Detection and Analysis of RNA Ribose 2′-O-Methylations: Challenges and Solutions



Abstract

1. Introduction
2. Classical Methods for 2′-O-Methylation Detection
2.1. General Methods of RNA Analytical Chemistry
2.2. Specific Detection Strategies
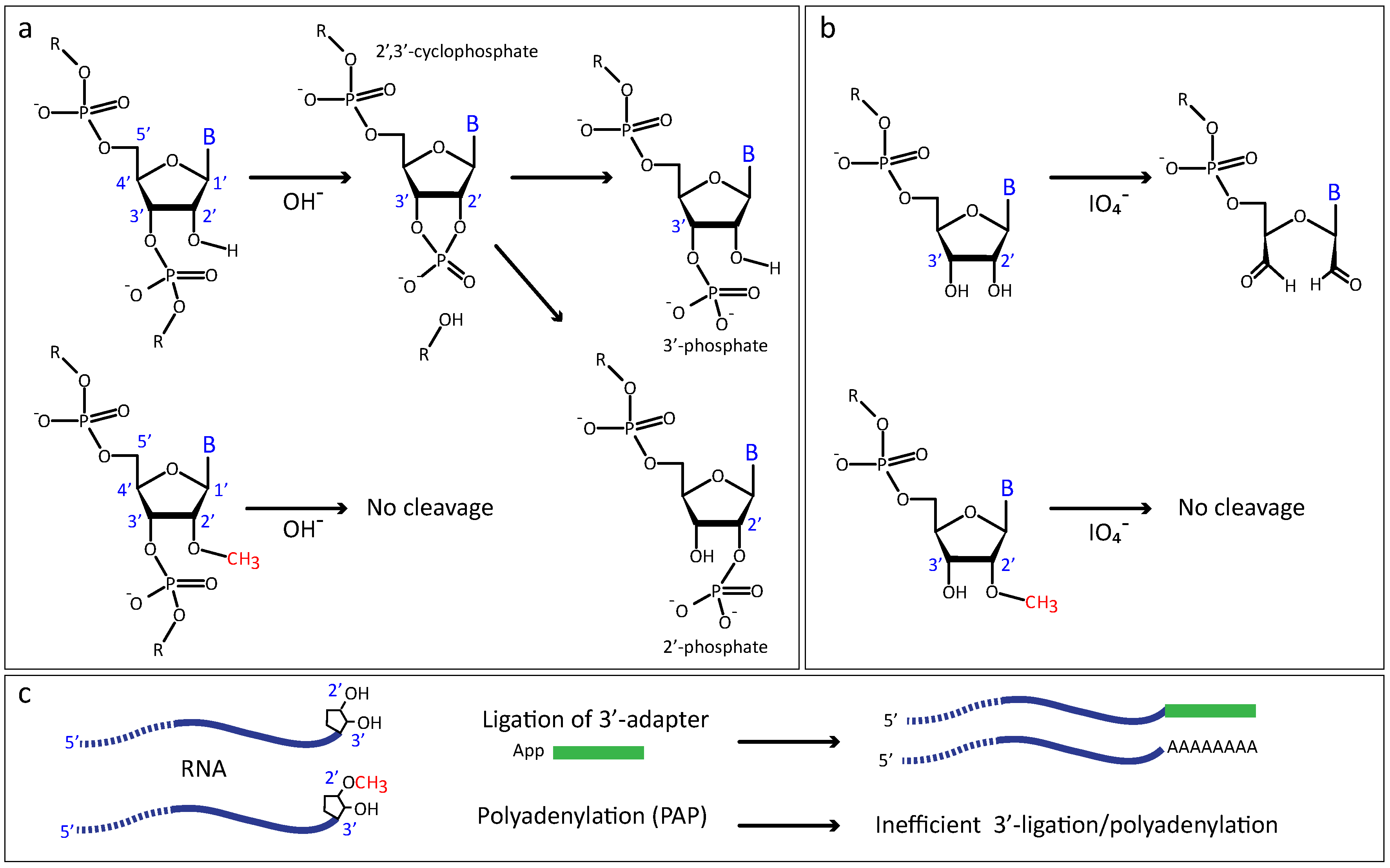
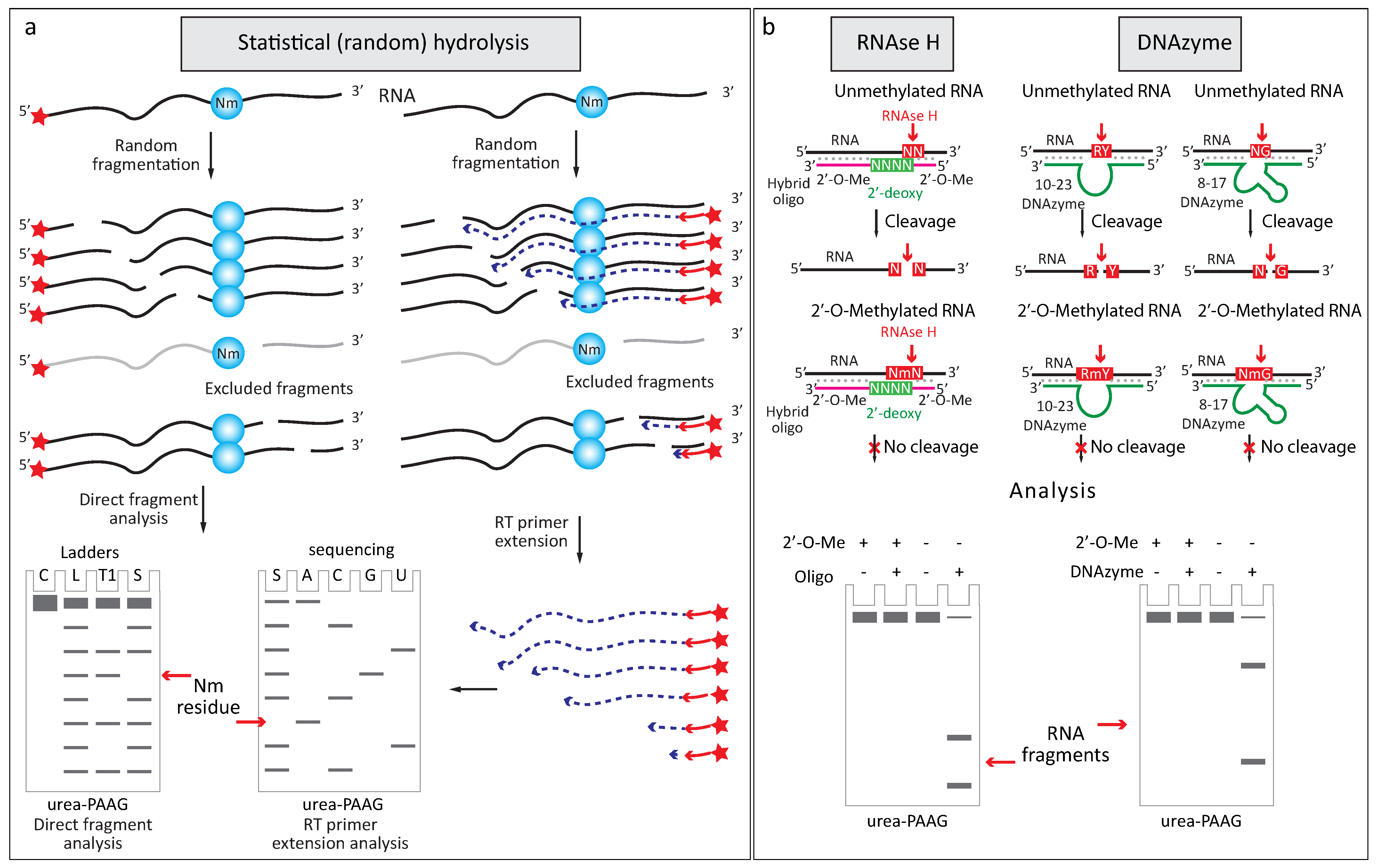
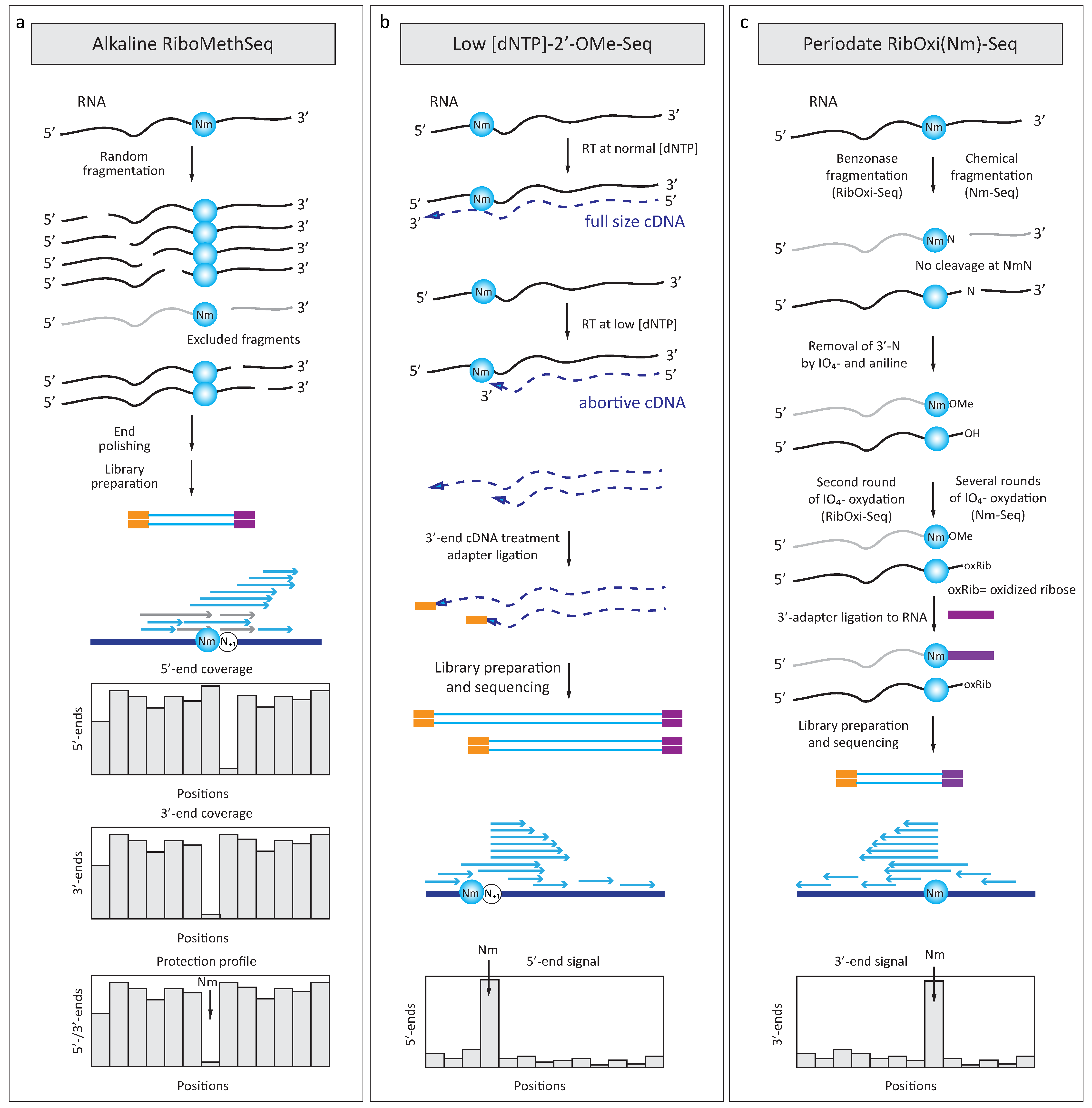
2.2.1. Increased Resistance to Alkaline or Enzymatic Hydrolysis
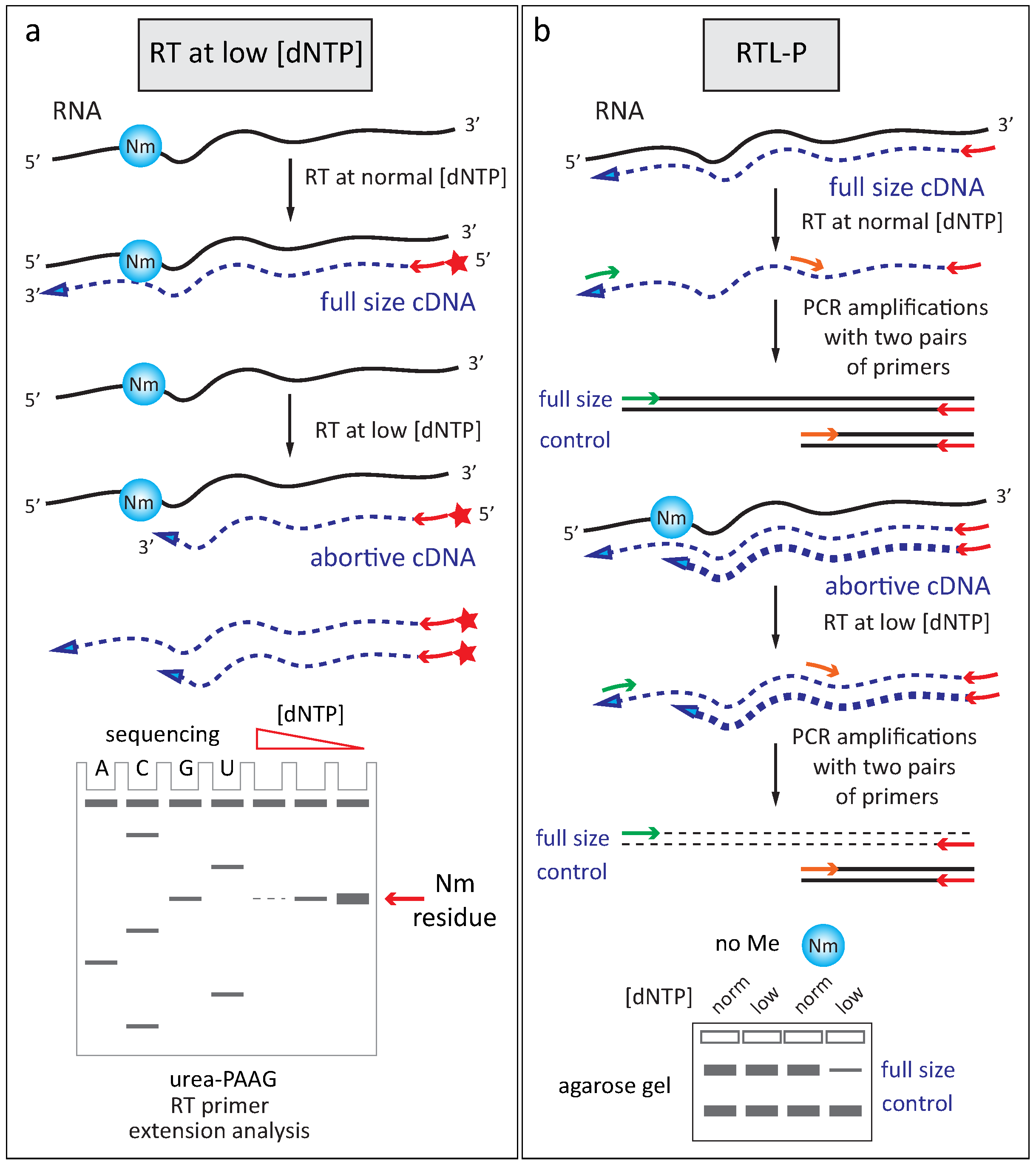
2.2.2. Reverse Transciptase Stop at Low Deoxynucleotide Triphosphates Concentration
2.2.3. Altered Enzymatic Activity with 2′-O-Me RNA 3′-Termini
3. Limitations of Classical Detection Methods
- The required amounts of input RNA are quite substantial and a purification step is generally indispensable, making analysis possible only for highly abundant RNAs.
- Quantification is difficult and all approaches are laborious, time consuming and do not allow high-throughput analyses.
4. Deep Sequencing-based Approaches
4.1. RiboMethSeq
4.2. 2′-OMe-Seq
4.3. RibOxi-Seq and Nm-Seq
5. Specific Features of Deep Sequencing Methods
5.1. Area of Applications
5.2. Specificity and Sensitivity of 2′-O-Methylation Detection
5.3. Required Amount of Input RNA
5.4. Required Depth of Sequencing
5.5. Quantification of the Methylation Level
5.6. Sequencing and Bioinformatics Issues
6. Conclusions
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Ayadi, L.; Galvanin, A.; Pichot, F.; Marchand, V.; Motorin, Y. RNA ribose methylation (2′-O-methylation): Occurrence, biosynthesis and biological functions. BBA Gene Regul. Mech. 2019, in press. [Google Scholar] [CrossRef]
- Kawai, G.; Ue, H.; Yasuda, M.; Sakamoto, K.; Hashizume, T.; McCloskey, J.A.; Miyazawa, T.; Yokoyama, S. Relation between functions and conformational characteristics of modified nucleosides found in tRNAs. Nucl. Acids Symp. Ser. 1991, 49–50. [Google Scholar]
- Kawai, G.; Yamamoto, Y.; Kamimura, T.; Masegi, T.; Sekine, M.; Hata, T.; Iimori, T.; Watanabe, T.; Miyazawa, T.; Yokoyama, S. Conformational rigidity of specific pyrimidine residues in tRNA arises from posttranscriptional modifications that enhance steric interaction between the base and the 2′-hydroxyl group. Biochemistry 1992, 31, 1040–1046. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, P.; Yathindra, N.; Sundaralingam, M. Effect of ribose O(2′)-methylation on the conformation of nucleosides and nucleotides. Biochim. Biophys. Acta 1974, 366, 115–123. [Google Scholar] [CrossRef]
- Monaco, P.L.; Marcel, V.; Diaz, J.-J.; Catez, F. 2′-O-Methylation of Ribosomal ribosomal RNA: Towards an epitranscriptomic control of translation? Biomolecules 2018, 8, 106. [Google Scholar] [CrossRef] [PubMed]
- Natchiar, S.K.; Myasnikov, A.G.; Hazemann, I.; Klaholz, B.P. Visualizing the role of 2′-OH rRNA methylations in the human ribosome structure. Biomolecules 2018, 8, 125. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Chen, X. Analysis of miRNA Modifications. Methods Mol. Biol. 2010, 592, 137–148. [Google Scholar] [PubMed]
- Munafó, D.B.; Robb, G.B. Optimization of enzymatic reaction conditions for generating representative pools of cDNA from small RNA. RNA 2010, 16, 2537–2552. [Google Scholar] [CrossRef] [PubMed]
- Dard-Dascot, C.; Naquin, D.; d’Aubenton-Carafa, Y.; Alix, K.; Thermes, C.; van Dijk, E. Systematic comparison of small RNA library preparation protocols for next-generation sequencing. BMC Genom. 2018, 19, 118. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Ebright, Y.W.; Yu, B.; Chen, X. HEN1 recognizes 21–24 nt small RNA duplexes and deposits a methyl group onto the 2′ OH of the 3′ terminal nucleotide. Nucl. Acids Res. 2006, 34, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Qu, S.; Sun, W.; Zeng, Z.; Liang, H.; Zhang, C.-Y.; Chen, X.; Zen, K. Direct quantification of 3′ terminal 2′-O-methylation of small RNAs by RT-qPCR. RNA 2018, 24, 1520–1529. [Google Scholar] [CrossRef] [PubMed]
- Maden, B.E. Mapping 2′-O-methyl groups in ribosomal RNA. Methods 2001, 25, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Baskin, F.; Dekker, C.A. A rapid and specific assay for sugar methylation in ribonucleic acid. J. Biol. Chem. 1967, 242, 5447–5449. [Google Scholar] [PubMed]
- Trim, A.R.; Parker, J.E. Nucleotide sequence in fourteen dinucleotides, modified by 2′-O-methylation, from yeast ribonucleic acid, determined by periodate degradation and by pentose analysis. Anal. Biochem. 1972, 46, 482–488. [Google Scholar] [CrossRef]
- Abbate, J.; Rottman, F. Gas chromatographic method for determination of 2′-O-methylation in RNA. Anal. Biochem. 1972, 47, 378–388. [Google Scholar] [CrossRef]
- Sardana, M.K.; Fuke, M. A rapid procedure to determine the content of 2′-O-methylation in RNA by homochromatography. Anal. Biochem. 1980, 103, 285–288. [Google Scholar] [CrossRef]
- Qiu, F.; McCloskey, J.A. Selective detection of ribose-methylated nucleotides in RNA by a mass spectrometry-based method. Nucl. Acids Res. 1999, 27, e20. [Google Scholar] [CrossRef] [PubMed]
- Kirpekar, F.; Hansen, L.H.; Rasmussen, A.; Poehlsgaard, J.; Vester, B. The archaeon Haloarcula marismortui has few modifications in the central parts of its 23S ribosomal RNA. J. Mol. Biol. 2005, 348, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Takeda, N.; Nakamura, M.; Yoshizumi, H.; Tatematsu, A. Detection of ribose-methylated nucleotides in Pyrodictium occultum tRNA by liquid chromatography—frit-fast atom bombardment mass spectrometry. J. Chromatogr. B Biomed. Sci. Appl. 1994, 660, 223–233. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, Y. Differentiation of 2′-O- and 3′-O-methylated ribonucleosides by tandem mass spectrometry. J. Am. Soc. Mass. Spectrom. 2006, 17, 1096–1099. [Google Scholar] [CrossRef] [PubMed]
- Kellner, S.; Burhenne, J.; Helm, M. Detection of RNA modifications. RNA Biol. 2010, 7, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Gaston, K.W.; Limbach, P.A. The identification and characterization of non-coding and coding RNAs and their modified nucleosides by mass spectrometry. RNA Biol. 2014, 11, 1568–1585. [Google Scholar] [CrossRef] [PubMed]
- Jora, M.; Lobue, P.A.; Ross, R.L.; Williams, B.; Addepalli, B. Detection of ribonucleoside modifications by liquid chromatography coupled with mass spectrometry. Biochim. Biophys. Acta Gene Regul. Mech. 2018. [Google Scholar] [CrossRef] [PubMed]
- Cavaillé, J.; Chetouani, F.; Bachellerie, J.P. The yeast Saccharomyces cerevisiae YDL112w ORF encodes the putative 2′-O-ribose methyltransferase catalyzing the formation of Gm18 in tRNAs. RNA 1999, 5, 66–81. [Google Scholar] [CrossRef] [PubMed]
- Bonnerot, C.; Pintard, L.; Lutfalla, G. Functional redundancy of Spb1p and a snR52-dependent mechanism for the 2′-O-ribose methylation of a conserved rRNA position in yeast. Mol. Cell 2003, 12, 1309–1315. [Google Scholar] [CrossRef]
- Pintard, L.; Lecointe, F.; Bujnicki, J.M.; Bonnerot, C.; Grosjean, H.; Lapeyre, B. Trm7p catalyses the formation of two 2′-O-methylriboses in yeast tRNA anticodon loop. EMBO J. 2002, 21, 1811–1820. [Google Scholar] [CrossRef] [PubMed]
- Brand, R.C.; Klootwijk, J.; Van Steenbergen, T.J.; De Kok, A.J.; Planta, R.J. Secondary methylation of yeast ribosomal precursor RNA. Eur. J. Biochem. 1977, 75, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Maden, B.E. Identification of the locations of the methyl groups in 18 S ribosomal RNA from Xenopus laevis and man. J. Mol. Biol. 1986, 189, 681–699. [Google Scholar] [CrossRef]
- Maden, B.E. Locations of methyl groups in 28S rRNA of Xenopus laevis and man. Clustering in the conserved core of molecule. J. Mol. Biol. 1988, 201, 289–314. [Google Scholar] [CrossRef]
- Yang, J.; Sharma, S.; Kötter, P.; Entian, K.-D. Identification of a new ribose methylation in the 18S rRNA of S. cerevisiae. Nucl. Acids Res. 2015, 43, 2342–2352. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Sharma, S.; Watzinger, P.; Hartmann, J.D.; Kötter, P.; Entian, K.-D. Mapping of complete set of ribose and base modifications of yeast rRNA by RP-HPLC and mung bean nuclease assay. PLoS ONE 2016, 11, e0168873. [Google Scholar] [CrossRef] [PubMed]
- Taoka, M.; Nobe, Y.; Yamaki, Y.; Yamauchi, Y.; Ishikawa, H.; Takahashi, N.; Nakayama, H.; Isobe, T. The complete chemical structure of Saccharomyces cerevisiae rRNA: Partial pseudouridylation of U2345 in 25S rRNA by snoRNA snR9. Nucl. Acids Res. 2016, 44, 8951–8961. [Google Scholar] [CrossRef] [PubMed]
- Taoka, M.; Nobe, Y.; Yamaki, Y.; Sato, K.; Ishikawa, H.; Izumikawa, K.; Yamauchi, Y.; Hirota, K.; Nakayama, H.; Takahashi, N.; et al. Landscape of the complete RNA chemical modifications in the human 80S ribosome. Nucl. Acids Res. 2018, 46, 9289–9298. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Lane, B.G. The alkali-stable dinucleotide sequences in 18S + 28S ribonucleates from wheat germ. Can. J. Biochem. 1964, 42, 1011–1021. [Google Scholar] [CrossRef] [PubMed]
- Trim, A.R.; Parker, J.E. Preparation, purification and analyses of thirteen alkali-stable dinucleotides from yeast ribonucleic acid. Biochem. J. 1970, 116, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Tycowski, K.T.; Smith, C.M.; Shu, M.D.; Steitz, J.A. A small nucleolar RNA requirement for site-specific ribose methylation of rRNA in Xenopus. Proc. Natl. Acad. Sci. USA 1996, 93, 14480–14485. [Google Scholar] [CrossRef] [PubMed]
- Kiss-László, Z.; Henry, Y.; Bachellerie, J.P.; Caizergues-Ferrer, M.; Kiss, T. Site-specific ribose methylation of preribosomal RNA: A novel function for small nucleolar RNAs. Cell 1996, 85, 1077–1088. [Google Scholar] [CrossRef]
- Motorin, Y.; Muller, S.; Behm-Ansmant, I.; Branlant, C. Identification of modified residues in RNAs by reverse transcription-based methods. Meth. Enzymol. 2007, 425, 21–53. [Google Scholar] [PubMed]
- Huang, C.; Karijolich, J.; Yu, Y.-T. Detection and quantification of RNA 2′-O-methylation and pseudouridylation. Methods 2016, 103, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.T.; Shu, M.D.; Steitz, J.A. A new method for detecting sites of 2′-O-methylation in RNA molecules. RNA 1997, 3, 324–331. [Google Scholar] [PubMed]
- Lapham, J.; Yu, Y.T.; Shu, M.D.; Steitz, J.A.; Crothers, D.M. The position of site-directed cleavage of RNA using RNase H and 2′-O-methyl oligonucleotides is dependent on the enzyme source. RNA 1997, 3, 950–951. [Google Scholar] [PubMed]
- Buchhaupt, M.; Peifer, C.; Entian, K.-D. Analysis of 2′-O-methylated nucleosides and pseudouridines in ribosomal RNAs using DNAzymes. Anal. Biochem. 2007, 361, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. A computational screen for methylation guide snoRNAs in yeast. Science 1999, 283, 1168–1171. [Google Scholar] [CrossRef] [PubMed]
- Maden, B.E.; Corbett, M.E.; Heeney, P.A.; Pugh, K.; Ajuh, P.M. Classical and novel approaches to the detection and localization of the numerous modified nucleotides in eukaryotic ribosomal RNA. Biochimie 1995, 77, 22–29. [Google Scholar] [CrossRef]
- Rebane, A.; Roomere, H.; Metspalu, A. Locations of several novel 2′-O-methylated nucleotides in human 28S rRNA. BMC Mol. Biol. 2002, 3, 1. [Google Scholar] [CrossRef]
- Higa, S.; Maeda, N.; Kenmochi, N.; Tanaka, T. Location of 2(‘)-O-methyl nucleotides in 26S rRNA and methylation guide snoRNAs in Caenorhabditis elegans. Biochem. Biophys. Res. Commun. 2002, 297, 1344–1349. [Google Scholar] [CrossRef]
- Piekna-Przybylska, D.; Decatur, W.A.; Fournier, M.J. The 3D rRNA modification maps database: With interactive tools for ribosome analysis. Nucl. Acids Res. 2008, 36, D178–D183. [Google Scholar] [CrossRef] [PubMed]
- Filippova, J.A.; Stepanov, G.A.; Semenov, D.V.; Koval, O.A.; Kuligina, E.V.; Rabinov, I.V.; Richter, V.A. Modified method of rRNA structure analysis reveals novel characteristics of box C/D RNA analogues. Acta Nat. 2015, 7, 64–73. [Google Scholar]
- Dong, Z.-W.; Shao, P.; Diao, L.-T.; Zhou, H.; Yu, C.-H.; Qu, L.-H. RTL-P: A sensitive approach for detecting sites of 2′-O-methylation in RNA molecules. Nucl. Acids Res. 2012, 40, e157. [Google Scholar] [CrossRef] [PubMed]
- Aschenbrenner, J.; Marx, A. Direct and site-specific quantification of RNA 2′-O-methylation by PCR with an engineered DNA polymerase. Nucl. Acids Res. 2016, 44, 3495–3502. [Google Scholar] [CrossRef] [PubMed]
- Saikia, M.; Dai, Q.; Decatur, W.A.; Fournier, M.J.; Piccirilli, J.A.; Pan, T. A systematic, ligation-based approach to study RNA modifications. RNA 2006, 12, 2025–2033. [Google Scholar] [CrossRef] [PubMed]
- Birkedal, U.; Christensen-Dalsgaard, M.; Krogh, N.; Sabarinathan, R.; Gorodkin, J.; Nielsen, H. Profiling of ribose methylations in RNA by high-throughput sequencing. Angew. Chem. Int. Ed. 2015, 54, 451–455. [Google Scholar] [CrossRef]
- Marchand, V.; Blanloeil-Oillo, F.; Helm, M.; Motorin, Y. Illumina-based RiboMethSeq approach for mapping of 2′-O-Me residues in RNA. Nucl. Acids Res. 2016, 44, e135. [Google Scholar] [CrossRef] [PubMed]
- Gumienny, R.; Jedlinski, D.J.; Schmidt, A.; Gypas, F.; Martin, G.; Vina-Vilaseca, A.; Zavolan, M. High-throughput identification of C/D box snoRNA targets with CLIP and RiboMeth-seq. Nucl. Acids Res. 2017, 45, 2341–2353. [Google Scholar] [CrossRef] [PubMed]
- Krogh, N.; Birkedal, U.; Nielsen, H. RiboMeth-seq: Profiling of 2′-O-Me in RNA. Methods Mol. Biol. 2017, 1562, 189–209. [Google Scholar] [PubMed]
- Incarnato, D.; Anselmi, F.; Morandi, E.; Neri, F.; Maldotti, M.; Rapelli, S.; Parlato, C.; Basile, G.; Oliviero, S. High-throughput single-base resolution mapping of RNA 2′-O-methylated residues. Nucl. Acids Res. 2017, 45, 1433–1441. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Pirnie, S.P.; Carmichael, G.G. High-throughput and site-specific identification of 2′-O-methylation sites using ribose oxidation sequencing (RibOxi-seq). RNA 2017, 23, 1303–1314. [Google Scholar] [CrossRef] [PubMed]
- Dai, Q.; Moshitch-Moshkovitz, S.; Han, D.; Kol, N.; Amariglio, N.; Rechavi, G.; Dominissini, D.; He, C. Nm-seq maps 2′-O-methylation sites in human mRNA with base precision. Nat. Methods 2017, 14, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.J.; Fei, Q.; Dai, Q.; Shi, H.; Dominissini, D.; Ma, L.; He, C. Single base resolution mapping of 2′-O-methylation sites in human mRNA and in 3′ terminal ends of small RNAs. Methods 2018. [Google Scholar] [CrossRef] [PubMed]
- Krogh, N.; Jansson, M.D.; Häfner, S.J.; Tehler, D.; Birkedal, U.; Christensen-Dalsgaard, M.; Lund, A.H.; Nielsen, H. Profiling of 2′-O-Me in human rRNA reveals a subset of fractionally modified positions and provides evidence for ribosome heterogeneity. Nucl. Acids Res. 2016, 44, 7884–7895. [Google Scholar] [CrossRef] [PubMed]
- Erales, J.; Marchand, V.; Panthu, B.; Gillot, S.; Belin, S.; Ghayad, S.E.; Garcia, M.; Laforêts, F.; Marcel, V.; Baudin-Baillieu, A.; et al. Evidence for rRNA 2′-O-methylation plasticity: Control of intrinsic translational capabilities of human ribosomes. Proc. Natl. Acad. Sci. USA 2017, 114, 12934–12939. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Liu, Y.; Rohde, C.; Pauli, C.; Gerloff, D.; Köhn, M.; Misiak, D.; Bäumer, N.; Cui, C.; Göllner, S.; et al. AML1-ETO requires enhanced C/D box snoRNA/RNP formation to induce self-renewal and leukaemia. Nat. Cell Biol. 2017, 19, 844–855. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Marchand, V.; Motorin, Y.; Lafontaine, D.L.J. Identification of sites of 2′-O-methylation vulnerability in human ribosomal RNAs by systematic mapping. Sci. Rep. 2017, 7, 11490. [Google Scholar] [CrossRef] [PubMed]
- Marchand, V.; Pichot, F.; Thüring, K.; Ayadi, L.; Freund, I.; Dalpke, A.; Helm, M.; Motorin, Y. Next-Generation Sequencing-Based RiboMethSeq Protocol for Analysis of tRNA 2′-O-Methylation. Biomolecules 2017, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Krogh, N.; Kongsbak-Wismann, M.; Geisler, C.; Nielsen, H. Substoichiometric ribose methylations in spliceosomal snRNAs. Org. Biomol. Chem. 2017, 15, 8872–8876. [Google Scholar] [CrossRef] [PubMed]
- Ayadi, L.; Motorin, Y.; Marchand, V. Quantification of 2′-O-Me residues in RNA using next-generation sequencing (Illumina RiboMethSeq Protocol). Methods Mol. Biol. 2018, 1649, 29–48. [Google Scholar] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RiboMethSeq [53] | 2′-OMe-Seq [56] | RibOxi-Seq [57] | (Nm-Seq) [58,59] | |
|---|---|---|---|---|
| Described applications | rRNA, tRNA, (snRNA) | rRNA | rRNA | rRNA, mRNA |
| RNA input | 10 ng (50 ng in routine) | 2 × 2 µg | 7.5 µg | 10 µg rRNA 10 µg polyA mRNA |
| Sequencing depth | ~1000 reads/RNA position (10–15 mln raw reads/sample) | 10–15 mln reads/sample | 10–15 mln reads/sample | 10–15 mln reads/sample |
| Quantification | Yes | Yes (relative only) | No | No |
| Sequencing mode used | Single read 50 nt | Paired end 2 × 75 nt |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Motorin, Y.; Marchand, V. Detection and Analysis of RNA Ribose 2′-O-Methylations: Challenges and Solutions. Genes 2018, 9, 642. https://doi.org/10.3390/genes9120642
Motorin Y, Marchand V. Detection and Analysis of RNA Ribose 2′-O-Methylations: Challenges and Solutions. Genes. 2018; 9(12):642. https://doi.org/10.3390/genes9120642
Chicago/Turabian StyleMotorin, Yuri, and Virginie Marchand. 2018. "Detection and Analysis of RNA Ribose 2′-O-Methylations: Challenges and Solutions" Genes 9, no. 12: 642. https://doi.org/10.3390/genes9120642
APA StyleMotorin, Y., & Marchand, V. (2018). Detection and Analysis of RNA Ribose 2′-O-Methylations: Challenges and Solutions. Genes, 9(12), 642. https://doi.org/10.3390/genes9120642