Ancient Ancestry Informative Markers for Identifying Fine-Scale Ancient Population Structure in Eurasians




Abstract
1. Introduction
1.1. Towards High-Resolution Population Models Using Ancient Samples
1.2. Next Generation Sequencing Technologies to Study Ancient DNA
1.3. The Problems of Ascertainment Bias and Population Stratification in Ancient DNA
1.4. The Use of Ancestry Informative Markers in Genetics
1.5. Ancient Ancestry Informative Markers to Define Ancient Population Structure
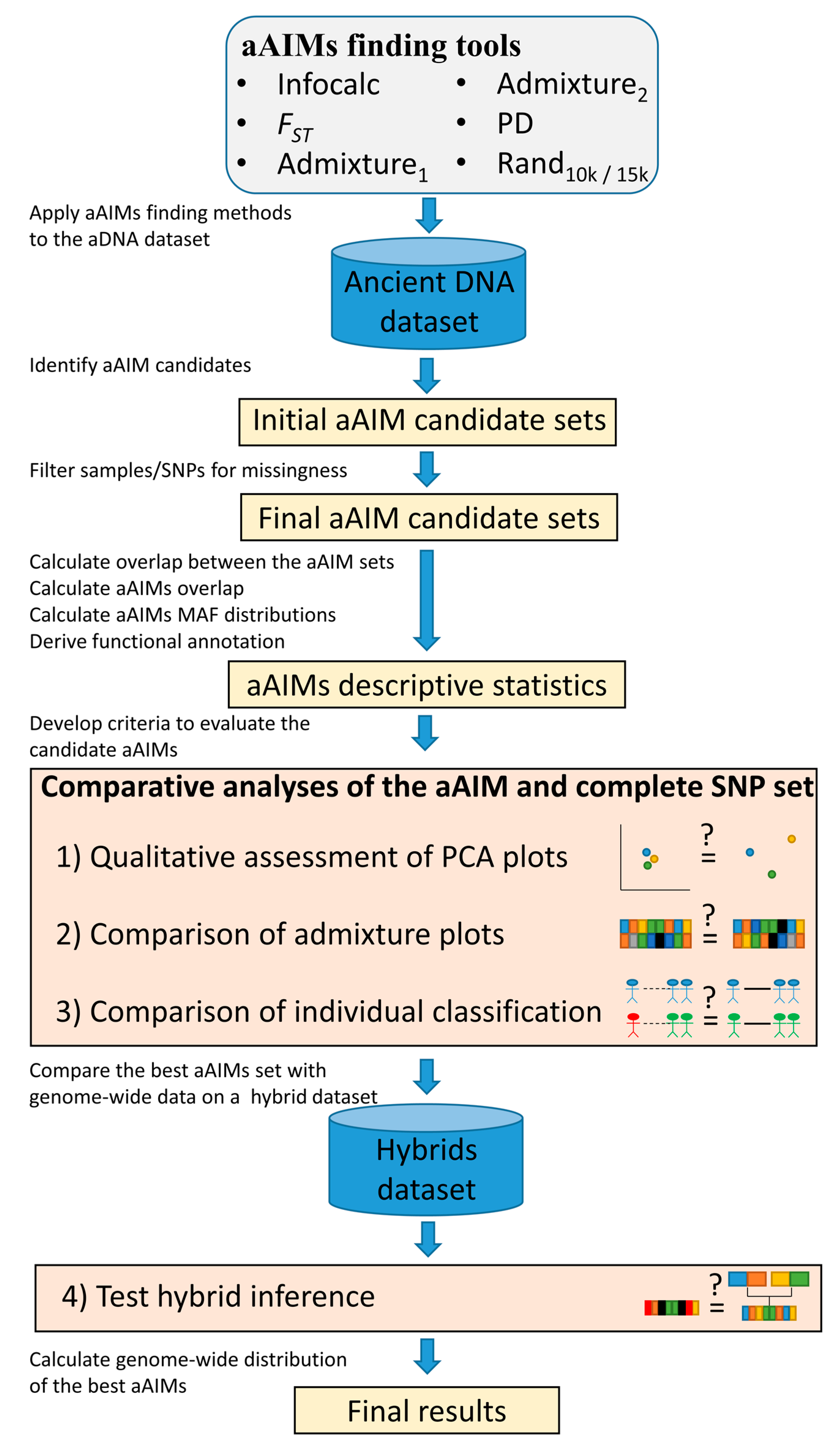
2. Materials and Methods
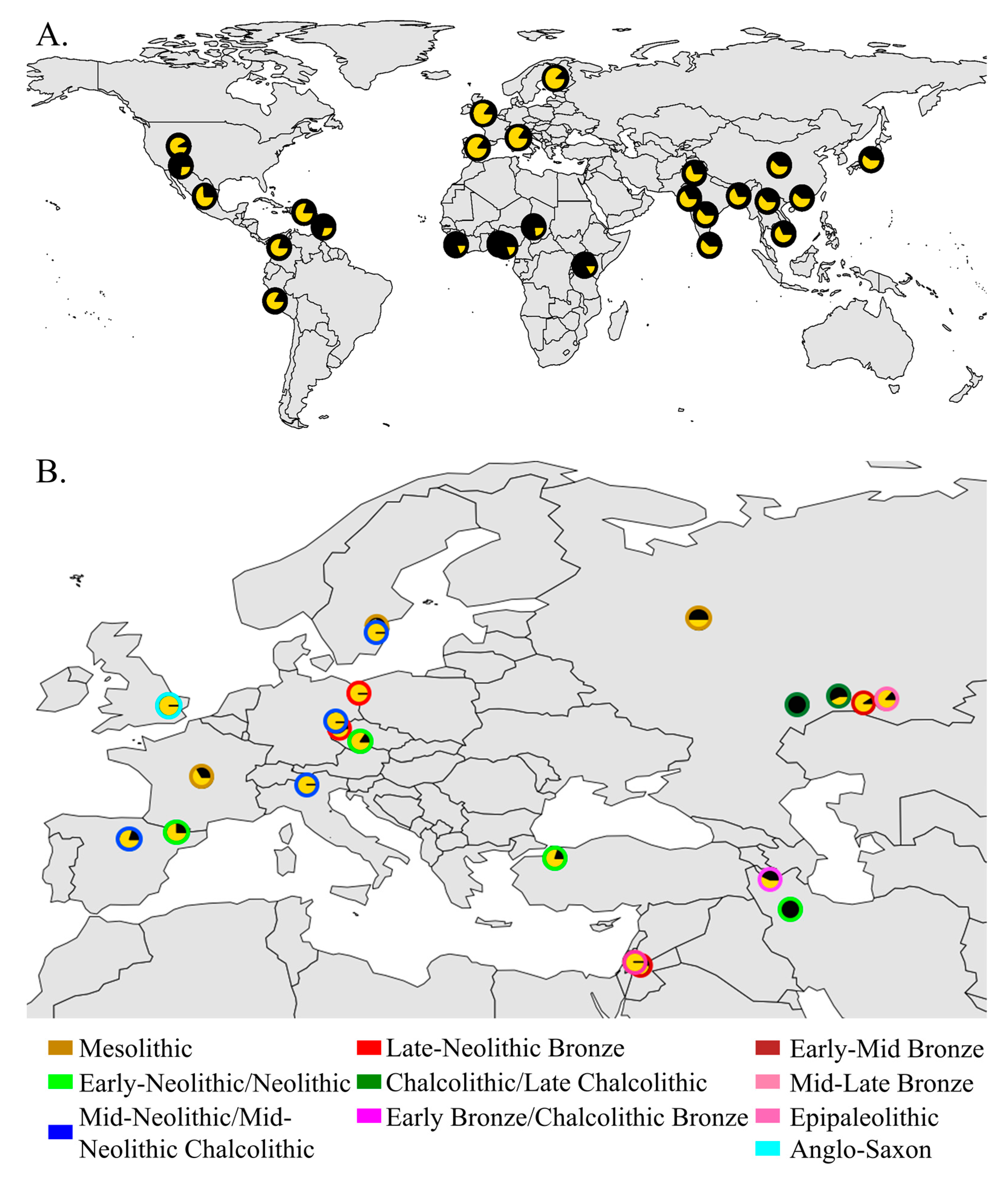
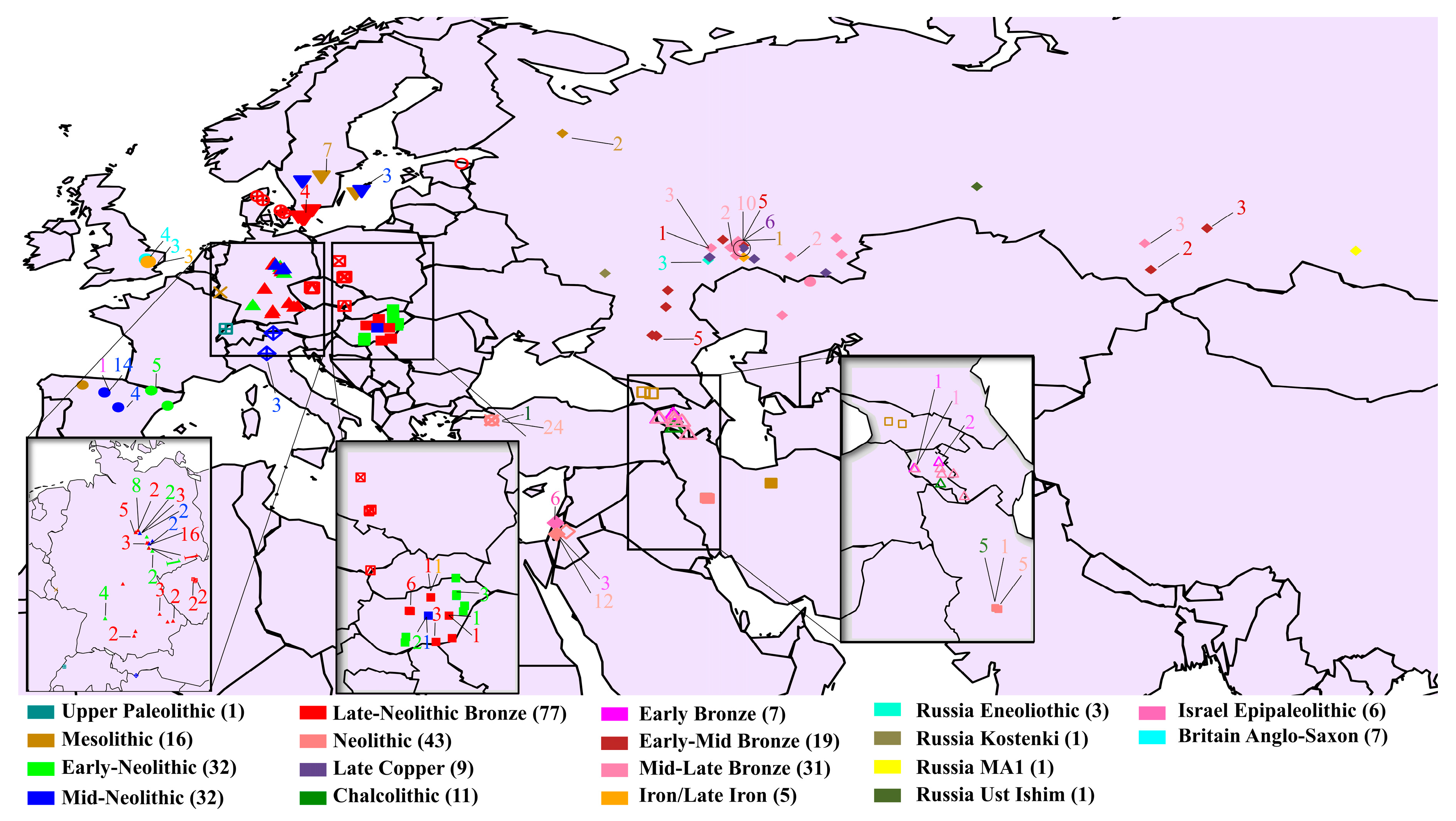
2.1. Ancient Data Collection
2.2. Data Analyses
2.2.1. The Genetic Structure Canvas of Ancient Eurasian Genomes
2.2.2. Identifying aAIMs Using Multiple Methods
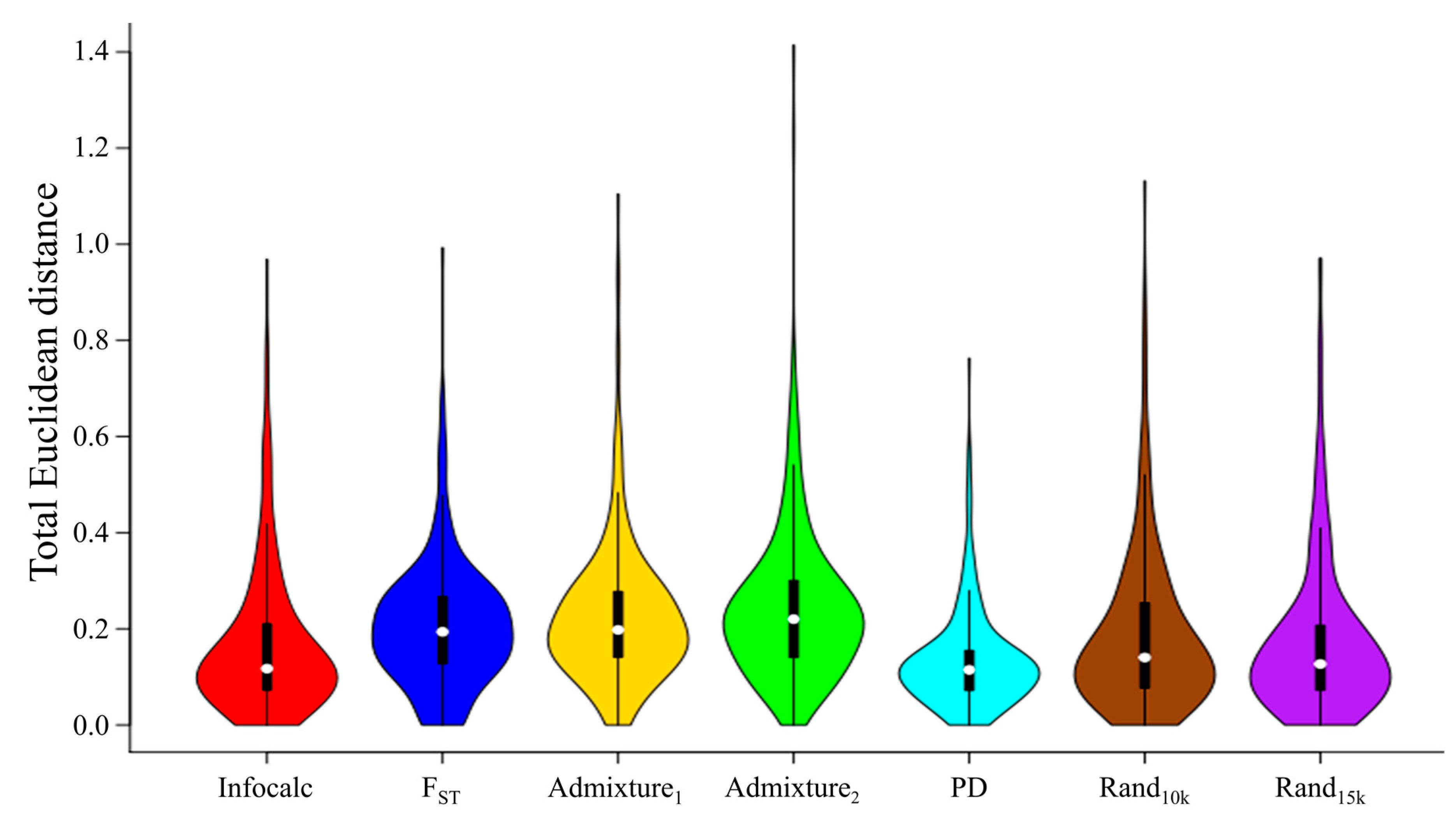
- Infocalc v1.1 [28], determines the amount of information that multiallelic markers provide of an individual’s ancestry by calculating the informativeness (I) of each marker separately and ranking the SNPs by their informativeness. Infocalc determines I based on the mathematical expression described in Rosenberg et al. (2003). We compared the performances (Figure 2) of the top 5000, 10,000, 15,000, and 20,000 most informative markers (results not shown). The 15,000 dataset outperformed all of the other datasets, and was selected for further analyses.
- FST. Wright’s fixation indices (FST) [30] measures the degree of differentiation among populations, which was potentially arising due to the genetic structure within populations. Given a set of populations (Table S1), we employed PLINK v1.9 [39] to estimate FST separately for all the markers using the --fst command alongside --within flag. Due to the high fragmentation of the data, FST values could only be calculated for 46% of the dataset. We compared the performances (Figure 2) of 5000, 10,000, 15,000, and 20,000 SNPs with the highest FST values (results not shown). The 15,000 dataset outperformed all of the other datasets, and was selected for further analyses.
- Admixture1. This method assumes that aAIMs have high allelic frequencies in certain subpopulations that drive the differentiation of admixture components. Analyzing ADMIXTURE’s output file (P file) for K = 10, we identified the markers (rows) that had high allele frequency (>0.9) in only one admixture component (columns). Comparing the number of high-MAF SNPs in all of the columns, we selected 9309 from the five columns with the highest number of such SNPs.
- Admixture2. This method assumes that aAIMs embody both high allelic frequencies in certain subpopulations, and that the high variance between these allelic frequencies differentiates the admixture components. Analyzing ADMIXTURE’s output file for K of 10, we identified 11,418 SNPs showing high variance (≥0.04) and a high allele frequency range (maxima–minima ≥ 0.65) between the admixture components.
- Principal Component-derived (PD). This method assumes that AIMs can replicate the population structure of subpopulations represented by the variation in the first two PCs. This is an interactive PC-based approach that identifies the smallest set of markers necessary to capture the population structure of a group of individuals, as captured by the complete SNP set (CSS). More specifically, for each population group (Table S1) in which at least 100 SNPs were available, we carried out PCA after all of the SNPs with high missingness (>0.05) were excised. If the population group had insufficient SNPs, we relaxed the missingness threshold by an additional 0.05, although 0.05 were sufficient for almost all of the groups. We then scored the SNPs by their informativeness, as in [42], and used the top 100 most informative SNPs to plot the individuals on a scatter plot using PC1 and PC2 as axes. We visually compared the plot to that obtained from the CSS (Figure S11). If the plots were dissimilar, we repeated the analysis using an additional 100 top-scored SNPs until either the plots exhibited high similarity or a threshold of 2000 SNPs was reached. In this manner, we identified the minimum number of the most informative SNPs that were needed to replicate the PCA results of the CSS. We were unable to complete the analyses for three populations due to the small number of individuals. The PD method is available on https://github.com/eelhaik/PCA-derived-aAIMs. On average, 861 SNPs were collated per population group. Overall, the dataset comprised 13,027 SNPs.
2.2.3. Classifying Individuals into Populations from Genomic Data
2.2.4. Assessing Admixture Mapping
3. Results
3.1. Depicting Ancient Population Structure
3.2. Identifying and Describing the Ancient Ancestry Informative Markers Candidates
3.3. Comparative Testing of Ancient Ancestry Informative Marker Candidates
3.4. Inference of Admixed Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Morozova, I.; Flegontov, P.; Mikheyev, A.S.; Bruskin, S.; Asgharian, H.; Ponomarenko, P.; Klyuchnikov, V.; ArunKumar, G.; Prokhortchouk, E.; Gankin, Y.; et al. Toward high-resolution population genomics using archaeological samples. DNA Res. 2016, 23, 295–310. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, S.; Perry, G.H. Harnessing ancient genomes to study the history of human adaptation. Nat. Rev. Genet. 2017, 18, 659–674. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, L.M.; Martiniano, R.; Murphy, E.M.; Teasdale, M.D.; Mallory, J.; Hartwell, B.; Bradley, D.G. Neolithic and Bronze Age migration to Ireland and establishment of the insular Atlantic genome. Proc. Natl. Acad. Sci. USA 2016, 113, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Patterson, N.J.; Moorjani, P.; Luo, Y.; Mallick, S.; Rohland, N.; Zhan, Y.; Genschoreck, T.; Webster, T.; Reich, D. Ancient admixture in Human history. Genetics 2012, 192, 1065–1093. [Google Scholar] [CrossRef] [PubMed]
- Mathieson, I.; Lazaridis, I.; Rohland, N.; Mallick, S.; Patterson, N.; Roodenberg, S.A.; Harney, E.; Stewardson, K.; Fernandes, D.; Novak, M.; et al. Genome-wide patterns of selection in 230 ancient Eurasians. Nature 2015, 528, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Hajdinjak, M.; Moldovan, O.T.; Constantin, S.; Mallick, S.; Skoglund, P.; Patterson, N.; Rohland, N.; Lazaridis, I.; Nickel, B.; et al. An early modern human from Romania with a recent Neanderthal ancestor. Nature 2015, 524, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Lazaridis, I.; Nadel, D.; Rollefson, G.; Merrett, D.C.; Rohland, N.; Mallick, S.; Fernandes, D.; Novak, M.; Gamarra, B.; Sirak, K.; et al. Genomic insights into the origin of farming in the ancient Near East. Nature 2016, 536, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Z.; Absher, D.M.; Tang, H.; Southwick, A.M.; Casto, A.M.; Ramachandran, S.; Cann, H.M.; Barsh, G.S.; Feldman, M.; Cavalli-Sforza, L.L.; et al. Worldwide human relationships inferred from genome-wide patterns of variation. Science 2008, 319, 1100–1104. [Google Scholar] [CrossRef] [PubMed]
- Elhaik, E.; Yusuf, L.; Anderson, A.I.J.; Pirooznia, M.; Arnellos, D.; Vilshansky, G.; Ercal, G.; Lu, Y.; Webster, T.; Baird, M.L.; et al. The Diversity of REcent and Ancient huMan (DREAM): A new microarray for genetic anthropology and genealogy, forensics, and personalized medicine. Genome Biol. Evol. 2017, 9, 3225–3237. [Google Scholar] [CrossRef] [PubMed]
- Elhaik, E.; Greenspan, E.; Staats, S.; Krahn, T.; Tyler-Smith, C.; Xue, Y.; Tofanelli, S.; Francalacci, P.; Cucca, F.; Pagani, L.; et al. The GenoChip: A new tool for genetic anthropology. Genome Biol. Evol. 2013, 5, 1021–1031. [Google Scholar] [CrossRef]
- Hublin, J.-J. The last Neanderthal. Proc. Natl. Acad. Sci. USA 2017, 114, 10520–10522. [Google Scholar] [CrossRef] [PubMed]
- Jones, S. The Archaeology of Ethnicity: Constructing Identities in the Past and Present; Routledge: London, UK, 1997. [Google Scholar]
- Albrechtsen, A.; Nielsen, F.C.; Nielsen, R. Ascertainment biases in SNP chips affect measures of population divergence. Mol. Biol. Evol. 2010, 27, 2534–2547. [Google Scholar] [CrossRef] [PubMed]
- Marchini, J.; Cardon, L.R.; Phillips, M.S.; Donnelly, P. The effects of human population structure on large genetic association studies. Nat. Genet. 2004, 36, 512–517. [Google Scholar] [CrossRef]
- Yusuf, S.; Wittes, J. Interpreting geographic variations in results of randomized, controlled trials. N. Engl. J. Med. 2016, 375, 2263–2271. [Google Scholar] [CrossRef]
- Elhaik, E.; Ryan, D.M. Pair Matcher (PaM): Fast model-based optimisation of treatment/case-control matches. Bioinformatics 2018. [Google Scholar] [CrossRef]
- Elhaik, E.; Tatarinova, T.; Chebotarev, D.; Piras, I.S.; Maria Calò, C.; De Montis, A.; Atzori, M.; Marini, M.; Tofanelli, S.; Francalacci, P.; et al. Geographic population structure analysis of worldwide human populations infers their biogeographical origins. Nat. Commun. 2014, 5, 1–12. [Google Scholar] [CrossRef]
- Phillips, C.; Parson, W.; Lundsberg, B.; Santos, C.; Freire-Aradas, A.; Torres, M.; Eduardoff, M.; Borsting, C.; Johansen, P.; Fondevila, M.; et al. Building a forensic ancestry panel from the ground up: The EUROFORGEN Global AIM-SNP set. Forensic Sci. Int. Genet. 2014, 11, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Kosoy, R.; Nassir, R.; Tian, C.; White, P.A.; Butler, L.M.; Silva, G.; Kittles, R.; Alarcon-Riquelme, M.E.; Gregersen, P.K.; Belmont, J.W.; et al. Ancestry informative marker sets for determining continental origin and admixture proportions in common populations in America. Hum. Mutat. 2009, 30, 69–78. [Google Scholar] [CrossRef]
- Qin, H.; Zhu, X. Power comparison of admixture mapping and direct association analysis in genome-wide association studies. Genet. Epidemiol. 2012, 36, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, F.B.; Cagnin, N.F.; Simioni, M.; Farias, A.A.; Torres, F.R.; Molck, M.C.; Araujo, T.K.; Gil-Da-Silva-Lopes, V.L.; Donadi, E.A.; Simões, A.L. Ancestry informative marker panel to estimate population stratification using genome-wide human array. Ann. Hum. Genet. 2017, 81, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Schork, N.J.; Wilhelmsen, K.C.; Ehlers, C.L. Whole genome sequence association and ancestry-informed polygenic profile of EEG alpha in a Native American population. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2017, 174, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Shriner, D. Overview of admixture mapping. Curr. Protoc. Hum. Genet. 2013, 76, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Kidd, K.K.; Speed, W.C.; Pakstis, A.J.; Furtado, M.R.; Fang, R.; Madbouly, A.; Maiers, M.; Middha, M.; Friedlaender, F.R.; Kidd, J.R. Progress toward an efficient panel of SNPs for ancestry inference. Forensic Sci. Int. Genet. 2014, 10, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Huckins, L.M.; Boraska, V.; Franklin, C.S.; Floyd, J.A.; Southam, L.; Boraska, V.; Franklin, C.S.; Floyd, J.A.; Thornton, L.M.; Huckins, L.M.; et al. Using ancestry-informative markers to identify fine structure across 15 populations of European origin. Eur. J. Hum. Genet. 2014, 22, 1190–1200. [Google Scholar] [CrossRef]
- Xu, S.; Huang, W.; Qian, J.; Jin, L. Analysis of genomic admixture in Uyghur and its implication in mapping strategy. Am. J. Hum. Genet. 2008, 82, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Pakstis, A.J.; Kang, L.; Liu, L.; Zhang, Z.; Jin, T.; Grigorenko, E.L.; Wendt, F.R.; Budowle, B.; Hadi, S.; Al Qahtani, M.S.; et al. Increasing the reference populations for the 55 AISNP panel: The need and benefits. Int. J. Leg. Med. 2017, 131, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, N.A.; Li, L.M.; Ward, R.; Pritchard, J.K. Informativeness of genetic markers for inference of ancestry. Am. J. Hum. Genet. 2003, 73, 1402–1422. [Google Scholar] [CrossRef]
- Kidd, J.R.; Friedlaender, F.R.; Speed, W.C.; Pakstis, A.J.; De La Vega, F.M.; Kidd, K.K. Analyses of a set of 128 ancestry informative single-nucleotide polymorphisms in a global set of 119 population samples. Investig. Genet. 2011, 2, 1. [Google Scholar] [CrossRef]
- Wright, S. Evolution and the Genetics of Populations. A Treatise in Three Volumes; University of Chicago Press: Chicago, IL, USA, 1968. [Google Scholar]
- Marshall, S.; Das, R.; Pirooznia, M.; Elhaik, E. Reconstructing Druze population history. Sci. Rep. 2016, 6, 35837. [Google Scholar] [CrossRef]
- Lazaridis, I.; Patterson, N.; Mittnik, A.; Renaud, G.; Mallick, S.; Kirsanow, K.; Sudmant, P.H.; Schraiber, J.G.; Castellano, S.; Lipson, M. Ancient human genomes suggest three ancestral populations for present-day Europeans. Nature 2014, 513, 409–413. [Google Scholar] [CrossRef]
- Marcus, J.H.; Novembre, J. Visualizing the geography of genetic variants. Bioinformatics 2017, 33, 594–595. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Durbin, R.M.; Abecasis, G.R.; Altshuler, D.L.; Auton, A.; Brooks, L.D.; Gibbs, R.A.; Hurles, M.E.; McVean, G.A. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [CrossRef]
- Paschou, P.; Ziv, E.; Burchard, E.G.; Choudhry, S.; Rodriguez-Cintron, W.; Mahoney, M.W.; Drineas, P. PCA-correlated SNPs for structure identification in worldwide human populations. PLoS Genet. 2007, 3, 1672–1686. [Google Scholar] [CrossRef]
- Das, R.; Wexler, P.; Pirooznia, M.; Elhaik, E. Localizing Ashkenazic Jews to primeval villages in the ancient Iranian lands of Ashkenaz. Genome Biol. Evol. 2016, 8, 1132–1149. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Wexler, P.; Pirooznia, M.; Elhaik, E. The Origins of Ashkenaz, Ashkenazic Jews, and Yiddish. Front. Genet. 2017, 8, 87. [Google Scholar] [CrossRef] [PubMed]
- Baughn, L.B.; Pearce, K.; Larson, D.; Polley, M.-Y.; Elhaik, E.; Baird, M.; Colby, C.; Benson, J.; Li, Z.; Asmann, Y.; et al. Differences in genomic abnormalities among African individuals with monoclonal gammopathies using calculated ancestry. Blood Cancer J. 2018, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Elhaik, E. In search of the Jüdische Typus: A proposed benchmark to test the genetic basis of Jewishness challenges notions of “Jewish biomarkers”. Front. Genet. 2016, 7, 141. [Google Scholar] [CrossRef] [PubMed]
- Callaway, E. Divided by DNA: The uneasy relationship between archaeology and ancient genomics. Nature 2018, 555, 573–576. [Google Scholar] [CrossRef]
- Bose, N.; Carlberg, K.; Sensabaugh, G.; Erlich, H.; Calloway, C. Target capture enrichment of nuclear SNP markers for massively parallel sequencing of degraded and mixed samples. Forensic Sci. Int. Genet. 2018, 34, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Bulbul, O.; Speed, W.C.; Gurkan, C.; Soundararajan, U.; Rajeevan, H.; Pakstis, A.J.; Kidd, K.K. Improving ancestry distinctions among Southwest Asian populations. Forensic Sci. Int. Genet. 2018, 35, 14–20. [Google Scholar] [CrossRef]
- López-Cortés, A.; Echeverría-Garcés, G.; Burgos, G.; Zambrano, A.; Cabrera-Andrade, A.; García-Cárdenas, J.; Salazar, C.; Leone, P.; Paz-y-Miño, C. Molecular analysis of ancestry informative markers (AIMs-INDELs) in a high altitude Ecuadorian mestizo population affected with breast cancer. Forensic Sci. Int. Genet. Suppl. Ser. 2017, 6, e231–e232. [Google Scholar] [CrossRef]
- Tian, C.; Hinds, D.A.; Shigeta, R.; Adler, S.G.; Lee, A.; Pahl, M.V.; Silva, G.; Belmont, J.W.; Hanson, R.L.; Knowler, W.C.; et al. A genomewide single-nucleotide-polymorphism panel for Mexican American admixture mapping. Am. J. Hum. Genet. 2007, 80, 1014–1023. [Google Scholar] [CrossRef] [PubMed]
- Paschou, P.; Lewis, J.; Javed, A.; Drineas, P. Ancestry informative markers for fine-scale individual assignment to worldwide populations. J. Med. Genet. 2010, 47, 835–847. [Google Scholar] [CrossRef]
- Arenas, M.; Francois, O.; Currat, M.; Ray, N.; Excoffier, L. Influence of admixture and paleolithic range contractions on current European diversity gradients. Mol. Biol. Evol. 2013, 30, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Elhaik, E. The missing link of Jewish European ancestry: Contrasting the Rhineland and the Khazarian hypotheses. Genome Biol. Evol. 2013, 5, 61–74. [Google Scholar] [CrossRef] [PubMed]
- McVean, G. A genealogical interpretation of principal components analysis. PLoS Genet. 2009, 5, e1000686. [Google Scholar] [CrossRef] [PubMed]
- Novembre, J.; Stephens, M. Interpreting principal component analyses of spatial population genetic variation. Nat. Genet. 2008, 40, 646–649. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Wiener, H.; Abebe, T.; Altaye, M.; Go, R.C.; Kercsmar, C.; Grabowski, G.; Martin, L.J.; Hershey, G.K.; Chakorborty, R.; et al. Comparison of measures of marker informativeness for ancestry and admixture mapping. BMC Genom. 2011, 12, 622. [Google Scholar] [CrossRef] [PubMed]
- Elhaik, E. Empirical distributions of FST from large-scale Human polymorphism data. PLoS ONE 2012, 7, e49837. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | n | CSS | PD | FST | Infocalc | Admixture1 | Admixture2 | Rand10k | Rand15k |
|---|---|---|---|---|---|---|---|---|---|
| Britain Iron Saxon | 10 | 10 (100) | 4 (40) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (10) | 3 (30) |
| Caucasus Chalcolithic Bronze | 22 | 21 (95) | 8 (36) | 0 (0) | 12 (55) | 6 (27) | 4 (18) | 13 (59) | 9 (41) |
| Caucasus Mesolithic Neolithic | 9 | 6 (67) | 7 (78) | 0 (0) | 6 (67) | 1 (11) | 7 (78) | 4 (44) | 4 (44) |
| Central EU Early Neolithic | 26 | 17 (65) | 14 (54) | 4 (15) | 18 (69) | 4 (15) | 5 (19) | 14 (54) | 18 (69) |
| Central EU Late Neolithic Bronze | 57 | 16 (28) | 17 (30) | 19 (33) | 19 (33) | 13 (23) | 21 (37) | 25 (44) | 21 (37) |
| Central EU Mid Neolithic Chalc | 6 | 2 (33) | 3 (50) | 0 (0) | 3 (50) | 3 (50) | 3 (50) | 2 (33) | 2 (33) |
| Central North EU Late Neol Bronz | 20 | 18 (90) | 9 (45) | 0 (0) | 6 (30) | 0 (0) | 5 (25) | 4 (20) | 6 (30) |
| Central Western EU Mesolithic | 3 | 3 (100) | 2 (67) | 0 (0) | 3 (100) | 0 (0) | 0 (0) | 1 (33) | 3 (100) |
| Italy Mid Neolithic Chalcolithic | 4 | 4 (100) | 3 (75) | 0 (0) | 1 (25) | 1 (25) | 0 (0) | 1 (25) | 1 (25) |
| Jordan Bronze | 3 | 3 (100) | 2 (67) | 0 (0) | 0 (0) | 2 (67) | 3 (100) | 1 (33) | 2 (67) |
| Levant Epipaleolithic Neolithic | 19 | 7 (37) | 6 (32) | 0 (0) | 9 (47) | 8 (42) | 7 (37) | 4 (21) | 7 (37) |
| Russia Chalcolithic | 3 | 2 (67) | 3 (100) | 0 (0) | 1 (33) | 0 (0) | 2 (67) | 1 (33) | 1 (33) |
| Russia Early Mid Bronze | 19 | 19 (100) | 15 (79) | 0 (0) | 10 (53) | 0 (0) | 18 (95) | 10 (53) | 14 (74) |
| Russia Late Chalcolithic | 9 | 6 (67) | 6 (67) | 0 (0) | 5 (56) | 0 (0) | 1 (11) | 3 (33) | 3 (33) |
| Russia Mesolithic | 3 | 2 (67) | 2 (67) | 0 (0) | 2 (67) | 0 (0) | 1 (33) | 2 (67) | 2 (67) |
| Russia Mid Late Bronze | 22 | 15 (68) | 16 (73) | 0 (0) | 7 (32) | 0 (0) | 0 (0) | 4 (18) | 6 (27) |
| Spain Early Neolithic | 6 | 4 (67) | 5 (83) | 0 (0) | 6 (100) | 4 (67) | 4 (67) | 4 (67) | 5 (83) |
| Spain Mid Neolithic Chalcolithic | 18 | 7 (39) | 6 (33) | 0 (0) | 7 (39) | 5 (28) | 3 (17) | 5 (28) | 5 (28) |
| Sweden Mesolithic | 8 | 8 (100) | 8 (100) | 0 (0) | 7 (88) | 4 (50) | 1 (13) | 6 (75) | 7 (88) |
| Sweden Mid Neolithic | 4 | 4 (100) | 1 (25) | 1 (25) | 2 (50) | 1 (25) | 0 (0) | 4 (100) | 2 (50) |
| Turkey Neolithic | 24 | 23 (96) | 18 (75) | 0 (0) | 12 (50) | 3 (13) | 6 (25) | 8 (33) | 11 (46) |
| 76 ± 5 | 61 ± 5 | 3 ± 2 | 50 ± 6 | 21 ± 5 | 33 ± 7 | 42 ± 5 | 50 ± 5 |
| Parental Population A | Parental Population B | # Hybrids | |||
|---|---|---|---|---|---|
| Britain Iron Saxon | Britain Iron Saxon | 6 | 0.026 | 0.212 | 0.208 |
| Britain Iron Saxon | Russia Late Chalcolithic | 9 | 0.009 | 0.610 | 0.601 |
| Britain Iron Saxon | Sweden Mesolithic | 9 | 0.051 | 0.344 | 0.337 |
| Britain Iron Saxon | Turkey Neolithic | 9 | 0.003 | 0.428 | 0.431 |
| Britain Iron Saxon | Spain Early Neolithic | 9 | 0.108 | 0.221 | 0.241 |
| Russia Late Chalcolithic | Russia Late Chalcolithic | 6 | 0.009 | 0.443 | 0.448 |
| Russia Late Chalcolithic | Sweden Mesolithic | 9 | 0.062 | 0.578 | 0.561 |
| Russia Late Chalcolithic | Turkey Neolithic | 9 | 0.063 | 0.661 | 0.633 |
| Russia Late Chalcolithic | Spain Early Neolithic | 9 | 0.101 | 0.520 | 0.491 |
| Sweden Mesolithic | Sweden Mesolithic | 6 | 0.000 | 0.384 | 0.384 |
| Sweden Mesolithic | Turkey Neolithic | 9 | 0.055 | 0.567 | 0.522 |
| Spain Early Neolithic | Sweden Mesolithic | 9 | 0.108 | 0.402 | 0.377 |
| Turkey Neolithic | Turkey Neolithic | 6 | 0.001 | 0.627 | 0.626 |
| Spain Early Neolithic | Turkey Neolithic | 9 | 0.092 | 0.483 | 0.493 |
| Spain Early Neolithic | Spain Early Neolithic | 6 | 0.041 | 0.197 | 0.172 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esposito, U.; Das, R.; Syed, S.; Pirooznia, M.; Elhaik, E. Ancient Ancestry Informative Markers for Identifying Fine-Scale Ancient Population Structure in Eurasians. Genes 2018, 9, 625. https://doi.org/10.3390/genes9120625
Esposito U, Das R, Syed S, Pirooznia M, Elhaik E. Ancient Ancestry Informative Markers for Identifying Fine-Scale Ancient Population Structure in Eurasians. Genes. 2018; 9(12):625. https://doi.org/10.3390/genes9120625
Chicago/Turabian StyleEsposito, Umberto, Ranajit Das, Syakir Syed, Mehdi Pirooznia, and Eran Elhaik. 2018. "Ancient Ancestry Informative Markers for Identifying Fine-Scale Ancient Population Structure in Eurasians" Genes 9, no. 12: 625. https://doi.org/10.3390/genes9120625
APA StyleEsposito, U., Das, R., Syed, S., Pirooznia, M., & Elhaik, E. (2018). Ancient Ancestry Informative Markers for Identifying Fine-Scale Ancient Population Structure in Eurasians. Genes, 9(12), 625. https://doi.org/10.3390/genes9120625