Population Genomic Analysis of North American Eastern Wolves (Canis lycaon) Supports Their Conservation Priority Status

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
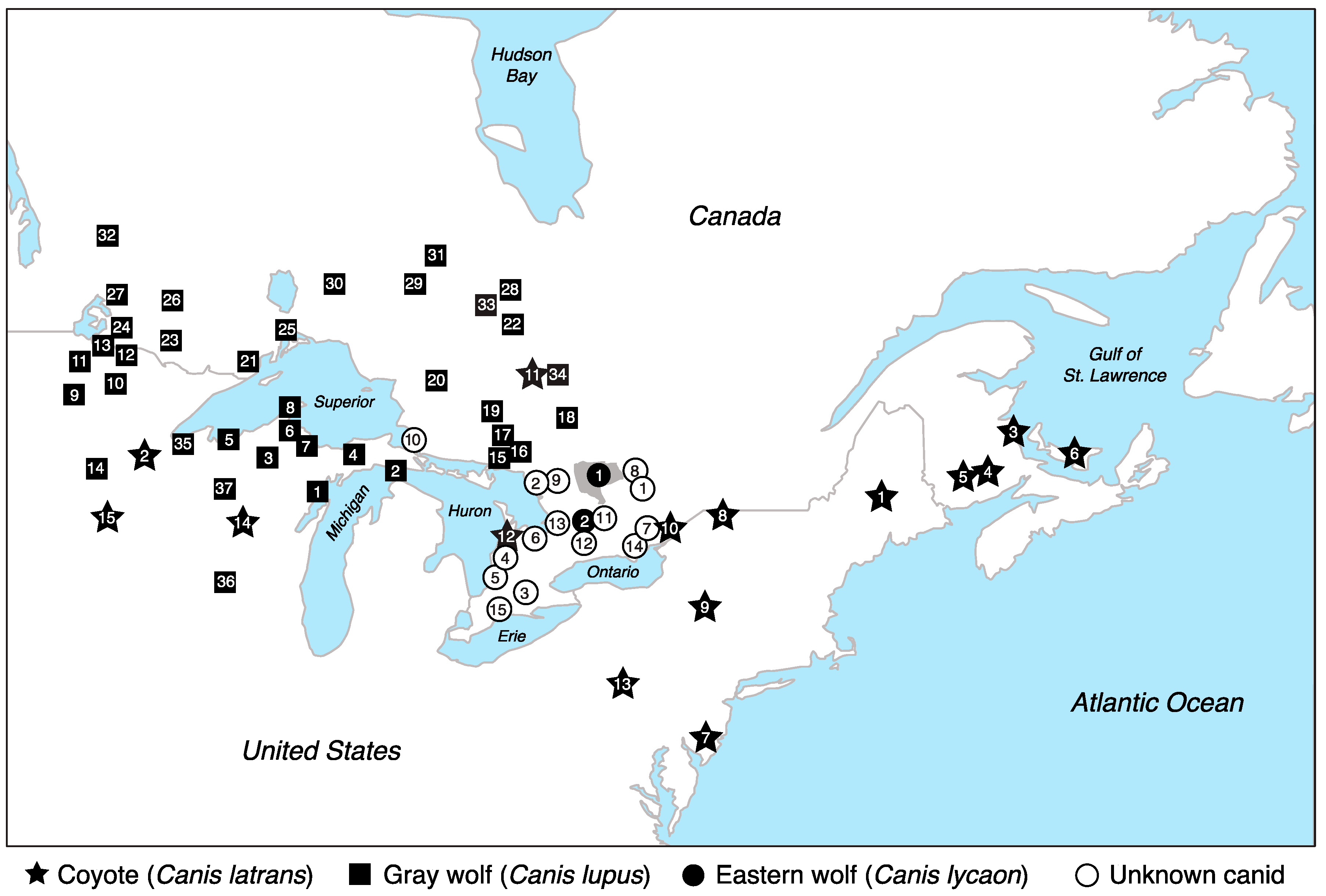
2.1. Sample Collection and DNA Extraction
2.2. Reduced Representation Sequencing and Data Processing
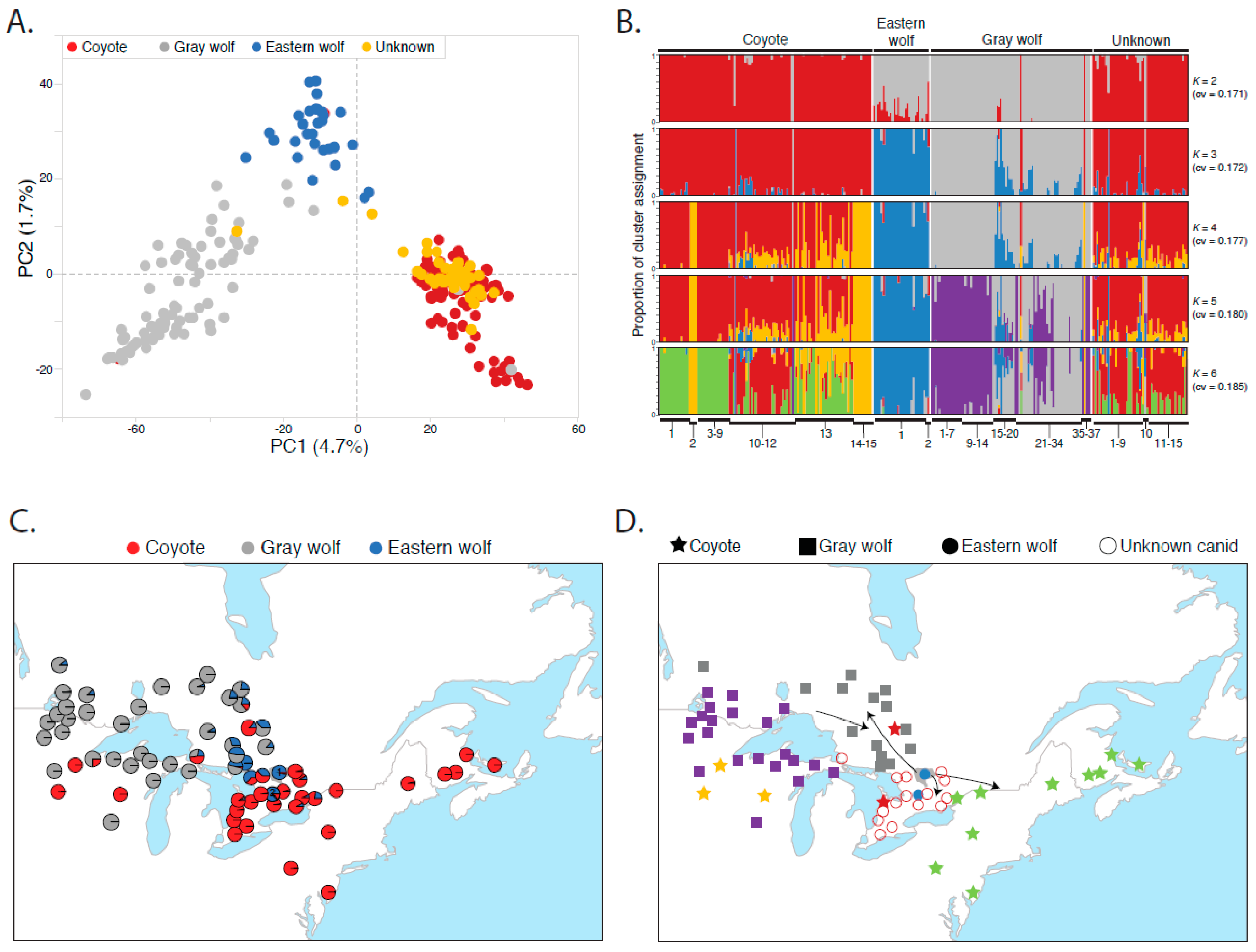
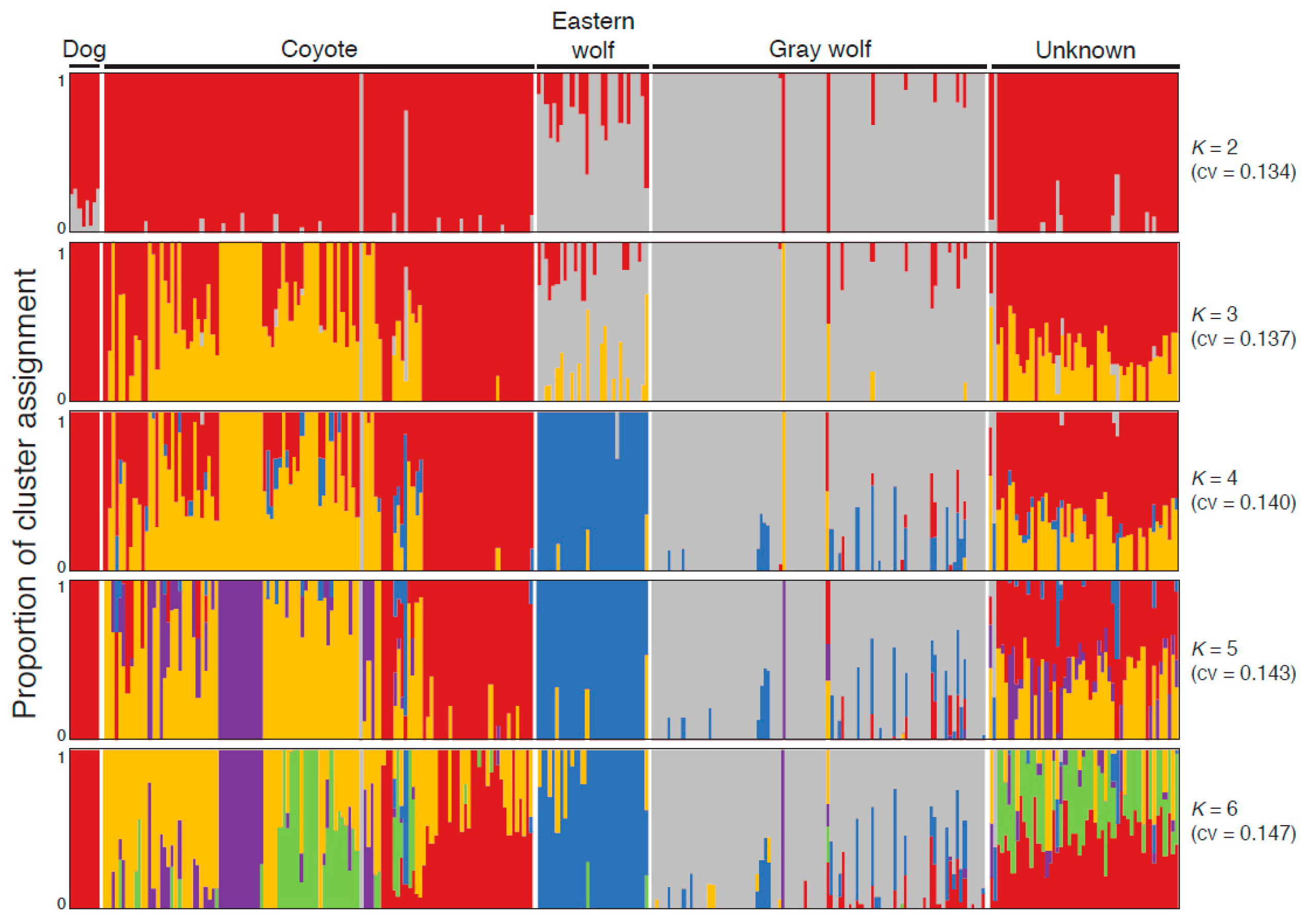
2.3. Clustering and Genetic Structure Analysis
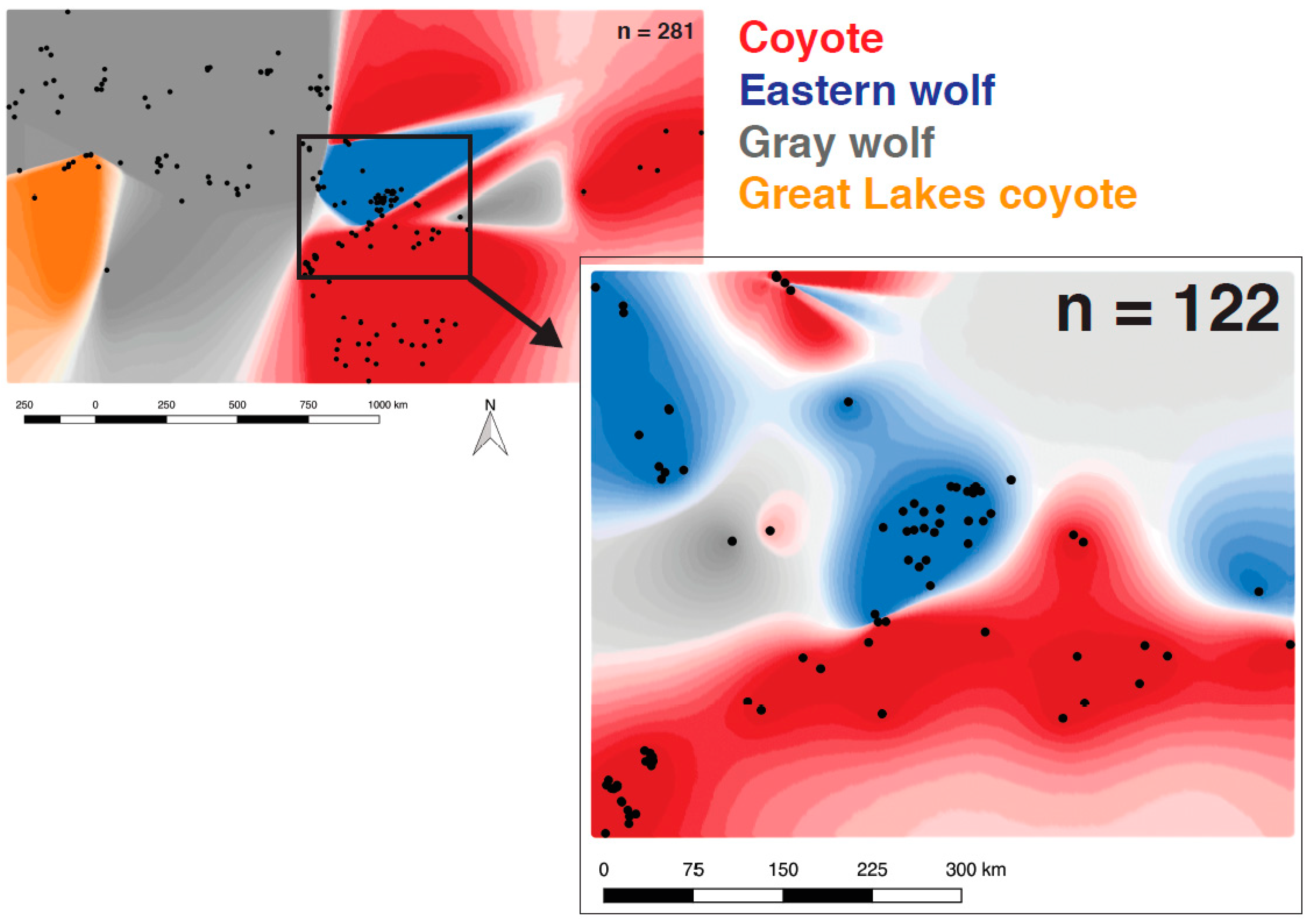
2.4. Spatially Explicit Bayesian Clustering
3. Results
3.1. Eastern Wolves Are a Genetically Isolated Population of Wolves
3.2. Canids of Unknown Taxonomic Affiliation Carry Predominantly Coyote Genetic Variation
3.3. Eastern Wolf Genetic Variation Is Geographically Surrounded by Coyote Populations
3.4. Eastern Wolves Are Spatially and Genetically Isolated
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix

References
- Kimura, M. Evolutionary rate at the molecular level. Nature 1968, 217, 624–626. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.; O’Hely, M. Captive breeding and the genetic fitness of natural populations. Conserv. Genet. 2001, 2, 363–378. [Google Scholar] [CrossRef]
- Yoder, A.D.; Poelstra, J.W.; Tiley, G.P.; Williams, R.C. Neutral theory is the foundation of conservation genetics. Mol. Biol. Evol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, B. Effective population size and patterns of molecular evolution and variation. Nat. Rev. Genet. 2009, 10, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Frankham, R.; Ballou, J.D.; Briscoe, D.A. Introduction to Conservation Genetics; Cambridge University Press: Cambridge, UK, 2002. [Google Scholar]
- Wakeley, J. Polymorphism and divergence for island-model species. Genetics 2003, 163, 411–420. [Google Scholar] [PubMed]
- Dudaniec, R.Y.; Schlotfeldt, B.E.; Bertozzi, T.; Donnellan, S.C.; Kleindorfer, S. Genetic and morphological divergence in island and mainland birds: Informing conservation priorities. Biol. Conserv. 2011, 144, 2902–2912. [Google Scholar] [CrossRef]
- Nosil, P.; Feder, J.L. Genomic divergence during speciation: Causes and consequences. Philos. Trans. R. Soc. B 2012, 367, 332–342. [Google Scholar] [CrossRef]
- Allendorf, F.W.; Leary, R.F.; Spruell, P.; Wenburg, J.K. The problems with hybrids: Setting conservation guidelines. TREE 2001, 16, 613–622. [Google Scholar] [CrossRef]
- Mallet, J. Hybridization as an invasion of the genome. Trends Ecol. Evol. 2005, 20, 229–237. [Google Scholar] [CrossRef]
- Crispo, E.; Moore, J.-S.; Lee-Yaw, J.A.; Gray, S.M.; Haller, B.C. Broken barriers: Human-induced changes to gene flow and introgression in animals. BioEssays 2011, 33, 508–518. [Google Scholar] [CrossRef]
- Grabenstein, K.C.; Taylor, S.A. Breaking barriers: Causes, consequences, and experimental utility of human-mediated hybridization. Trends Ecol. Evol. 2017, 33, 198–212. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, S.J.; Mayr, E. Bureaucratic mischief: Recognizing endangered species and subspecies. Science 1991, 251, 1187–1188. [Google Scholar] [CrossRef] [PubMed]
- Jackiw, R.N.; Mandil, G.; Hager, H.A. A framework to guide the conservation of species hybrids based on ethical and ecological considerations. Conserv. Biol. 2015, 29, 1040–1051. [Google Scholar] [CrossRef] [PubMed]
- Waples, R.S.; Kays, R.; Fredrickson, R.J.; Pacifici, K.; Mills, L.S. Is the red wolf a listable unit under the US Endangered Species Act? J. Hered. 2018, 109, 585–597. [Google Scholar] [CrossRef] [PubMed]
- COSEWIC (Committee on the Status of Endangered Wildlife in Canada) COSEWIC Annual Report. 2008. Available online: http://www.sararegistry.gc.ca/sar/assessment/cosewic_annual_e.cfm (accessed on 14 July 2018).
- Rhymer, J.M.; Simberloff, D. Extinction by hybridization and introgression. Ann. Rev. Ecol. Syst. 1996, 27, 83–109. [Google Scholar] [CrossRef]
- Gow, J.L.; Peichel, C.L.; Taylor, E.B. Contrasting hybridization rates between sympatric three-spined sticklebacks highlight the fragility of reproductive barriers between evolutionarily young species. Mol. Ecol. 2006, 15, 739–752. [Google Scholar] [CrossRef]
- Taylor, E.B.; Boughman, J.W.; Groenenboom, M.; Sniatynski, M.; Schluter, D.; Gow, J.L. Speciation in reverse: Morphological and genetic evidence of the collapse of a three-spined stickleback (Gasterosteus aculeatus) species pair. Mol. Ecol. 2006, 15, 343–355. [Google Scholar] [CrossRef]
- Seehausen, O.; Takimoto, G.; Roy, D.; Jokela, J. Speciation reversal and biodiversity dynamics with hybridization in changing environments. Mol. Ecol. 2008, 17, 30–44. [Google Scholar] [CrossRef]
- Mee, J.A.; Taylor, E.B. The cybrid invasion: Widespread postglacial dispersal by Phoxinus (Pisces: Cyprinidae) cytoplasmic hybrids. Can. J. Zool. 2012, 90, 577–584. [Google Scholar] [CrossRef]
- Quilodrán, C.S.; Montoya-Burgos, J.I.; Currat, M. Modelling interspecific hybridization with genome exclusion to identify conservation actions: The case of native and invasive Pelophylax waterfrogs. Evol. Appl. 2015, 8, 199–210. [Google Scholar] [CrossRef]
- Quilodrán, C.S.; Currat, M.; Montoya-Burgos, J.I. Effect of hybridization with genome exclusion on extinction risk. Conserv. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Koepfli, K.-P.; Pollinger, J.; Godinho, R.; Robinson, J.; Lea, A.; Hendricks, S.; Schweizer, R.M.; Thalmann, O.; Silva, P.; Fan, Z.; et al. Genome-wide evidence reveals that African and Eurasian golden jackals are distinct species. Curr. Biol. 2015, 25, 2158–2165. [Google Scholar] [CrossRef]
- Viranta, S.; Atickem, A.; Werdelin, L.; Stenseth, N.C. Rediscovering a forgotten canid species. BMC Zool. 2017, 2, 6. [Google Scholar] [CrossRef]
- Lovette, I.J.; Clegg, S.M.; Smith, T.B. Limited utility of mtDNA markers for determining connectivity among breeding and overwintering locations in three Neotropical migrant birds. Conserv. Biol. 2004, 18, 156–166. [Google Scholar] [CrossRef]
- Rubinoff, D. Utility of mitochondrial DNA barcodes in species conservation. Conserv. Biol. 2006, 20, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.B.; Freyer, C.; Elson, J.L.; Larsson, N.G. Purifying selection of mtDNA and its implications for understanding evolution and mitochondrial disease. Nat. Rev. Genet. 2008, 9, 657–662. [Google Scholar] [CrossRef]
- Wilson, P.J.; Grewal, S.; Lawford, I.D.; Heal, J.N.M.; Granacki, A.G.; Pennock, D.; Theberge, J.B.; Theberge, M.T.; Voight, D.R.; Waddell, W.; et al. DNA profiles of the eastern Canadian wolf and the red wolf provide evidence for a common evolutionary history independent of the gray wolf. Can. J. Zool. 2000, 78, 2156–2166. [Google Scholar] [CrossRef]
- Kyle, C.J.; Johnson, A.R.; Patterson, B.R.; Wilson, P.J.; Shami, K.; Grewal, S.K.; White, B.N. Genetic nature of eastern wolves: Past, present and future. Conserv. Genet. 2006, 7, 273–287. [Google Scholar] [CrossRef]
- Koblmüller, S.; Nord, M.; Wayne, R.K.; Leonard, J.A. Origin and status of the Great Lakes wolf. Mol. Ecol. 2009, 18, 2313–2326. [Google Scholar] [CrossRef]
- Kays, R.; Curtis, A.; Kirchman, J.J. Rapid adaptive evolution of northeastern coyotes via hybridization with wolves. Biol. Lett. 2010, 6, 89–93. [Google Scholar] [CrossRef]
- Rutledge, L.Y.; Wilson, P.J.; Klütsch, C.F.C.; Patterson, B.R.; White, B.N. Conservation genomics in perspective: A holistic approach to understanding Canis evolution in North America. Biol. Conserv. 2012, 155, 186–192. [Google Scholar] [CrossRef]
- von Holdt, B.M.; Pollinger, J.P.; Earl, D.A.; Knowles, J.C.; Boyko, A.R.; Parker, H.; Geffen, E.; Pilot, M.; Jedrzejewski, W.; Jedrzejewska, B.; et al. A genome-wide perspective on the evolutionary history of enigmatic wolf-like canids. Genome Res. 2011, 21, 1294–1305. [Google Scholar] [CrossRef] [PubMed]
- Rutledge, L.Y.; Devillard, S.; Boone, J.Q.; Hohenlohe, P.A.; White, B.N. RAD sequencing and genomic simulations resolve hybrid origins within North American Canis. Biol. Lett. 2015, 11, 20150303. [Google Scholar] [CrossRef] [PubMed]
- von Holdt, B.M.; Cahill, J.A.; Fan, Z.; Gronau, I.; Robinson, J.; Pollinger, J.P.; Shapiro, B.; Wall, J.; Wayne, R.K. Whole-genome sequence analysis shows that two endemic species of North American wolf are admixtures of the coyote and gray wolf. Sci. Adv. 2016, 2, e1501714. [Google Scholar] [CrossRef] [PubMed]
- Rutledge, L.Y.; Garroway, C.J.; Loveless, K.M.; Patterson, B.R. Genetic differentiation of eastern wolves in Algonquin Park despite bridging gene flow between coyotes and gray wolves. Heredity 2010, 10, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Benson, J.F.; Patterson, B.R.; Wheeldon, T.J. Spatial genetic and morphologic structure of wolves and coyotes in relation to environmental heterogeneity in a Canis hybrid zone. Mol. Ecol. 2012, 21, 5934–5954. [Google Scholar] [CrossRef] [PubMed]
- Rutledge, L.Y.; Desy, G.; Fryxell, J.M.; Middel, K.; White, B.N.; Patterson, B.R. Patchy distribution and low effective size raise concern for an at-risk top predator. Divers. Distrib. 2017, 23, 79–89. [Google Scholar] [CrossRef]
- Benson, J.F.; Patterson, B.R.; Mahoney, P.J. A protected area influences genotype-specific survival and structure of a Canis hybrid zone. Ecology 2014, 95, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Benson, J.F.; Patterson, B.R. Inter-specific territoriality in a Canis hybrid zone: Spatial segregation between wolves, coyotes, and hybrids. Oecologia 2013, 173, 1539–1550. [Google Scholar] [CrossRef] [PubMed]
- COSEWIC (Committee on the Status of Endangered Wildlife in Canada). COSEWIC Assessment and Status Report on the Eastern Wolf Canis spp. cf. lycaon in Canada. 2015. Available online: https://www.registrelep-sararegistry.gc.ca/default.asp?lang=En&n=65C48F31-1 (accessed on 14 July 2018).
- COSSARO (Committee on the Status of Species at Risk in Ontario). Ontario Species at Risk Evaluation Report for Algonquin Wolf (Canis sp.), An Evolutionarily Significant and Distinct Hybrid with Canis lycaon, C. latrans, and C. lupus Ancestry. 2016. Available online: https://www.ontario.ca/page/algonquin-wolf (accessed on 15 October 2018).
- Parker, G.R. Eastern Coyote: The Story of Its Success; Nimbus: Halifax, NS, Canada, 1995. [Google Scholar]
- Wilson, P.; Grewal, S.; Mallory, F.; White, B. Genetic characterization of hybrid wolves across Ontario. J. Hered. 2009, 100, S80–S89. [Google Scholar] [CrossRef]
- Bozarth, C.A.; Hailer, F.; Rockwood, L.L.; Edwards, C.W.; Maldonado, J.E. Coyote colonization of northern Virginia and admixture with Great Lakes wolves. J. Mammal. 2011, 92, 1070–1080. [Google Scholar] [CrossRef]
- Ali, O.A.; O’Rourke, S.M.; Amish, S.J.; Meek, M.H.; Luikart, G.; Jeffres, C.; Miller, M.R. RAD capture (Rapture): Flexible and efficient sequence-based genotyping. Genetics 2016, 202, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An analysis tool set for population genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef] [PubMed]
- Lunter, G.; Goodson, M. Stampy: A statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 2011, 21, 936–939. [Google Scholar] [CrossRef] [PubMed]
- Lindblad-Toh, K.; Wade, C.; Mikkelsen, T.S.; Karlsson, E.K.; Jaffe, D.B.; Kamal, M.; Clamp, M.; Chang, J.L.; Kulbokas, E.J.; Zody, M.C.; et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 2005, 438, 803–819. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The sequence alignment/map (SAM) format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole- genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Szpiech, Z.A.; Jakobsson, M.; Rosenberg, N.A. ADZE: A rarefaction approach for counting alleles private to combinations of populations. Bioinformatics 2008, 24, 2498–2504. [Google Scholar] [CrossRef]
- Abraham, G.; Inouye, M. Fast principal component analysis of large-scale genome-wide data. PLoS ONE 2014, 9, e93766. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet. 2010, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar] [PubMed]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Dominant markers and null alleles. Mol. Ecol. Notes 2007, 7, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Hubisz, M.J.; Falush, D.; Stephens, M.; Pritchard, J.K. Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resour. 2009, 9, 1322–1332. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Swofford, D.L. PAUP*. Phylogenetic Analysis Using Parsimony (* and Other Methods); Sinauer Associates: Sunderland, MA, USA, 2003. [Google Scholar]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree. v.1.4.0. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 8 September 2012).
- Pickrell, J.K.; Pritchard, J.K. Inference of population splits and mixture from genome-wide allele frequency data. PLoS Genet. 2012, 8, e1002967. [Google Scholar] [CrossRef]
- Guillot, G.; Mortier, F.; Estoup, A. Geneland: a computer package for landscape genetics. Mol. Ecol. Resour. 2005, 5, 712–715. [Google Scholar] [CrossRef]
- Jon, T.; Schnute, N.B.; Haigh, R. PBSmapping: Mapping Fisheries Data and Spatial Analysis Tools. R Package Version 2.70.4. Available online: https://CRAN.R-project.org/package=PBSmapping (accessed on 13 September 2018).
- Guillot, G. Inference of structure in subdivided populations at low levels of genetic differentiation. The correlated allele frequencies model revisited. Bioinformatics 2008, 24, 2222–2228. [Google Scholar] [CrossRef]
- QGIS Development Team. QGIS Geographic Information System. Open Source Geospatial Foundation Project. Available online: http://qgis.org (accessed on 13 September 2018).
- Sears, H.J.; Theberge, J.B.; Theberge, M.T.; Thornton, I.; Campbell, G.D. Landscape influence on Canis morphological and ecological variation in a coyote-wolf C. lupus x latrans hybrid zone, southeastern Ontario. Can. Field Nat. 2003, 117, 589–600. [Google Scholar] [CrossRef]
- Turner, I.M.; Corlett, R.T. The conservation value of small, isolated fragments of lowland tropical rain forest. TREE 1996, 11, 330–333. [Google Scholar] [CrossRef]
- Stockwell, C.A.; Hendry, A.P.; Kinnison, M.T. Contemporary evolution meets conservation biology. Trends Ecol. Evol. 2003, 18, 94–101. [Google Scholar] [CrossRef]
- Rutledge, L.Y.; White, B.N.; Row, J.R.; Patterson, B.R. Intense harvesting of eastern wolves facilitated hybridization with coyotes. Ecol. Evol. 2012, 2, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Gese, E.M.; Knowlton, F.F.; Adams, J.R.; Beck, K.; Fuller, T.K.; Murray, D.L.; Steury, T.D.; Stoskopf, M.K.; Waddell, W.T.; Waits, L.P. Managing hybridization of a recovering endangered species: The red wolf Canis rufus as a case study. Curr. Zool. 2015, 61, 191–205. [Google Scholar] [CrossRef]
- Bohling, J.H.; Waits, L.P. Factors influencing red wolf-coyote hybridization in eastern North Carolina, USA. Biol. Conserv. 2015, 184, 108–116. [Google Scholar] [CrossRef]
- Hinton, J.W.; Brzeski, K.E.; Rabon, D.R.; Chamberlain, M.C. Effects of anthropogenic mortality on critically endangered red wolf Canis rufus breeding pairs: Implications for red wolf recovery. Oryx 2017, 51, 174–181. [Google Scholar] [CrossRef]
- Arnold, M. Genetic exchange and species concepts. In Divergence with Genetic Exchange; Arnold, M., Ed.; Oxford Press: Oxford, UK, 2016; pp. 24–34. [Google Scholar]
- Hamilton, J.A.; Miller, J.M. Adaptive introgression as a resource for management and genetic conservation in a changing environment. Conserv. Biol. 2016, 30, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.A.; Sgrò, C.M. Climate change and evolutionary adaptation. Nature 2011, 470, 479–485. [Google Scholar] [CrossRef] [PubMed]
- vonHoldt, B.M.; Brzeski, K.E.; Wilcove, D.S.; Rutledge, L.Y. Redefining the role of admixture and genomics in species conservation. Conserv. Lett. 2018, 11, e12371. [Google Scholar] [CrossRef]
- Parmesean, C.; Yohe, G. A globally coherent fingerprint of climate change impacts across natural systems. Nature 2003, 421, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Barlow, A.; Cahill, J.A.; Hartmann, S.; Theunert, C.; Xenikoudakis, G.; Fortes, G.G.; Paijmans, J.L.A.; Rabeder, G.; Frischauf, C.; Grandal-d’Anglade, A.; et al. Partial genomic survival of cave bears in living brown bears. Nat. Ecol. Evol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, E.G.; Elmhagen, B.; Glen, A.S.; Letnic, M.; Ludwig, G.; McDonald, R.A. Ecosystem restoration with teeth: What role for predators. Trends Ecol. Evol. 2012, 27, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Benson, J.F.; Loveless, K.M.; Rutledge, L.Y.; Patterson, B.R. Ungulate predation and ecological roles of wolves and coyotes in eastern North America. Ecol. Appl. 2017, 27, 718–733. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heppenheimer, E.; Harrigan, R.J.; Rutledge, L.Y.; Koepfli, K.-P.; DeCandia, A.L.; Brzeski, K.E.; Benson, J.F.; Wheeldon, T.; Patterson, B.R.; Kays, R.; et al. Population Genomic Analysis of North American Eastern Wolves (Canis lycaon) Supports Their Conservation Priority Status. Genes 2018, 9, 606. https://doi.org/10.3390/genes9120606
Heppenheimer E, Harrigan RJ, Rutledge LY, Koepfli K-P, DeCandia AL, Brzeski KE, Benson JF, Wheeldon T, Patterson BR, Kays R, et al. Population Genomic Analysis of North American Eastern Wolves (Canis lycaon) Supports Their Conservation Priority Status. Genes. 2018; 9(12):606. https://doi.org/10.3390/genes9120606
Chicago/Turabian StyleHeppenheimer, Elizabeth, Ryan J. Harrigan, Linda Y. Rutledge, Klaus-Peter Koepfli, Alexandra L. DeCandia, Kristin E. Brzeski, John F. Benson, Tyler Wheeldon, Brent R. Patterson, Roland Kays, and et al. 2018. "Population Genomic Analysis of North American Eastern Wolves (Canis lycaon) Supports Their Conservation Priority Status" Genes 9, no. 12: 606. https://doi.org/10.3390/genes9120606
APA StyleHeppenheimer, E., Harrigan, R. J., Rutledge, L. Y., Koepfli, K.-P., DeCandia, A. L., Brzeski, K. E., Benson, J. F., Wheeldon, T., Patterson, B. R., Kays, R., Hohenlohe, P. A., & Von Holdt, B. M. (2018). Population Genomic Analysis of North American Eastern Wolves (Canis lycaon) Supports Their Conservation Priority Status. Genes, 9(12), 606. https://doi.org/10.3390/genes9120606