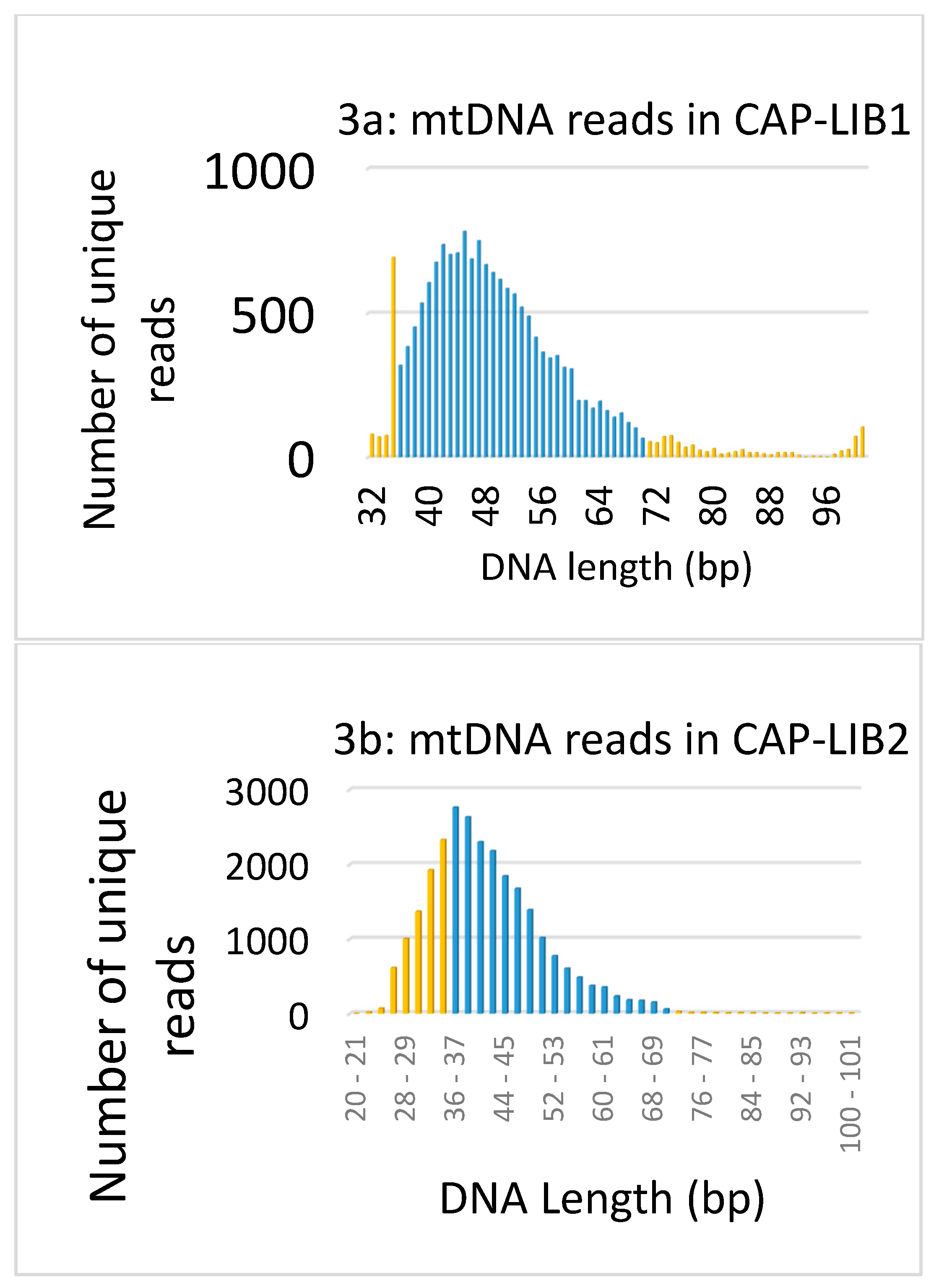
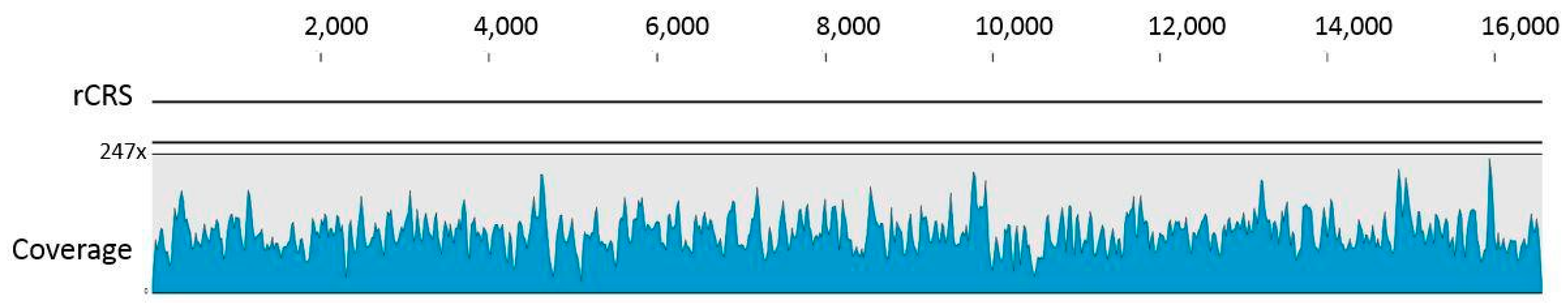
3.1. Data Authentication Based on Captured mtDNA Reads
3.2. Quantification of Deamination
3.3. Contamination
3.4. Mitochondrial Haplotype
3.5. Shotgun Sequencing
3.6. Biological Sex Determination
4. Discussion
4.1. Origins of the Mummy
4.2. Eurasian mtDNA Haplogroups in Ancient Egyptians
4.3. Perspectives for Forensic Laboratories
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Freed, R.E.; Berman, L.M.; Doxey, D.M.; Picardo, N.S. The Secrets of Tomb 10A: Egypt 2000 BC; MFA Publications: Boston, MA, USA, 2009. [Google Scholar]
- Willems, H.O. The nomarchs of the Hare nome and early Middle Kingdom history. In Proceedings of the Fourth International Congress of Egyptology, Munich, Germany, 26 August–1 September 1985. [Google Scholar]
- De Meyer, M.; Dils, P. Fowl for the Governor: the tomb of governor Djehutinakht IV or V at Dayr al-Barshā reinvestigated. Part I. J. Egyptian Archaeol. 2012, 98, 55–72. [Google Scholar] [CrossRef]
- Roth, A.M.; Roehrig, C.H. The Bersha procession: A new reconstruction. J. Mus. Fine Arts 1989, 1, 31–40. [Google Scholar]
- Museum of Fine Arts, Boston. Available online: http://www.mfa.org/collections/object/front-side-panel-of-outer-coffin-of-djehutynakht-142815 (accessed on 26 February 2018).
- De Meyer, M.; Linseele, V.; Vereecken, S.; Williams, L.J. Fowl for the governor. The tomb of governor Djehutinakht IV or V at Dayr al- Barsha reinvestigated. Part 2: Pottery, human remains, and faunal remains. J. Egyptian Archaeol. 2014, 100, 67–87. [Google Scholar] [CrossRef]
- Gupta, R.; Markowitz, Y.; Berman, L.; Chapman, P. High-resolution imaging of an ancient Egyptian mummified head: New insights into the mummification process. Am. J. Neuroradiol. 2008, 29, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R., Anthropologist at the Federal Bureau of Investigation. Personal communication, March 2017.
- Poinar, H.N.; Hoss, M.; Bada, J.L.; Pääbo, S. Amino acid racemization and the preservation of ancient DNA. Science 1996, 272, 864–866. [Google Scholar] [CrossRef] [PubMed]
- Pääbo, S. Molecular cloning of Ancient Egyptian mummy DNA. Nature 1985, 314, 644–645. [Google Scholar] [CrossRef] [PubMed]
- Van der Kuyl, A.C.; Dekker, J.; Attia, M.A.M.; Iskander, N.; Perizonius, W.R.K.; Goudsmit, J. DNA from ancient Egyptian monkey bones. Ancient DNA Newsletters 1994, 2, 19–20. [Google Scholar]
- Smith, C.I.; Chamberlain, A.T.; Riley, M.S.; Stringer, C.; Collins, M.J. The thermal history of human fossils and the likelihood of successful DNA amplification. J. Hum. Evol. 2003, 45, 203–217. [Google Scholar] [CrossRef]
- Lindahl, T.; Nyberg, B. Rate of depurination of native deoxyribonucleic acid. Biochemistry 1972, 11, 3610–3618. [Google Scholar] [CrossRef] [PubMed]
- Marota, I.; Basile, C.; Ubaldi, M.; Rollo, F. DNA decay rate in papyri and human remains from Egyptian archaeological sites. Am. J. Phys. Anthropol. 2002, 117, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.T.; Barnes, I.; Collins, M.J.; Smith, C.; Eklund, J.; Goudsmit, J.; Poinar, H.; Cooper, A. Long-term survival of ancient DNA in Egypt: Response to Zink and Nerlich. Am. J. Phys. Anthropol. 2005, 128, 110–114. [Google Scholar] [CrossRef] [PubMed]
- ForenSeqTM DNA Siganture Prep kit (TG-450–1001); Illumina, Inc.: San Diego, CA, USA, 2017.
- Precision ID Kits (Identity panel; ancestry panel; mtDNA panel); ThermoFisher: Waltham, MA, USA, 2017.
- Nunes, F., Assistant Professor of Clinical Pediatrics (Indiana University School of Medicine). Personal communication, January 2017.
- Hofreiter, M.; Jaenicke, V.; Serre, D.; von Haeseler, A.; Pääbo, S. DNA sequences from multiple amplifications reveal artifacts induced by cytosine deamination in ancient DNA. Nucleic Acids Res. 2001, 29, 4793–4799. [Google Scholar] [CrossRef] [PubMed]
- Stiller, M.; Green, R.E.; Ronan, M.; Simons, J.F.; Du, L.; He, W.; Egholm, M.; Rothberg, J.M.; Keates, S.G.; Ovodov, N.D.; et al. Patterns of nucleotide misincorporations during enzymatic amplification and direct large-scale sequencing of ancient DNA. Proc. Natl. Acad. Sci. USA 2006, 103, 13578–13584. [Google Scholar] [CrossRef] [PubMed]
- Briggs, A.W.; Stenzel, U.; Johnson, P.L.; Green, R.E.; Kelso, J.; Prufer, K.; Meyer, M.; Krause, J.; Ronan, M.T.; Lachmann, M.; et al. Patterns of damage in genomic DNA sequences from a Neandertal. Proc. Natl. Acad. Sci. USA 2007, 104, 14616–14621. [Google Scholar] [CrossRef] [PubMed]
- Loreille, O.M.; Parr, R.L.; McGregor, K.A.; Fitzpatrick, C.M.; Lyon, C.; Yang, D.Y.; Speller, C.F.; Grimm, M.R.; Grimm, M.J.; Irwin, J.A.; et al. Integrated DNA and fingerprint analyses in the identification of 60-year-old mummified human remains discovered in an Alaskan glacier. J. Forensic Sci. 2010, 55, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Dabney, J.; Knapp, M.; Glocke, I.; Gansauge, M.T.; Weihmann, A.; Nickel, B.; Valdiosera, C.; Garcia, N.; Pääbo, S.; Arsuaga, J.L.; et al. Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proc. Natl. Acad. Sci. USA 2013, 110, 15758–15763. [Google Scholar] [CrossRef] [PubMed]
- Korlevic, P.; Gerber, T.; Gansauge, M.T.; Hajdinjak, M.; Nagel, S.; Aximu-Petri, A.; Meyer, M. Reducing microbial and human contamination in DNA extractions from ancient bones and teeth. Biotechniques 2015, 59, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Rohland, N.; Harney, E.; Mallick, S.; Nordenfelt, S.; Reich, D. Partial uracil-DNA-glycosylase treatment for screening of ancient DNA. Phil. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20130624. [Google Scholar] [CrossRef] [PubMed]
- Rohland, N.; Reich, D. Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Res. 2012, 22, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Andrews, R.M.; Kubacka, I.; Chinnery, P.F.; Lightowlers, R.N.; Turnbull, D.M.; Howell, N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 1999, 23, 147. [Google Scholar] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Skoglund, P.; Storå, J.; Götherström, A.; Jakobsson, M. Accurate sex identification of ancient human remains using DNA shotgun sequencing. J. Archaeol. Sci. 2013, 40, 4477–4482. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Picard. Available online: http://broadinstitute.github.io/picard (accessed on 26 February 2018).
- Meyer, M.; Kircher, M.; Gansauge, M.T.; Li, H.; Racimo, F.; Mallick, S.; Schraiber, J.G.; Jay, F.; Prufer, K.; de Filippo, C.; et al. A high-coverage genome sequence from an archaic Denisovan individual. Science 2012, 338, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Arsuaga, J.L.; de Filippo, C.; Nagel, S.; Aximu-Petri, A.; Nickel, B.; Martínez, I.; Gracia, A.; Bermúdez de Castro, J.M.; Carbonell, E.; et al. Nuclear DNA sequences from the Middle Pleistocene Sima de los Huesos hominins. Nature 2016, 531, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, H.; Ginolhac, A.; Schubert, M.; Johnson, P.L.; Orlando, L. MapDamage2.0: Fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 2013, 29, 1682–1684. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Mittnik, A.; Johnson, P.L.F.; Bos, K.; Lari, M.; Bollongino, R.; Sun, C.; Giemsch, L.; Schmitz, R.; Burger, J.; et al. A revised timescale for human evolution based on ancient mitochondrial genomes. Curr. Biol. 2013, 23, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Van Oven, M. PhyloTree Build 17: Growing the human mitochondrial DNA tree. Forensic Sci. Int. Genet. Suppl. Ser. 2015, 5, e392–e394. [Google Scholar] [CrossRef]
- Weissensteiner, H.; Pacher, D.; Kloss-Brandstatter, A.; Forer, L.; Specht, G.; Bandelt, H.J.; Kronenberg, F.; Salas, A.; Schonherr, S. HaploGrep 2: Mitochondrial haplogroup classification in the era of high-throughput sequencing. Nucleic Acids Res. 2016, 44, W58–W63. [Google Scholar] [CrossRef] [PubMed]
- Gamba, C.; Jones, E.R.; Teasdale, M.D.; McLaughlin, R.L.; Gonzalez-Fortes, G.; Mattiangeli, V.; Domboroczki, L.; Kovari, I.; Pap, I.; Anders, A.; et al. Genome flux and stasis in a five millennium transect of European prehistory. Nat. Commun. 2014, 5, 5257. [Google Scholar] [CrossRef] [PubMed]
- Lazaridis, I.; Patterson, N.; Mittnik, A.; Renaud, G.; Mallick, S.; Kirsanow, K.; Sudmant, P.H.; Schraiber, J.G.; Castellano, S.; Lipson, M.; et al. Ancient human genomes suggest three ancestral populations for present-day Europeans. Nature 2014, 513, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Haak, W.; Lazaridis, I.; Patterson, N.; Rohland, N.; Mallick, S.; Llamas, B.; Brandt, G.; Nordenfelt, S.; Harney, E.; Stewardson, K.; et al. Massive migration from the steppe was a source for Indo-European languages in Europe. Nature 2015, 522, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.R.; Gonzalez-Fortes, G.; Connell, S.; Siska, V.; Eriksson, A.; Martiniano, R.; McLaughlin, R.L.; Gallego Llorente, M.; Cassidy, L.M.; Gamba, C.; et al. Upper Palaeolithic genomes reveal deep roots of modern Eurasians. Nat. Commun. 2015, 6, 8912. [Google Scholar] [CrossRef] [PubMed]
- Mathieson, I.; Lazaridis, I.; Rohland, N.; Mallick, S.; Patterson, N.; Roodenberg, S.A.; Harney, E.; Stewardson, K.; Fernandes, D.; Novak, M.; et al. Genome-wide patterns of selection in 230 ancient Eurasians. Nature 2015, 528, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Olalde, I.; Schroeder, H.; Sandoval-Velasco, M.; Vinner, L.; Lobon, I.; Ramirez, O.; Civit, S.; Garcia Borja, P.; Salazar-Garcia, D.C.; Talamo, S.; et al. A common genetic origin for early farmers from Mediterranean Cardial and Central European LBK Cultures. Mol. Biol. Evol. 2015, 32, 3132–3142. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, M.; Steinrucken, M.; Harris, K.; Schiffels, S.; Rasmussen, S.; DeGiorgio, M.; Albrechtsen, A.; Valdiosera, C.; Avila-Arcos, M.C.; Malaspinas, A.S.; et al. Genomic evidence for the Pleistocene and recent population history of Native Americans. Science 2015, 349, aab3884. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, H.; Avila-Arcos, M.C.; Malaspinas, A.S.; Poznik, G.D.; Sandoval-Velasco, M.; Carpenter, M.L.; Moreno-Mayar, J.V.; Sikora, M.; Johnson, P.L.; Allentoft, M.E.; et al. Genome-wide ancestry of 17th-century enslaved Africans from the Caribbean. Proc. Natl. Acad. Sci. USA 2015, 112, 3669–3673. [Google Scholar] [CrossRef] [PubMed]
- Kilinc, G.M.; Omrak, A.; Ozer, F.; Gunther, T.; Buyukkarakaya, A.M.; Bicakci, E.; Baird, D.; Donertas, H.M.; Ghalichi, A.; Yaka, R.; et al. The demographic development of the first farmers in Anatolia. Curr. Biol. 2016, 26, 2659–2666. [Google Scholar] [CrossRef] [PubMed]
- Martiniano, R.; Caffell, A.; Holst, M.; Hunter-Mann, K.; Montgomery, J.; Muldner, G.; McLaughlin, R.L.; Teasdale, M.D.; van Rheenen, W.; Veldink, J.H.; et al. Genomic signals of migration and continuity in Britain before the Anglo-Saxons. Nat. Commun. 2016, 7, 10326. [Google Scholar] [CrossRef] [PubMed]
- Omrak, A.; Gunther, T.; Valdiosera, C.; Svensson, E.M.; Malmstrom, H.; Kiesewetter, H.; Aylward, W.; Stora, J.; Jakobsson, M.; Gotherstrom, A. Genomic evidence establishes Anatolia as the source of the European neolithic gene pool. Curr. Biol. 2016, 26, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Skoglund, P.; Posth, C.; Sirak, K.; Spriggs, M.; Valentin, F.; Bedford, S.; Clark, G.R.; Reepmeyer, C.; Petchey, F.; Fernandes, D.; et al. Genomic insights into the peopling of the Southwest Pacific. Nature 2016, 538, 510–513. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Fortes, G.; Jones, E.R.; Lightfoot, E.; Bonsall, C.; Lazar, C.; Grandal-d’Anglade, A.; Garralda, M.D.; Drak, L.; Siska, V.; Simalcsik, A.; et al. Paleogenomic evidence for multi-generational mixing between Neolithic farmers and Mesolithic hunter-gatherers in the Lower Danube basin. Curr. Biol. 2017, 27, 1801. [Google Scholar] [CrossRef] [PubMed]
- Hedenstierna-Jonson, C.; Kjellstrom, A.; Zachrisson, T.; Krzewinska, M.; Sobrado, V.; Price, N.; Gunther, T.; Jakobsson, M.; Gotherstrom, A.; Stora, J. A female Viking warrior confirmed by genomics. Am. J. Phys. Anthropol. 2017, 164, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Juras, A.; Chylenski, M.; Krenz-Niedbala, M.; Malmstrom, H.; Ehler, E.; Pospieszny, L.; Lukasik, S.; Bednarczyk, J.; Piontek, J.; Jakobsson, M.; et al. Investigating kinship of Neolithic post-LBK human remains from Krusza Zamkowa, Poland using ancient DNA. Forensic Sci. Int. Genet. 2017, 26, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Kennett, D.J.; Plog, S.; George, R.J.; Culleton, B.J.; Watson, A.S.; Skoglund, P.; Rohland, N.; Mallick, S.; Stewardson, K.; Kistler, L.; et al. Archaeogenomic evidence reveals prehistoric matrilineal dynasty. Nat. Commun. 2017, 8, 14115. [Google Scholar] [CrossRef] [PubMed]
- Lindo, J.; Achilli, A.; Perego, U.A.; Archer, D.; Valdiosera, C.; Petzelt, B.; Mitchell, J.; Worl, R.; Dixon, E.J.; Fifield, T.E.; et al. Ancient individuals from the North American Northwest Coast reveal 10,000 years of regional genetic continuity. Proc. Natl. Acad. Sci. USA 2017, 114, 4093–4098. [Google Scholar] [CrossRef] [PubMed]
- Sikora, M.; Seguin-Orlando, A.; Sousa, V.C.; Albrechtsen, A.; Korneliussen, T.; Ko, A.; Rasmussen, S.; Dupanloup, I.; Nigst, P.R.; Bosch, M.D.; et al. Ancient genomes show social and reproductive behavior of early Upper Paleolithic foragers. Science 2017, 358, 659–662. [Google Scholar] [CrossRef] [PubMed]
- Mittnik, A.; Wang, C.C.; Svoboda, J.; Krause, J. A Molecular Approach to the Sexing of the Triple Burial at the Upper Paleolithic Site of Dolni Vestonice. PLoS ONE 2016, 11, e0163019. [Google Scholar] [CrossRef] [PubMed]
- Skoglund, P.; Northoff, B.H.; Shunkov, M.V.; Derevianko, A.P.; Pääbo, S.; Krause, J.; Jakobsson, M. Separating endogenous ancient DNA from modern day contamination in a Siberian Neandertal. Proc. Natl. Acad. Sci. USA 2014, 111, 2229–2234. [Google Scholar] [CrossRef] [PubMed]
- Enk, J.; Rouillard, J.M.; Poinar, H. Quantitative PCR as a predictor of aligned ancient DNA read counts following targeted enrichment. BioTechniques 2013, 55, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, S.; Krause, J.; Guschanski, K.; Savolainen, V.; Pääbo, S. Temporal patterns of nucleotide misincorporations and DNA fragmentation in ancient DNA. PLoS ONE 2012, 7, e34131. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, L.M.; Martiniano, R.; Murphy, E.M.; Teasdale, M.D.; Mallory, J.; Hartwell, B.; Bradley, D.G. Neolithic and Bronze Age migration to Ireland and establishment of the insular Atlantic genome. Proc. Natl. Acad. Sci. USA 2016, 113, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Hofmanova, Z.; Kreutzer, S.; Hellenthal, G.; Sell, C.; Diekmann, Y.; Diez-Del-Molino, D.; van Dorp, L.; Lopez, S.; Kousathanas, A.; Link, V.; et al. Early farmers from across Europe directly descended from Neolithic Aegeans. Proc. Natl. Acad. Sci. USA 2016, 113, 6886–6891. [Google Scholar] [CrossRef] [PubMed]
- Molto, J.E.; Loreille, O.; Mallott, E.K.; Malhi, R.S.; Fast, S.; Daniels-Higginbotham, J.; Marshall, C.; Parr, R. Complete Mitochondrial Genome Sequencing of a Burial from a Romano-Christian Cemetery in the Dakhleh Oasis, Egypt: Preliminary Indications. Genes 2017, 8, 262. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Varela, R.; Gunther, T.; Krzewinska, M.; Stora, J.; Gillingwater, T.H.; MacCallum, M.; Arsuaga, J.L.; Dobney, K.; Valdiosera, C.; Jakobsson, M.; et al. Genomic analyses of Pre-European conquest human remains from the Canary Islands reveal close affinity to modern North Africans. Curr. Biol. 2017, 27, 3396–3402. [Google Scholar] [CrossRef] [PubMed]
- Schlebusch, C.M.; Malmstrom, H.; Gunther, T.; Sjodin, P.; Coutinho, A.; Edlund, H.; Munters, A.R.; Vicente, M.; Steyn, M.; Soodyall, H.; et al. Southern African ancient genomes estimate modern human divergence to 350,000 to 260,000 years ago. Science 2017, 358, 652–655. [Google Scholar] [CrossRef] [PubMed]
- Siska, V.; Jones, E.R.; Jeon, S.; Bhak, Y.; Kim, H.M.; Cho, Y.S.; Kim, H.; Lee, K.; Veselovskaya, E.; Balueva, T.; et al. Genome-wide data from two early Neolithic East Asian individuals dating to 7700 years ago. Sci. Adv. 2017, 3, e1601877. [Google Scholar] [CrossRef] [PubMed]
- Matisoo-Smith, E.A.; Gosling, A.L.; Boocock, J.; Kardailsky, O.; Kurumilian, Y.; Roudesli-Chebbi, S.; Badre, L.; Morel, J.P.; Sebai, L.L.; Zalloua, P.A. A European mitochondrial haplotype identified in ancient Phoenician remains from Carthage, North Africa. PLoS ONE 2016, 11, e0155046. [Google Scholar] [CrossRef] [PubMed]
- Stevanovitch, A.; Gilles, A.; Bouzaid, E.; Kefi, R.; Paris, F.; Gayraud, R.P.; Spadoni, J.L.; El-Chenawi, F.; Beraud-Colomb, E. Mitochondrial DNA sequence diversity in a sedentary population from Egypt. Ann. Hum. Genet. 2004, 68, 23–39. [Google Scholar] [CrossRef] [PubMed]
- Saunier, J.L.; Irwin, J.A.; Strouss, K.M.; Ragab, H.; Sturk, K.A.; Parsons, T.J. Mitochondrial control region sequences from an Egyptian population sample. Forensic Sci. Int. Genet. 2009, 3, e97–e103. [Google Scholar] [CrossRef] [PubMed]
- Kujanova, M.; Pereira, L.; Fernandes, V.; Pereira, J.B.; Cerny, V. Near eastern neolithic genetic input in a small oasis of the Egyptian Western Desert. Am. J. Phys. Anthropol. 2009, 140, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Pagani, L.; Schiffels, S.; Gurdasani, D.; Danecek, P.; Scally, A.; Chen, Y.; Xue, Y.; Haber, M.; Ekong, R.; Oljira, T.; et al. Tracing the route of modern humans out of Africa by using 225 human genome sequences from Ethiopians and Egyptians. Am. J. Hum. Genet. 2015, 96, 986–991. [Google Scholar] [CrossRef] [PubMed]
- Elmadawy, M.A.; Nagai, A.; Gomaa, G.M.; Hegazy, H.M.; Shaaban, F.E.; Bunai, Y. Investigation of mtDNA control region sequences in an Egyptian population sample. Leg. Med. 2013, 15, 338–341. [Google Scholar] [CrossRef] [PubMed]
- EMPOP (https://empop.online/). Population EMP00351; EMPOP: Ismailia, Egypt, 2017. [Google Scholar]
- Schuenemann, V.J.; Peltzer, A.; Welte, B.; van Pelt, W.P.; Molak, M.; Wang, C.C.; Furtwangler, A.; Urban, C.; Reiter, E.; Nieselt, K.; et al. Ancient Egyptian mummy genomes suggest an increase of Sub-Saharan African ancestry in post-Roman periods. Nat. Commun. 2017, 8, 15694. [Google Scholar] [CrossRef] [PubMed]
- Doxey, D.M. Funerary beliefs and practices in the Middle Kingdom. In The Secrets of Tomb 10A; MFA Publications: New York, NY, USA, 2009; pp. 39–64. [Google Scholar]
- Ikram, S.; Dodson, A. The Mummy in Ancient Egypt: Equipping the Dead for Eternity; Thames & Hudson Ltd.: London, UK, 1998. [Google Scholar]
- Aufderheide, A.C. The Scientific Study of Mummies; Cambridge University Press: Cambridge, UK, 2003. [Google Scholar]
- Bard, K.A. An Introduction to the Archaeology of Ancient Egypt; Wiley-Blackwell: Hoboken, NJ, USA, 2015. [Google Scholar]
- De Meyer, M. The tomb of Henu at Deir el-Bersha. Egyptian Archaeol. 2007, 31, 20–24. [Google Scholar]
- Khairat, R. Next generation sequencing of DNA extracted from mummified tissue. Ph.D. Thesis, Universität Tübingen, Tübingen, Germany, 2013. [Google Scholar]
- Morris, A.G.; Heinze, A.; Chan, E.K.; Smith, A.B.; Hayes, V.M. First ancient mitochondrial human genome from a prepastoralist southern African. Genome Biol. Evol. 2014, 6, 2647–2653. [Google Scholar] [CrossRef] [PubMed]
- Gallego Llorente, M.; Jones, E.R.; Eriksson, A.; Siska, V.; Arthur, K.W.; Arthur, J.W.; Curtis, M.C.; Stock, J.T.; Coltorti, M.; Pieruccini, P.; et al. Ancient Ethiopian genome reveals extensive Eurasian admixture throughout the African continent. Science 2015, 350, 820–822. [Google Scholar] [CrossRef] [PubMed]
- Bramanti, B.; Thomas, M.G.; Haak, W.; Unterlaender, M.; Jores, P.; Tambets, K.; Antanaitis-Jacobs, I.; Haidle, M.N.; Jankauskas, R.; Kind, C.J.; et al. Genetic discontinuity between local hunter-gatherers and central Europe’s first farmers. Science 2009, 326, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Posth, C.; Renaud, G.; Mittnik, A.; Drucker, D.G.; Rougier, H.; Cupillard, C.; Valentin, F.; Thevenet, C.; Furtwangler, A.; Wissing, C.; et al. Pleistocene mitochondrial genomes suggest a single major dispersal of non-Africans and a late glacial population turnover in Europe. Curr. Biol. 2016, 26, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Bietak, M. Egypt and the Levant in the Egyptian World; Wilkinson, T., Ed.; Routledge: New York, NY, USA, 2007; pp. 417–448. [Google Scholar]
- Picardo, N.S. Middle Kingdom History, Politics and Social Organization. In The Secrets of Tomb 10A; MFA Publications: New York, NY, USA, 2009; pp. 17–38. [Google Scholar]
- Wilkinson, T. Early Dynastic Egypt; Routledge: New York, NY, USA, 2001. [Google Scholar]
- Skoglund, P.; Thompson, J.C.; Prendergast, M.E.; Mittnik, A.; Sirak, K.; Hajdinjak, M.; Salie, T.; Rohland, N.; Mallick, S.; Peltzer, A.; et al. Reconstructing Prehistoric African Population Structure. Cell 2017, 171, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Drosou, K.; Price, C.; Brown, T.A. The kinship of two 12th Dynasty mummies revealed by ancient DNA sequencing. J. Arch. Sci. 2018, 17, 793–797. [Google Scholar] [CrossRef]
- Fregel, R.; Mendez, F.L.; Bokbot, Y.; Martin-Soca, D.; Camalich-Massieu, M.D.; Ávila-Arcos, M.C.; Underhill, P.A.; Shapiro, B.; Wojcik, G.; Rodríguez-Santos, F.J.; et al. Neolithization of North Africa involved 1 the migration of people from both the Levant and Europe. bioRxiv 2018. [Google Scholar] [CrossRef]
- Fazi, A.; Gobeski, B.; Foran, D. Development of two highly sensitive forensic sex determination assays based on human DYZ1 and Alu repetitive DNA elements. Electrophoresis 2014, 35, 3028–3035. [Google Scholar] [CrossRef] [PubMed]
- Loreille, O.; Koshinsky, H.; Fofanov, V.Y.; Irwin, J.A. Application of next generation sequencing technologies to the identification of highly degraded unknown soldiers’ remains. Forensic Sci. Int. Genet. Suppl. Ser. 2011, 3, e540–e541. [Google Scholar] [CrossRef]
- Templeton, J.E.; Brotherton, P.M.; Llamas, B.; Soubrier, J.; Haak, W.; Cooper, A.; Austin, J.J. DNA capture and next-generation sequencing can recover whole mitochondrial genomes from highly degraded samples for human identification. Invest. Genet. 2013, 4, 26. [Google Scholar] [CrossRef] [PubMed]
- Eduardoff, M.; Xavier, C.; Strobl, C.; Casas-Vargas, A.; Parson, W. Optimized mtDNA control region primer extension capture analysis for forensically relevant samples and highly compromised mtDNA of different age and origin. Genes 2017, 8, 237. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.; Sturk-Andreaggi, K.; Daniels-Higginbotham, J.; Oliver, R.S.; Barritt-Ross, S.; McMahon, T.P. Performance evaluation of a mitogenome capture and Illumina sequencing protocol using non-probative, case-type skeletal samples: Implications for the use of a positive control in a next-generation sequencing procedure. Forensic Sci. Int. Genet. 2017, 31, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Parson, W.; Huber, G.; Moreno, L.; Madel, M.B.; Brandhagen, M.D.; Nagl, S.; Xavier, C.; Eduardoff, M.; Callaghan, T.C.; Irwin, J.A. Massively parallel sequencing of complete mitochondrial genomes from hair shaft samples. Forensic Sci. Int. Genet. 2015, 15, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Pakstis, A.J.; Speed, W.C.; Fang, R.; Hyland, F.C.; Furtado, M.R.; Kidd, J.R.; Kidd, K.K. SNPs for a universal individual identification panel. Hum. Genet. 2010, 127, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, J.J.; Phillips, C.; Borsting, C.; Balogh, K.; Bogus, M.; Fondevila, M.; Harrison, C.D.; Musgrave-Brown, E.; Salas, A.; Syndercombe-Court, D.; et al. A multiplex assay with 52 single nucleotide polymorphisms for human identification. Electrophoresis 2006, 27, 1713–1724. [Google Scholar] [CrossRef] [PubMed]
- Kidd, K.K.; Pakstis, A.J.; Speed, W.C.; Grigorenko, E.L.; Kajuna, S.L.; Karoma, N.J.; Kungulilo, S.; Kim, J.J.; Lu, R.B.; Odunsi, A.; et al. Developing a SNP panel for forensic identification of individuals. For. Sci. Int. 2006, 164, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Lazaridis, I.; Nadel, D.; Rollefson, G.; Merrett, D.C.; Rohland, N.; Mallick, S.; Fernandes, D.; Novak, M.; Gamarra, B.; Sirak, K.; et al. Genomic insights into the origin of farming in the ancient Near East. Nature 2016, 536, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Posth, C.; Hajdinjak, M.; Petr, M.; Mallick, S.; Fernandes, D.; Furtwangler, A.; Haak, W.; Meyer, M.; Mittnik, A.; et al. The genetic history of Ice Age Europe. Nature 2016, 534, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Kanzawa-Kiriyama, H.; Kryukov, K.; Jinam, T.A.; Hosomichi, K.; Saso, A.; Suwa, G.; Ueda, S.; Yoneda, M.; Tajima, A.; Shinoda, K.I.; et al. A partial nuclear genome of the Jomons who lived 3000 years ago in Fukushima, Japan. J. Hum. Genet. 2017, 62, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Gargis, A.S.; Kalman, L.; Lubin, I.M. Assuring the Quality of Next-Generation Sequencing in Clinical Microbiology and Public Health Laboratories. J. Clinic. Microbiol. 2016, 54, 2857–2865. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total # Reads | # Unique Reads Mapped to hg19 | # Unique Reads Mapped to the mtGenome | ||
|---|---|---|---|---|
| Cap-Lib1 | RB | 428,192 | 560 | 0 |
| NC | 47,414 | 121 | 0 | |
| Cap2-Lib2 | RB | 744,228 | 570 | 10 |
| NC | 250,282 | 128 | 7 |
| Sequence Statistics | FBI Shotgun-Lib1 | HMS Shotgun-Lib2 | |
|---|---|---|---|
| a | Number of raw paired reads | 164,451,485 | 266,162,607 |
| b | Number of reads mapped to the human genome hg19 and rCRS | 3,692,504 | 19,485,309 |
| c | Percentage of endogenous human DNA | 2.24% | 6.57% |
| d | Number of unique human reads with Q >30 | 1,595,239 | 7,691,326 |
| e | Average coverage hg19 Average coverage mtGenome | 0.02× 4.21× | 0.09× 8.93× |
| f | Number of unique mapped human reads with signs of damage (PMDtools score >3) | 518,381 | 344,995 |
| Samples | Lib1 | Sex | Lib2 | Sex |
|---|---|---|---|---|
| Mapped reads | 1,595,239 | 7,691,326 | ||
| Mapped to X | 37,605 | 176,181 | ||
| Mapped to Y | 3732 | 16,469 | ||
| RY | 0.090 | 0.0855 | ||
| 95% CI | 0.087–0.093 | ♂ | 0.084–0.087 | ♂ |
| RX | 0.45 | 0.433 | ||
| 95% CI | 0.429–0.471 | ♂ | 0.4–0.466 | ♂ |
| Samples | Lib1 | Sex | Lib2 | Sex |
|---|---|---|---|---|
| Mapped reads | 518,381 | 344,995 | ||
| Mapped to X | 11,512 | 6688 | ||
| Mapped to Y | 1133 | 637 | ||
| RY | 0.0896 | 0.087 | ||
| 95% CI | 0.0846–0.0946 | ♂ | 0.0805–0.0934 | ♂ |
| RX | 0.4216 | 0.3638 | ||
| 95% CI | 0.3987–0.4447 | ♂ | 0.3261–0.4015 | ♂ |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loreille, O.; Ratnayake, S.; Bazinet, A.L.; Stockwell, T.B.; Sommer, D.D.; Rohland, N.; Mallick, S.; Johnson, P.L.F.; Skoglund, P.; Onorato, A.J.; et al. Biological Sexing of a 4000-Year-Old Egyptian Mummy Head to Assess the Potential of Nuclear DNA Recovery from the Most Damaged and Limited Forensic Specimens. Genes 2018, 9, 135. https://doi.org/10.3390/genes9030135
Loreille O, Ratnayake S, Bazinet AL, Stockwell TB, Sommer DD, Rohland N, Mallick S, Johnson PLF, Skoglund P, Onorato AJ, et al. Biological Sexing of a 4000-Year-Old Egyptian Mummy Head to Assess the Potential of Nuclear DNA Recovery from the Most Damaged and Limited Forensic Specimens. Genes. 2018; 9(3):135. https://doi.org/10.3390/genes9030135
Chicago/Turabian StyleLoreille, Odile, Shashikala Ratnayake, Adam L. Bazinet, Timothy B. Stockwell, Daniel D. Sommer, Nadin Rohland, Swapan Mallick, Philip L.F. Johnson, Pontus Skoglund, Anthony J. Onorato, and et al. 2018. "Biological Sexing of a 4000-Year-Old Egyptian Mummy Head to Assess the Potential of Nuclear DNA Recovery from the Most Damaged and Limited Forensic Specimens" Genes 9, no. 3: 135. https://doi.org/10.3390/genes9030135
APA StyleLoreille, O., Ratnayake, S., Bazinet, A. L., Stockwell, T. B., Sommer, D. D., Rohland, N., Mallick, S., Johnson, P. L. F., Skoglund, P., Onorato, A. J., Bergman, N. H., Reich, D., & Irwin, J. A. (2018). Biological Sexing of a 4000-Year-Old Egyptian Mummy Head to Assess the Potential of Nuclear DNA Recovery from the Most Damaged and Limited Forensic Specimens. Genes, 9(3), 135. https://doi.org/10.3390/genes9030135