Rediscovery of Red Wolf Ghost Alleles in a Canid Population Along the American Gulf Coast

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. DNA Sequencing and Bioinformatic Processing
2.2. Population Structure Analyses
2.3. Private Allele Sharing Analyses
2.4. Mitochondrial DNA Sequence Analysis
3. Results
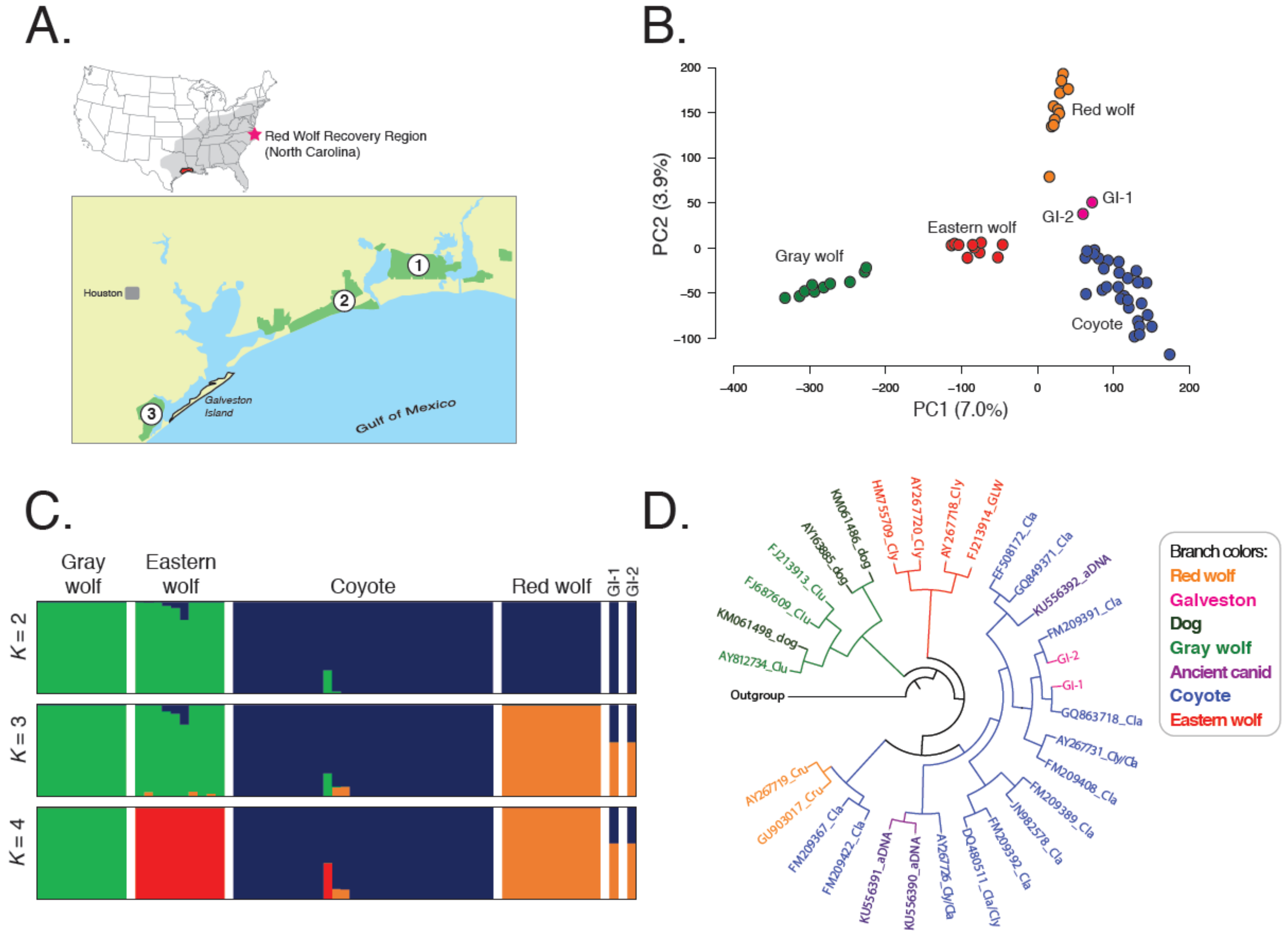
3.1. Galveston Island Canids Carry Red Wolf Genetic Signatures
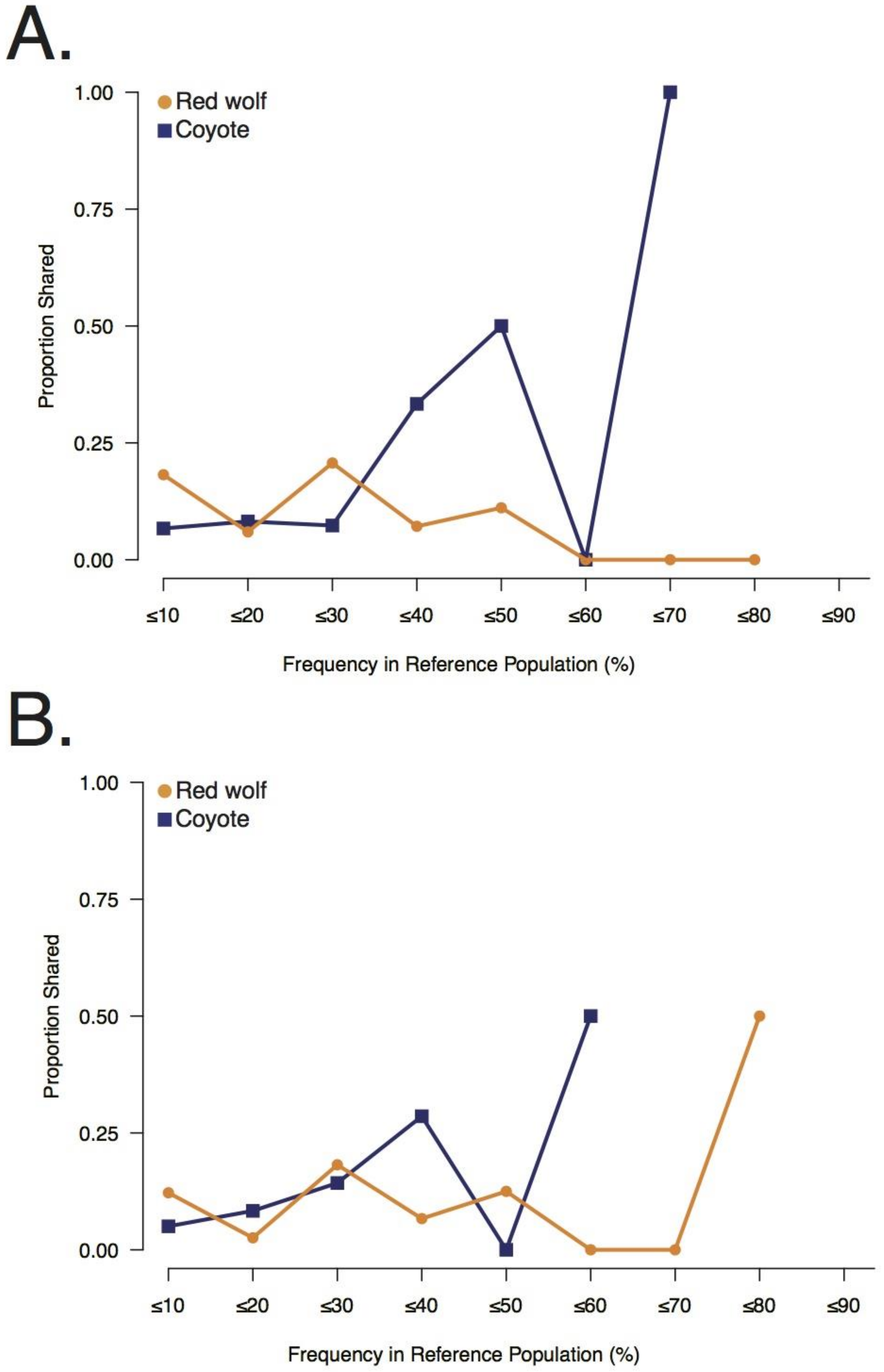
3.2. Galveston Canids Carry Red Wolf Private Alleles
3.3. Galveston Canids Carry Coyote mitochondrial DNA Haplotypes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McCarley, H.; Carley, C.J. Recent Changes in the Distribution and Status of Wild Red Wolves Canis rufus; Endangered Species Report No. 4; U.S. Fish and Wildlife Service: Albuquerque, NM, USA, 1979.
- Paradiso, J.L. Recent records of red wolves from the Gulf Coast of Texas. Southwest. Nat. 1965, 10, 318–319. [Google Scholar] [CrossRef]
- Pimlott, D.H.; Joslin, P.W. The status and distribution of the red wolf. T. N. Am. Wildl. Nat. Res. 1968, 33, 373–389. [Google Scholar]
- Nowak, R.W. Report on the red wolf. Def. Wildl. News 1970, 45, 82–94. [Google Scholar]
- Carley, C.J. Activities and Findings of the Red Wolf Recovery Program from Late 1973 to July 1, 1975; U.S. Fish and Wildlife Service: Albuquerque, NM, USA, 1975.
- Phillips, M.K.; Parker, W.T. Red wolf recovery: A progress report. Conserv. Biol. 1988, 2, 139–141. [Google Scholar] [CrossRef]
- Hedrick, P.W.; Fredrickson, J. Captive breeding and the reintroduction of Mexican and red wolves. Mol. Ecol. 2008, 17, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Fritts, S.H.; Bangs, E.E.; Fontaine, J.A.; Johnson, M.R.; Phillips, M.K.; Koch, E.D.; Gunson, J.R. Planning and implementing a reintroduction of wolves to Yellowstone National Park and central Idaho. Restor. Ecol. 1997, 5, 7–27. [Google Scholar] [CrossRef]
- Hedrick, P.W.; Miller, P.S.; Geffen, E.; Wayne, R.K. Genetic evaluation of the three captive Mexican wolf lineages. Zoo. Biol. 1997, 16, 47–69. [Google Scholar] [CrossRef]
- Endangered and Threatened Wildlife and Plants; Proposed Replacement of the Regulations for the Nonessential Experimental Population of Red Wolves in Northeastern North Carolina; Department of the Interior Fish and Wildlife Service Federal Register: Washington DC, USA, 2018; Volume 83, No. 125.
- Bohling, J.H.; Waits, L.P. Factors influencing red wolf–coyote hybridization in eastern North Carolina, USA. Biol. Cons. 2015, 184, 108–116. [Google Scholar] [CrossRef]
- Hinton, J.W.; Brzeski, K.E.; Rabon, D.R.; Chamberlain, M.J. Effects of anthropogenic mortality on critically endangered red wolf Canis rufus breeding pairs: Implications for red wolf recovery. Oryx 2015, 51, 174–181. [Google Scholar] [CrossRef]
- Weller, E. Red Wolf (Canis rufus). (United States Fish and Wildlife Service 5-Year Review. 2018. Available online: https://www.fws.gov/southeast/wildlife/mammal/red-wolf/# (accessed on 8 January 2018).
- Fredrickson, R.J.; Hedrick, P.W. Dynamics of hybridization and introgression in red wolves and coyotes. Conserv. Biol. 2006, 20, 1272–1283. [Google Scholar] [CrossRef]
- McCarley, H. The taxonomic status of wild Canis (Canidae) in the south central United States. Southwest. Nat. 1962, 7, 227–235. [Google Scholar] [CrossRef]
- Riley, G.A.; McBride, R.T. A survey of the red wolf (Canis rufus). U.S. Fish Wildl. Serv. Spec. Sci. Rep.-Wildl. 1972, 162, 1–15. [Google Scholar]
- Hailer, F.; Leonard, J.A. Hybridization among three native North American Canis species in a region of natural sympatry. PLoS ONE 2008, 3, e3333. [Google Scholar] [CrossRef] [PubMed]
- Hinton, J.W.; Chamberlain, M.J. Morphometrics of Canis taxa in eastern North Carolina. J. Mammal. 2014, 95, 855–861. [Google Scholar] [CrossRef]
- Giordano, M.R.; Pace, R.M., III. Morphometrics and movement patterns of coyote-like Canids in a southwest Louisiana marsh complex. Proc. Annu. Conf. Southeast. Assoc. Fish Wildl. Agencies 2000, 54, 424–435. [Google Scholar]
- Hinton, J.W.; Gittleman, J.L.; van Manen, F.T.; Chamberlain, M.J. Size-assortative choice and mate availability influences hybridization between red wolves (Canis rufus) and coyotes (Canis latrans). Ecol. Evol. 2018, 8, 3927–3940. [Google Scholar] [CrossRef]
- Ali, O.A.; O’Rourke, S.M.; Amish, S.J.; Meek, M.H.; Luikart, G.; Jeffres, C.; Miller, M.R. RAD Capture (Rapture): Flexible and efficient sequence-based genotyping. Genetics 2015, 202, 389–400. [Google Scholar] [CrossRef]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An analysis tool set for population genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef]
- Lindblad-Toh, K.; Wade, C.M.; Mikkelsen, T.S.; Karlsson, E.K.; Jaffe, D.B.; Kamal, M.; Clamp, M.; Chang, J.L.; Kulbokas, E.J., III; Zody, M.C.; et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 2005, 438, 803–819. [Google Scholar] [CrossRef]
- Lunter, G.; Goodson, M. Stampy: A statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 2011, 21, 936–939. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analysis. Am. J. Hum Genet. 2007, 81, 559–579. [Google Scholar] [CrossRef] [PubMed]
- Szpiech, Z.A.; Jakobsson, M.; Rosenberg, N.A. ADZE: A rarefaction approach for counting alleles private to combinations of populations. Bioinformatics 2008, 24, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Abraham, G.; Inouye, M. Fast principal component analysis of large-scale genome-wide data. PLoS ONE 2014, 9, e93766. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Gonder, M.K.; Locatelli, S.; Ghobrial, L.; Mitchell, M.W.; Kujawski, J.T.; Lankester, F.J.; Stewart, C.B.; Tishkoff, S.A. Evidence from Cameroon reveals differences in the genetic structure and histories of chimpanzee populations. Proc. Natl. Acad. Sci. USA 2011, 108, 4766–4771. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Girón, C.G.; et al. Ensembl 2018. Nucleic Acids Res. 2018, 46, D754–D761. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.A.; Wayne, R.K.; Wheeler, J.; Valadez, R.; Guillén, S.; Vilà, C. DNA evidence for Old World origin of New World dogs. Science 2002, 298, 1613–1616. [Google Scholar] [CrossRef]
- Brzeski, K.E.; DeBiasse, M.B.; Rabon, D.R., Jr.; Chamberlain, M.K.; Taylor, S.S. Mitochondrial DNA variation in southeastern Pre-Columbian canids. J. Hered. 2016, 107, 287–293. [Google Scholar] [CrossRef]
- Vilà, C.; Amorim, I.R.; Leonard, J.A.; Posada, D.; Castroviejo, J.; Petrucci-Fonseca, F.; Crandall, K.A.; Ellegren, H.; Wayne, R.K. Mitochondrial DNA phylogeography and population history of the grey wolf Canis lupus. Mol. Ecol. 1999, 8, 2089–2103. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUTi and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Koblmüller, S.; Wayne, R.K.; Leonard, J.A. Impact of Quarternary climate changes and interspecific competition on the demographic history of a highly mobile generalist carnivore, the coyote. Biol. Lett. 2012, 8, 644–647. [Google Scholar] [CrossRef] [PubMed]
- Haig, S.M.; Allendorf, F.W. The Endangered Species Act at Thirty: Volume 2: Conserving Biodiversity in Human-Dominated Landscapes; Scott, D.D., Davis, F.W., Eds.; Island Press: Washington, DC, USA, 2006; pp. 150–163. [Google Scholar]
- Becker, M.; Gruenheit, N.; Steel, M.; Voelckel, C.; Deusch, O.; Heenan, P.B.; McLenachan, P.A.; Kardailsky, O.; Leigh, J.W.; Lockhard, P.J. Hybridization may facilitate in situ survival of endemic species through periods of climate change. Nat. Clim. Chang. 2013, 3, 1039–1043. [Google Scholar] [CrossRef]
- Jackiw, R.N.; Mandil, G.; Hager, H.A. A framework to guide the conservation of species hybrids based on ethical and ecological considerations. Conserv. Biol. 2015, 29, 1040–1051. [Google Scholar] [CrossRef]
- Waples, R.; Kays, R.; Fredrickson, R.J.; Pacifici, K.; Mills, L.S. Is the red wolf a listable unit under the US Endangered Species Act? J. Hered. 2018, 109, 585–597. [Google Scholar] [CrossRef]
- Amador, C.; Hayes, B.J.; Daetwyler, H.D. Genomic selection for recovery of original genetic background from hybrids of endangered and common breeds. Evol. Appl. 2014, 7, 227–237. [Google Scholar] [CrossRef]
- Miller, J.M.; Quinzin, M.C.; Poulakakis, N.; Gibbs, J.P.; Beheregaray, L.B.; Garrick, R.C.; Russello, M.A.; Ciofi, C.; Edwards, D.L.; Hunter, E.A.; et al. Identification of genetically important individuals of the rediscovered Floreana Galápagos giant tortoise (Chelonoidis elephantopus) provide founders for Species Restoration Program. Sci. Rep. 2017, 7, 11471. [Google Scholar] [CrossRef]
- Allendorf, F.A.; Leary, R.F.; Spruell, P.; Wenburg, J.K. The problems with hybrids: Setting conservation guidelines. Trends Ecol. Evol. 2001, 16, 613–622. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Group | Sampling Location * | n | HO | HE | AR * | NPA | PAR ** |
|---|---|---|---|---|---|---|---|
| Coyote | Southeastern USA | 29 | 0.085 | 0.101 | 1.52 | 2,686 | 0.28 |
| Gray wolf | Yellowstone National Park | 10 | 0.072 | 0.076 | 1.27 | 368 | 0.10 |
| Eastern wolf | Algonquin Provincial Park | 10 | 0.079 | 0.087 | 1.36 | 332 | 0.11 |
| Red wolf (captive) | Point Defiance Zoo and Aquarium | 11 | 0.051 | 0.061 | 1.17 | 191 | 0.04 |
| Reference Group | GI-1 | GI-2 | ||||||
|---|---|---|---|---|---|---|---|---|
| NPA | SPA (Count) | SPA (%) | SPAr | NPA | SPA (count) | SPA (%) | SPAr | |
| Coyote | 2632 | 184 | 6.99% | 0.0102 | 2439 | 138 | 5.66% | 0.0093 |
| Gray wolf | 362 | 12 | 3.31% | 0.0035 | 335 | 10 | 2.99% | 0.0036 |
| Eastern wolf | 329 | 12 | 3.65% | 0.0045 | 303 | 8 | 2.64% | 0.0039 |
| Red wolf | 188 | 21 | 11.17% | 0.0059 | 171 | 14 | 8.19% | 0.0063 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heppenheimer, E.; Brzeski, K.E.; Wooten, R.; Waddell, W.; Rutledge, L.Y.; Chamberlain, M.J.; Stahler, D.R.; Hinton, J.W.; VonHoldt, B.M. Rediscovery of Red Wolf Ghost Alleles in a Canid Population Along the American Gulf Coast. Genes 2018, 9, 618. https://doi.org/10.3390/genes9120618
Heppenheimer E, Brzeski KE, Wooten R, Waddell W, Rutledge LY, Chamberlain MJ, Stahler DR, Hinton JW, VonHoldt BM. Rediscovery of Red Wolf Ghost Alleles in a Canid Population Along the American Gulf Coast. Genes. 2018; 9(12):618. https://doi.org/10.3390/genes9120618
Chicago/Turabian StyleHeppenheimer, Elizabeth, Kristin E. Brzeski, Ron Wooten, William Waddell, Linda Y. Rutledge, Michael J. Chamberlain, Daniel R. Stahler, Joseph W. Hinton, and Bridgett M. VonHoldt. 2018. "Rediscovery of Red Wolf Ghost Alleles in a Canid Population Along the American Gulf Coast" Genes 9, no. 12: 618. https://doi.org/10.3390/genes9120618
APA StyleHeppenheimer, E., Brzeski, K. E., Wooten, R., Waddell, W., Rutledge, L. Y., Chamberlain, M. J., Stahler, D. R., Hinton, J. W., & VonHoldt, B. M. (2018). Rediscovery of Red Wolf Ghost Alleles in a Canid Population Along the American Gulf Coast. Genes, 9(12), 618. https://doi.org/10.3390/genes9120618