1. Introduction
Shed hairs are one of the most commonly encountered evidence types [
1], but also among the most limited in terms of DNA quantity and quality. By some estimates, shed hair represents up to 90% of the hair samples collected at crime scenes [
2,
3]. Unfortunately, nuclear DNA (nuDNA) is generally too low in quantity and quality to permit successful short tandem repeat (STR) typing.
The difficulty of successful STR typing of shed hairs is attributed primarily to the keratinization process, which degrades cellular organelles and nucleic acids [
4]. Not only do the general enzymatic activities associated with keratinization result in DNA degradation, but also nuclear DNA, in particular, is specifically targeted for destruction [
5,
6,
7]. Nevertheless, some nuclear DNA is known to persist in telogen hairs, albeit at low quantities and highly variable qualities [
4]. Though a number of studies describe the presence, and successful PCR amplification, of autosomal STR markers from telogen hairs [
2,
8,
9,
10,
11,
12], STR typing is not routinely pursued in forensic laboratories for a number of reasons. For one, reduced size STR amplicons, ranging from ~60–150 bp, are generally required to achieve amplification success. Yet, since only a relatively small number of reduced sized amplicons can be multiplexed with currently employed capillary electrophoresis technologies, data recovery is somewhat limited. Second, elevated cycle numbers, often between 30 and 40, are generally required to amplify the low quantities of nuclear DNA to detectable levels. However, even when reduced size amplicons and elevated cycle numbers are employed, amplification success is still inconsistent, and resulting STR profiles are often incomplete. In addition, commonly employed quantitative PCR (qPCR) assays rarely yield enough information to adequately inform downstream analysis. The qPCR assays routinely implemented are simply not sensitive enough for the low levels of nuclear DNA present in telogen hairs [
12]. More recent studies have shown that direct amplification of telogen hairs (i.e., amplification without preliminary DNA extraction), when combined with elevated cycles, can result in full STR profiles approximately 20% of the time [
12]. However, it is difficult or impossible to predict a priori if a hair will yield probative DNA data.
It is also the case that success rates in routine forensic practice are likely lower. Most research studies are necessarily based on freshly, or relatively recently, collected hair samples. While general patterns can obviously be ascertained from such samples, the variability and difficulty of aged and/or damaged casework samples is nearly impossible to accurately represent. Indeed, in those studies for which hairs recovered from actual crime scenes were included, the evidence hairs performed worse than the recently collected hairs [
10,
13]. Given that amplification success cannot be predicted in advance, and that amplification success rates are generally low, it simply does not make sense from a practical perspective to exhaust limited sample material on a testing modality unlikely to yield probative results. Instead, mitochondrial DNA (mtDNA) is routinely sought in these cases due to its abundance relative to nuclear DNA. Though mtDNA profiles do not offer the discriminatory power of nuclear DNA profiles, mtDNA is recovered in 92.5% of telogen hair cases [
14].
Because of the difficulty of recovering nuclear DNA from hair, studies conducted to characterize and better understand the state of DNA in hair have largely focused on the more accessible mtDNA molecule. At a broad level, Melton et al., [
14] showed that with increasing age of the hair specimen, the likelihood of obtaining a full hypervariable region I/hypervariable region II (HVI/HVII) profile decreased. The same pattern was observed in a systematic study by Gilbert et al. [
15]. Additional studies, based on the size of recoverable PCR amplicons, have shown progressive degradation of mtDNA along the hair shaft [
16,
17,
18] with DNA quality deteriorating rapidly within a few millimeters of the root [
10].
Some of the most recent information on the overall quantity and quality of DNA in telogen hair shafts has come from studies employing next-generation sequencing (NGS). Because NGS-based shotgun sequencing is not dependent on pre-defined amplicons, the sequence data reflect the endogenous size of the DNA. Generally speaking, these studies show that DNA preservation in aged hair is overwhelmingly poor [
19,
20,
21,
22]. For example, the average mtDNA size of a 4000-year-old paleo Eskimo sample was 76 bp [
19], and mtDNA averaged only 61 bp (range between 48 to 73 bp) [
22] in 111 human hairs collected between 1920 and 1970.
Preliminary studies in our laboratory showed that in shotgun libraries of two freshly collected single shed hairs, 99.93 and 99.88% of the reads mapping to the human genome were nuclear DNA, and the remaining 0.07% and 0.12% were mitochondrial DNA [
23]. These results are consistent with those of other shotgun sequencing studies of aged hairs. In particular, Rasmussen et al. [
20] found that ~80% of the reads produced from a 1.5 g sample of 4000-year-old hair were human sequences and that only 0.13% of the human reads were mtDNA sequences. The remainder, 99.87%, were nuclear DNA sequences.
Here, we aim to confirm that, despite a high level of degradation, nuclear DNA comprises the vast majority of total human DNA in hair shafts. In addition, we further characterize the quantity and quality of both mitochondrial and nuclear DNA that can be recovered from single shed telogen hairs regularly encountered in forensic casework.
2. Materials and Methods
All tested samples were rootless hair shafts collected with informed consent from the donors under FBI Institutional Review Board approved project #417-17 (
Table 1 and
Table 2).
2.1. Types of Hair
2.1.1. Recent hairs
The hairs referred to as recent hairs (R series) were cut or collected less than 6 years before the date of the DNA testing and stored primarily at 4 °C. Recent hairs were collected from hairbrushes or by finger combing from random portions of the head. Prior to DNA extraction, the root (proximal) ends of these hairs were cut (~1 cm) and removed. Six recently collected hair samples were used to characterize total DNA (mitochondrial and nuclear) content in rootless shed hairs and approximately 5 to 6.5 cm was extracted for any given hair (
Table 2).
2.1.2. Aged Hairs
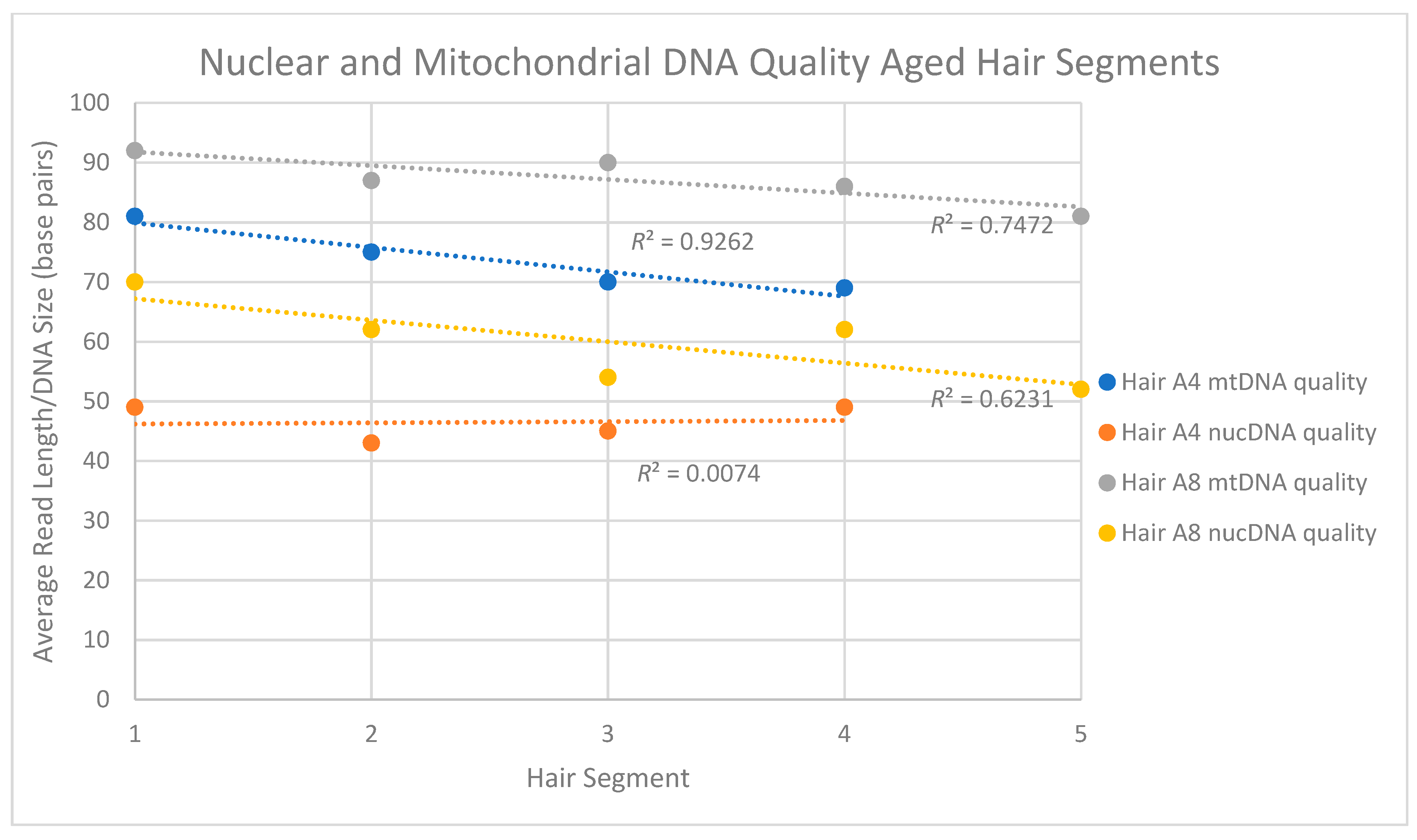
The aged hairs (A series) were taken from hair cuttings that were ~40 to 60 years old and had been stored at room temperature. Because the samples were hair cuttings, the lengths of the hairs at the time of cutting, and thus the distances from the scalp of the tested segments, were unknown. Furthermore, it was not possible to easily identify the proximal and distal end of these hairs. Samples A1, A2, A6 and A7 were children’s hairs that originated from three individuals whose mtDNA genome (mtGenome) profiles were known. Both hair types were used to evaluate: (1) mtDNA and nuclear DNA content, (2) mtDNA quantity and quality along the length of the shaft, (3) the possibility of complete mtGenome sequence recovery and 4) nuclear DNA quantity and quality along the length of the shaft.
2.1.3. Segmented Hairs
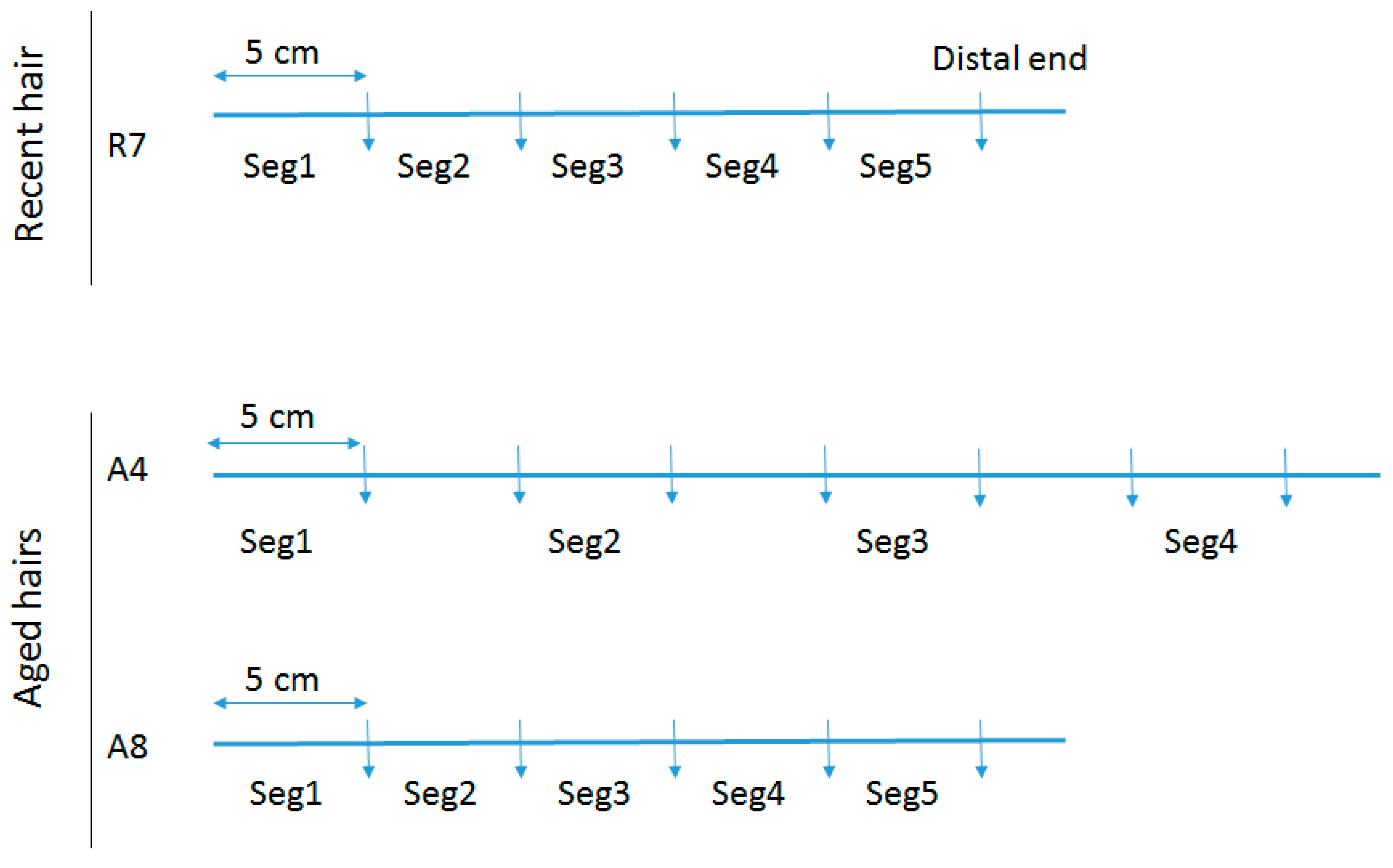
To assess total DNA quantity and quality along the length of individual hair shafts, a recent untreated hair that had been stored refrigerated for 4 years (R7) and two separate aged hairs (A4 & A8) were tested. Following removal of the root (if necessary), the hairs were cut into 5 cm segments. For R7, the mtGenome profile of the donor was known and five segments were tested (see
Figure 1), with Segment 1 representing the segment closest to the root end. For A4, seven segments were cut but only four were tested. For A8, five continuous segments were tested. Samples A4 and A8 are from a >50 cm adult braid that was cut at least 40 years ago and for which no reference profile was available.
2.2. Extraction
The protocols for washing and digesting the hair samples are presented in Protocols,
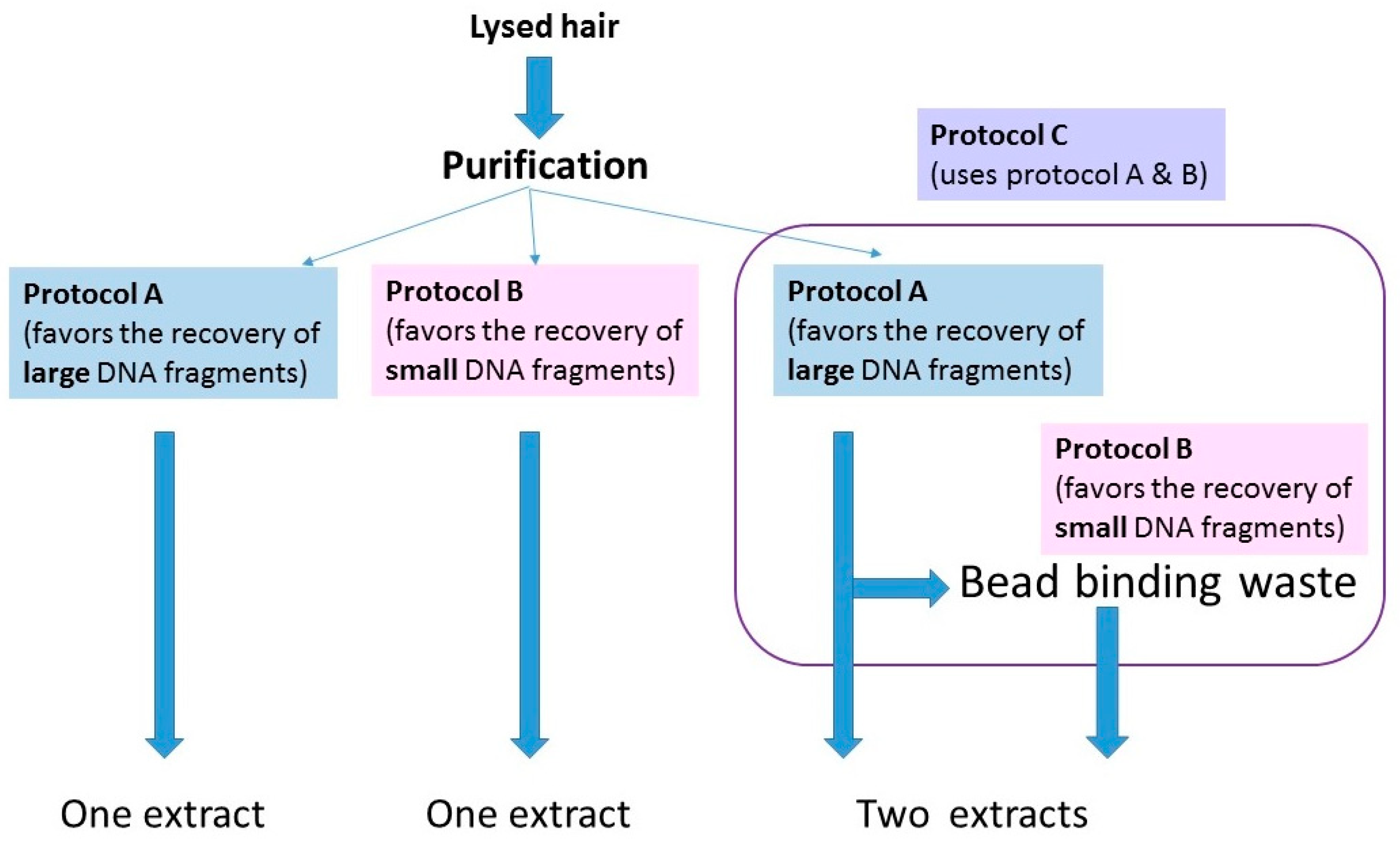
Supplementary Materials. Once the hairs were fully digested, lysates were purified with one of three protocols (
Figure 2). The purification methods were being tested for a different study, but essentially served as replicate extractions for those hairs purified with multiple protocols. The protocols are summarized below:
- (1)
A protocol adapted from a Qiagen user-developed method [
24,
25]) that has been employed in the FBI Laboratory’s routine casework since 2014 (referred to from here on as protocol A; see Protocols in the
Supplementary Materials).
- (2)
A protocol based on Allentoft et al., [
26] that employs a binding buffer that favors the recovery of small (<100 bp) DNA fragments.
- (3)
A combined protocol that follows protocol A until the step at which the lysate and silica beads are on the magnet. For this protocol, (protocol C in Protocols,
Supplementary Materials), the bead binding waste solution was retained and mixed with the binding buffer from protocol B. It was subsequently purified and eluted with MinElute columns (Qiagen, Germantown, MD, USA).
Table 2 summarizes the purification protocol used for each hair sample tested in this study. Generally speaking, protocol A was used for the purification of recently collected hairs while protocol B was used with aged hairs for which DNA was expected to be degraded.
2.3. Quantification
Quantity of mtDNA was assessed by qPCR of the DNA extracts. For the recent hair extracts, quality of the mtDNA was assessed in one of two ways: by a mtDNA qPCR assay developed by Kavlick [
27] on a 7500 Real Time PCR system (Thermo Fisher Scientific, Waltham, MA, USA) or by sequence read length for the aged hair extracts.
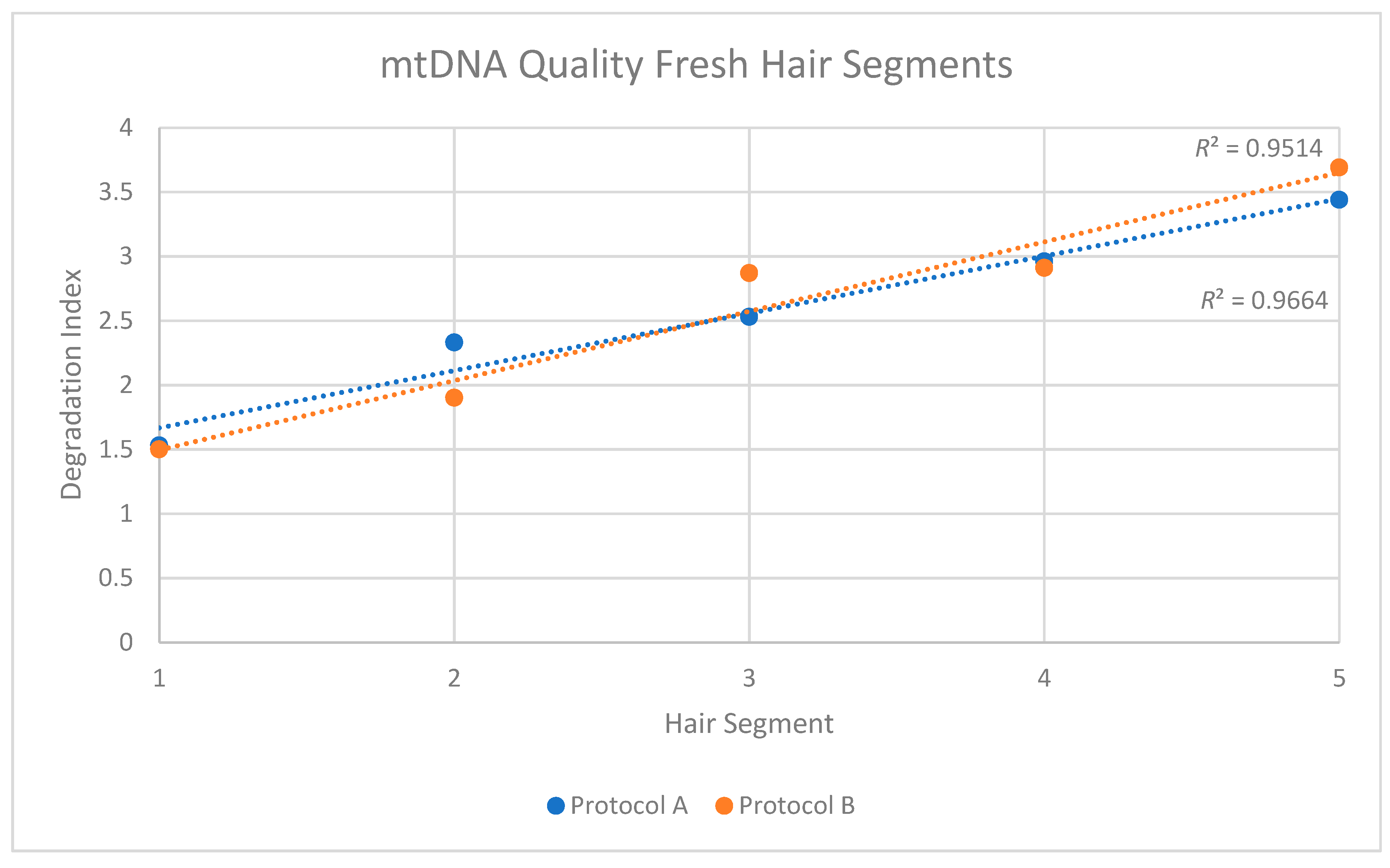
For the mtDNA quantification, two microliters of each extract and reagent blank (RB) were amplified in duplicate with a qPCR assay that incorporates a DNA degradation index. With this assay, degradation is assessed based on the ratio of large (≥316 bp) and small (≥105 bp) mtDNA fragments. The larger the degradation index, the lower the number of mtDNA fragments ≥316 bp relative to fragments between 105 bp–316 bp. For example, a degradation index of 1 or lower indicates no mtDNA degradation (all quantified fragments ≥316 bp), while a degradation index >1 indicates that fewer 316 bp or larger fragments are present in the extract than 105 bp–316 bp fragments [
27]. An undetermined degradation index indicates that all the human mtDNA is degraded to a size smaller than 316 bp.
Sequence read lengths could not be used to assess endogenous mtDNA quality in the recent hair extracts because, following qPCR, the DNA required shearing to ensure successful library preparation and sequencing. This is due to the presence of large mtDNA fragments that have been shown to exist in freshly collected hair shafts [
23]. Conversely, qPCR could not be used to assess mtDNA quality in the aged hairs because the degradation index, which is dependent on the amplification of DNA fragments of 105 and 316 bp in size, could not be recovered from most of the aged hair segments.
Nuclear DNA quantification of the recent hair extracts using the Quantifiler Trio DNA quantification kit (Thermo Fisher Scientific) was attempted for a number of samples, but the results were too low to be useful (<0.5 pg/µL).
2.4. Library Preparation
Following quantitation, aged hair DNA extracts were used for library preparation. For recent hairs, to ensure successful downstream library preparation and sequencing, DNA was fragmented prior to library preparation. Shearing was performed with the Fragmentase enzyme present in the KAPA HyperPlus Library Preparation Kit (Kapa Biosystems, Wilmington, MA, USA) for 30 min. Extracts were then purified with a Qiagen MinElute PCR purification kit, and eluted in 50 µL of H2O.
50 µL of each extract or RB was then converted to an Illumina library using the NEBNext Ultra II kit (NEB, Ipswich, MA, USA) and looped adapters from the NEBNext Multiplex oligos for Illumina kit (NEB). The libraries of the aged hairs were prepared according to the manufacturer’s instructions with the exception of the ligation which was performed overnight at 7 °C. Following ligation, the looped adapters were converted into Y-shaped adapters and the libraries purified with Ampure XP beads (Beckman coulter, Sykesville, MD, USA). All libraries were dual-indexed with indexed primers from the NEBNext Multiplex oligos for Illumina kit and subsequently amplified for 25 cycles with the NEBNext Ultra II Q5 PCR kit. Purification was performed using Ampure XP beads.
2.5. Sequencing
All libraries were shotgun sequenced on an Illumina MiSeq FGX instrument with a 300 cycles v2 cartridge and 2 × 150 cycles + 2 × 8 cycles for the indexes for the aged hairs and a v3 2 × 300 cycles + 2 × 8 cycles for the recent hairs.
2.6. Data Analysis
Read mapping and consensus variant calling were performed with the CLC Genomics Workbench software, version 10.0.1 (CLC Bio, Qiagen). All reads were trimmed and overlapping pairs merged (see details in CLC workflow,
Supplementary Materials). The default Genomics Workbench mapping and alignment parameters, which included a length fraction of 85% and a similarity fraction of 97%, as well as insertion/deletion (indel) and mismatch costs of 3, were used for all samples. Alignments to the mtGenome were performed using the revised Cambridge Reference Sequence (rCRS, [
28]). MtGenome variant calling was performed using the Fixed Ploidy variant caller.
Alignments to the human genome were performed using the human reference genome sequence build hg38. The percentages of reads mapping to both the mitochondrial and nuclear genomes were determined based on summary data and mapping statistics produced by the CLC software.
5. Conclusions
In an effort to directly characterize the quantity and quality of mtDNA and nuclear DNA present in the types of limited, aged, and degraded shed hair specimens often encountered in forensic casework, shotgun sequencing was performed on rootless telogen hairs. The results, based on direct observation of the endogenous molecules, revealed that:
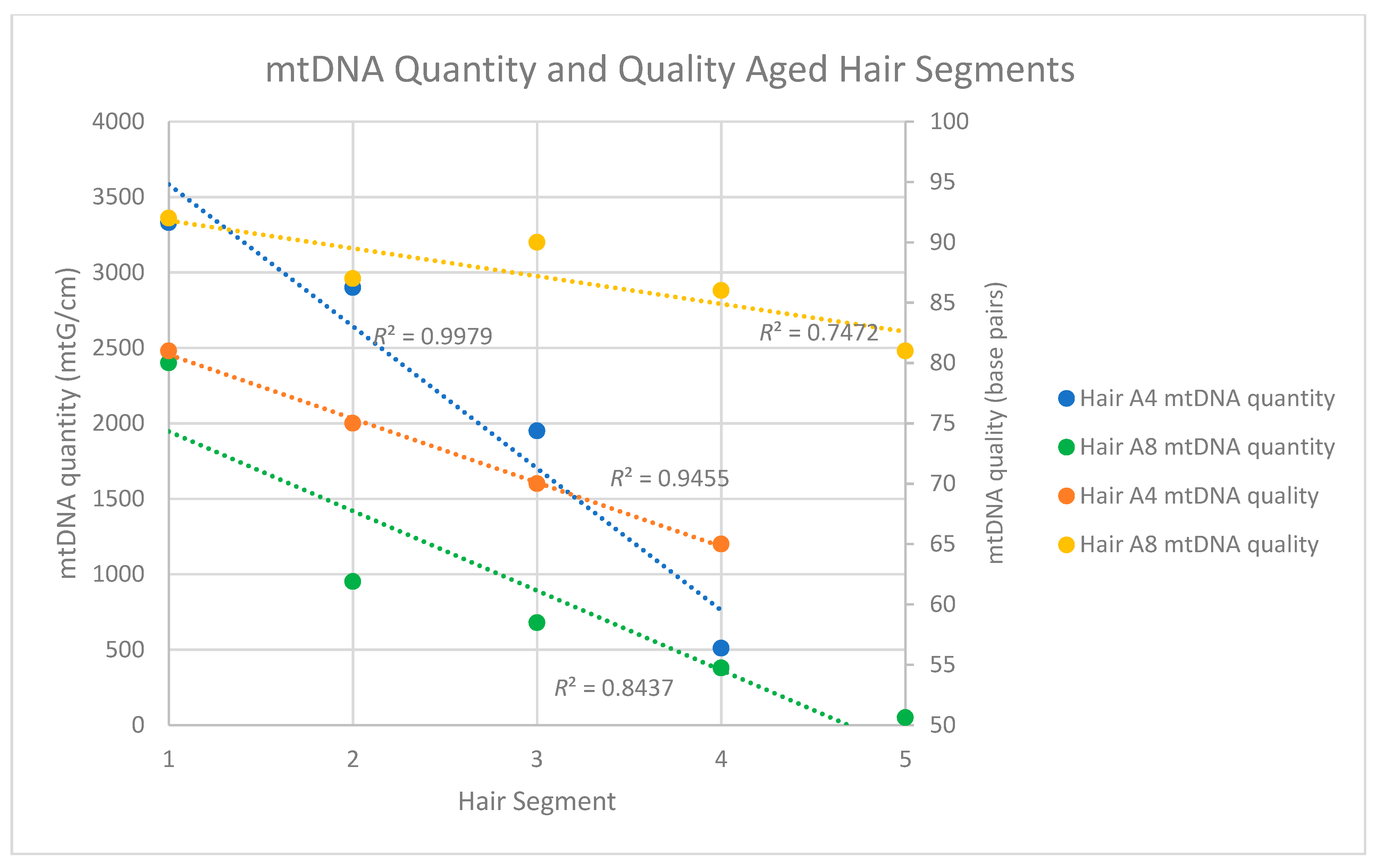
mtDNA quantity and quality decline along the length of the hair shaft,
mtDNA fragments are generally larger than nuclear DNA fragments in the same hair or hair segment,
complete mtGenomes can be recovered from aged hair shafts with shotgun sequencing data along (i.e., no enrichment)
nuDNA quality tends to decrease along the length of the hair shaft,
both nuclear and mitochondrial DNA fragment sizes in the aged hairs were generally <80 bp (too small for routinely employed targeted PCR amplicons) and
nuclear DNA was not only recovered but comprised the vast majority of DNA in any given hair sample.
Clearly, the relative sizes and copy numbers of the two genomes play a critical role in the recovery of informative DNA profiles from telogen hair samples (for reference, the percentage of nuclear DNA in a cell with 1000 mtGenomes is 99.997%), but our results show that in the types of specimens that have historically failed to yield mtDNA control region data with existing Sanger protocols, not only could complete mitochondrial genomes be developed, but also informative nuclear DNA data could be recovered. Further development of assays that accommodate the small size of the nuclear DNA may allow for more routine recovery of discriminatory nuclear DNA profiles from such samples. A better understanding of DNA quantity and quality, for nuclear DNA in particular, should promote further development of cost-effective forensic assays that can generate more discriminatory information from not only hair, but also other samples harboring extremely degraded DNA.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}