Towards a Central Role of ISL1 in the Bladder Exstrophy–Epispadias Complex (BEEC): Computational Characterization of Genetic Variants and Structural Modelling

 ,
,  ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sequence Homology-Based Single Nucleotide Polymorphism Prediction Using PolyPhen-2 & CADD
2.2. Identification of Conserved Residues and Sequence Motifs Using Consurf
2.3. Computational 3D Structural Modelling
2.4. Prediction of Functional Consequences of Non-Coding Variants and Prediction of Long Noncoding RNA Targets
2.5. Generation of Long Noncoding RNA Secondary Structures
2.6. Protein-Protein Interaction Network Analysis
3. Results
3.1. Characterization of Genetic Variants and Associated Long Noncoding RNA
3.2. Structural Modeling of ISL1 Wild Type Protein and the ISL1 Variant, p.Ala46Gly
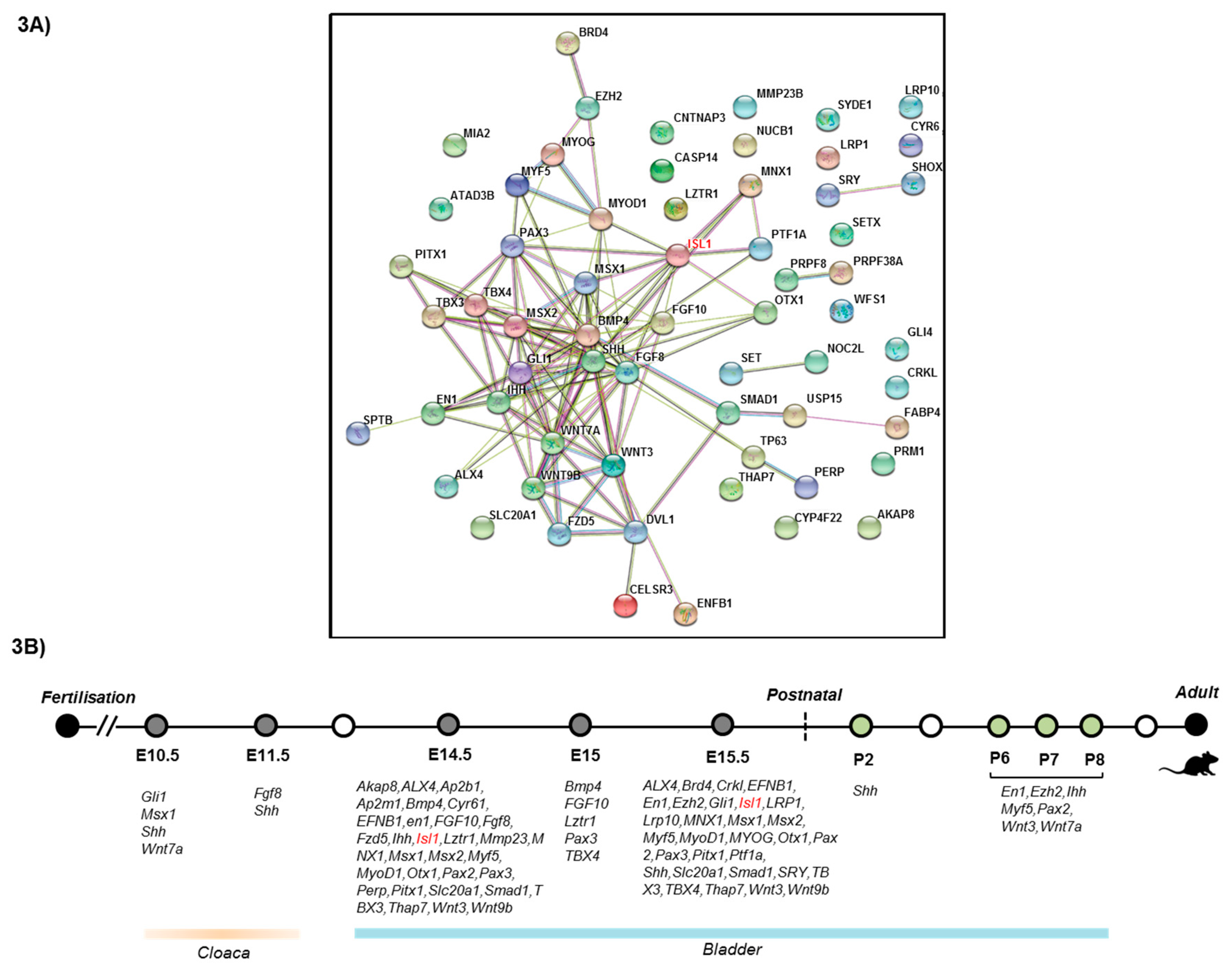
3.3. Protein–Protein InteractionsNetwork Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ebert, A.K.; Reutter, H.; Ludwig, M.; Rosch, W.H. The exstrophy-epispadias complex. Orphanet J. Rare Dis. 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Gearhart, J.P; Jeffs, R.D. Exstrophy-epispadias complex and bladder anomalies. In Campbell’s Urology, 7th ed.; Camp, P.C., Retik, A.B., Vaughan, E.D., Wein, A.J., Eds.; WB Saunders Co: Philadelphia, PA, USA, 1998; pp. 1939–1990. [Google Scholar]
- Draaken, M.; Knapp, M.; Pennimpede, T.; Schmidt, J.M.; Ebert, A.K.; Rosch, W.; Stein, R.; Utsch, B.; Hirsch, K.; Boemers, T.M.; et al. Genome-wide association study and meta-analysis identify ISL1 as genome-wide significant susceptibility gene for bladder exstrophy. PLoS Genet. 2015, 11, e1005024. [Google Scholar] [CrossRef]
- Arkani, S.; Cao, J.; Lundin, J.; Nilsson, D.; Kallman, T.; Barker, G.; Holmdahl, G.; Clementsson Kockum, C.; Matsson, H.; Nordenskjold, A. Evaluation of the ISL1 gene in the pathogenesis of bladder exstrophy in a Swedish cohort. Hum Genome Var. 2018, 5, 18009. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Knapp, M.; Suzuki, K.; Kajioka, D.; Schmidt, J.M.; Winkler, J.; Yilmaz, O.; Pleschka, M.; Cao, J.; Kockum, C.C.; et al. ISL1 is a major susceptibility gene for classic bladder exstrophy and a regulator of urinary tract development. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Gay, F.; Anglade, I.; Gong, Z.; Salbert, G. The LIM/homeodomain protein islet-1 modulates estrogen receptor functions. Mol. Endocrinol. 2000, 14, 1627–1648. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Ramensky, V.; Bork, P.; Sunyaev, S. Human non-synonymous SNPs: Server and survey. Nucleic Acids Res. 2002, 30, 3894–3900. [Google Scholar] [CrossRef] [PubMed]
- Dakal, T.C.; Kala, D.; Dhiman, G.; Yadav, V.; Krokhotin, A.; Dokholyan, N.V. Predicting the functional consequences of non-synonymous single nucleotide polymorphisms in IL8 gene. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O'Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazy, H.; Erez, E.; Martz, E.; Pupko, T.; Ben-Tal, N. Consurf 2010: Calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010, 38, W529–W533. [Google Scholar] [CrossRef]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Dakal, T.C.; Kumar, R.; Ramotar, D. Computational structural modeling of human organic cation transporters. Comput. Biol. Chem. 2017, 68, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Thornton, J.M. PROCHECK: Validation of protein structure coordinates. In International Tables of Crystallography, Volume F. Crystallography of Biological Macromolecules; Rossmann, M.G., Arnold, E., Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2001; pp. 722–725. [Google Scholar]
- Ritchie, G.R.; Dunham, I.; Zeggini, E.; Flicek, P. Functional annotation of noncoding sequence variants. Nat. Methods 2014, 11, 294–296. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhang, Z.; Bailey, T.L.; Perkins, A.C.; Tallack, M.R.; Xu, Z.; Liu, H. Prediction of novel long non-coding RNAs based on RNA-Seq data of mouse Klf1 knockout study. BMC Bioinformatics 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Wang, G.; Chen, L.; Jiang, J.; Liu, L.; Li, N.; Zhao, J.; Sun, X.; Zhou, P. Genome-wide analysis of long non-coding RNAs at early stage of skin pigmentation in goats (Capra hircus). BMC Genomics 2016, 17, 67. [Google Scholar] [CrossRef]
- Wang, Y.; Xue, S.; Liu, X.; Liu, H.; Hu, T.; Qiu, X.; Zhang, J.; Lei, M. Analyses of long non-coding RNA and mRNA profiling using RNA sequencing during the pre-implantation phases in pig endometrium. Sci. Rep. 2016, 6, 20238. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDtools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Li, J.; Ma, W.; Zeng, P.; Wang, J.; Geng, B.; Yang, J.; Cui, Q. LncTar: A tool for predicting the RNA targets of long noncoding RNAs. Brief Bioinform. 2015, 16, 806–812. [Google Scholar] [CrossRef]
- Hofacker, I.L. Vienna RNA secondary structure server. Nucleic Acids Res. 2003, 31, 3429–3431. [Google Scholar] [CrossRef]
- Von Mering, C.; Jensen, L.J.; Kuhn, M.; Chaffron, S.; Doerks, T.; Kruger, B.; Snel, B.; Bork, P. STRING 7--recent developments in the integration and prediction of protein interactions. Nucleic Acids Res. 2007, 35, D358–D362. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Finger, J.H.; Smith, C.M.; Hayamizu, T.F.; McCright, I.J.; Xu, J.; Law, M.; Shaw, D.R.; Baldarelli, R.M.; Beal, J.S.; Blodgett, O.; et al. The mouse gene expression database (GXD): 2017 update. Nucleic Acids Res. 2017, 45, D730–D736. [Google Scholar] [CrossRef] [PubMed]
- Gadd, M.S.; Jacques, D.A.; Nisevic, I.; Craig, V.J.; Kwan, A.H.; Guss, J.M.; Matthews, J.M. A structural basis for the regulation of the LIM-homeodomain protein islet 1 (Isl1) by intra- and intermolecular interactions. J. Biol. Chem. 2013, 288, 21924–21935. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, R.; Matsumaru, D.; Nakagata, N.; Miyagawa, S.; Suzuki, K.; Kitazawa, S.; Yamada, G. The hedgehog signal induced modulation of bone morphogenetic protein signaling: An essential signaling relay for urinary tract morphogenesis. PLoS ONE 2012, 7, e42245. [Google Scholar] [CrossRef] [PubMed]
- Baranowska Korberg, I.; Hofmeister, W.; Markljung, E.; Cao, J.; Nilsson, D.; Ludwig, M.; Draaken, M.; Holmdahl, G.; Barker, G.; Reutter, H.; et al. WNT3 involvement in human bladder exstrophy and cloaca development in zebrafish. Hum. Mol. Genet. 2015, 24, 5069–5078. [Google Scholar] [CrossRef]
- Xia, J.; Zeng, M.; Zhu, H.; Chen, X.; Weng, Z.; Li, S. Emerging role of hippo signalling pathway in bladder cancer. J. Cell. Mol. Med. 2018, 22, 4–15. [Google Scholar] [CrossRef]
- Shimomura, H.; Sanke, T.; Hanabusa, T.; Tsunoda, K.; Furuta, H.; Nanjo, K. Nonsense mutation of islet-1 gene (Q310X) found in a type 2 diabetic patient with a strong family history. Diabetes 2000, 49, 1597–1600. [Google Scholar] [CrossRef]
- Holm, P.; Rydlander, B.; Luthman, H.; Kockum, I. Interaction and association analysis of a type 1 diabetes susceptibility locus on chromosome 5q11-q13 and the 7q32 chromosomal region in Scandinavian families. Diabetes 2004, 53, 1584–1591. [Google Scholar] [CrossRef]
- Cai, C.L.; Liang, X.; Shi, Y.; Chu, P.H.; Pfaff, S.L.; Chen, J.; Evans, S. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev. Cell. 2003, 5, 877–889. [Google Scholar] [CrossRef]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, A.; Dakal, T.C.; Ludwig, M.; Fröhlich, H.; Mathur, R.; Reutter, H. Towards a Central Role of ISL1 in the Bladder Exstrophy–Epispadias Complex (BEEC): Computational Characterization of Genetic Variants and Structural Modelling. Genes 2018, 9, 609. https://doi.org/10.3390/genes9120609
Sharma A, Dakal TC, Ludwig M, Fröhlich H, Mathur R, Reutter H. Towards a Central Role of ISL1 in the Bladder Exstrophy–Epispadias Complex (BEEC): Computational Characterization of Genetic Variants and Structural Modelling. Genes. 2018; 9(12):609. https://doi.org/10.3390/genes9120609
Chicago/Turabian StyleSharma, Amit, Tikam Chand Dakal, Michael Ludwig, Holger Fröhlich, Riya Mathur, and Heiko Reutter. 2018. "Towards a Central Role of ISL1 in the Bladder Exstrophy–Epispadias Complex (BEEC): Computational Characterization of Genetic Variants and Structural Modelling" Genes 9, no. 12: 609. https://doi.org/10.3390/genes9120609
APA StyleSharma, A., Dakal, T. C., Ludwig, M., Fröhlich, H., Mathur, R., & Reutter, H. (2018). Towards a Central Role of ISL1 in the Bladder Exstrophy–Epispadias Complex (BEEC): Computational Characterization of Genetic Variants and Structural Modelling. Genes, 9(12), 609. https://doi.org/10.3390/genes9120609