Review of Ocular Manifestations of Joubert Syndrome

 ,
,
Abstract
1. Introduction
2. Epidemiology
3. Clinical Classifications
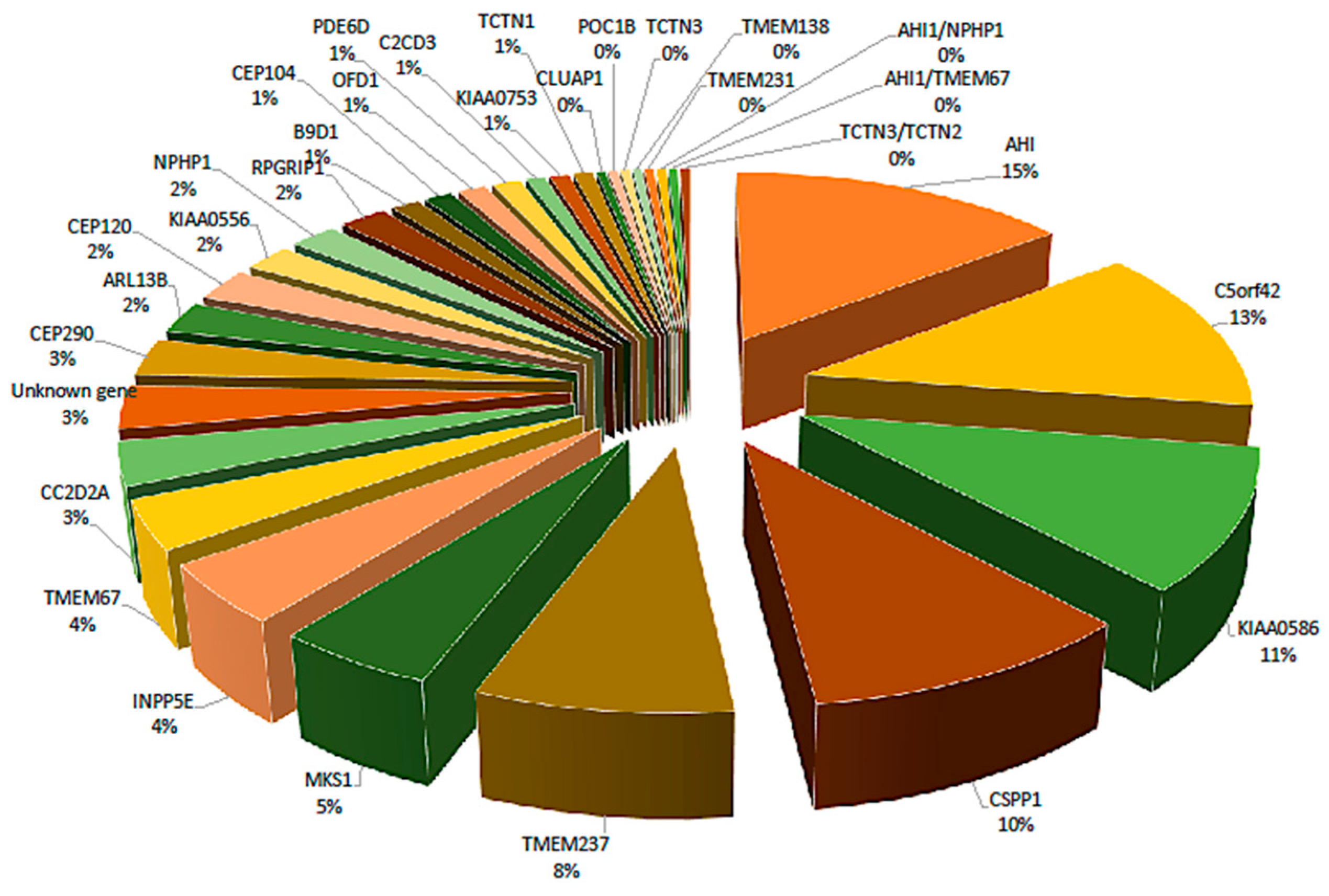
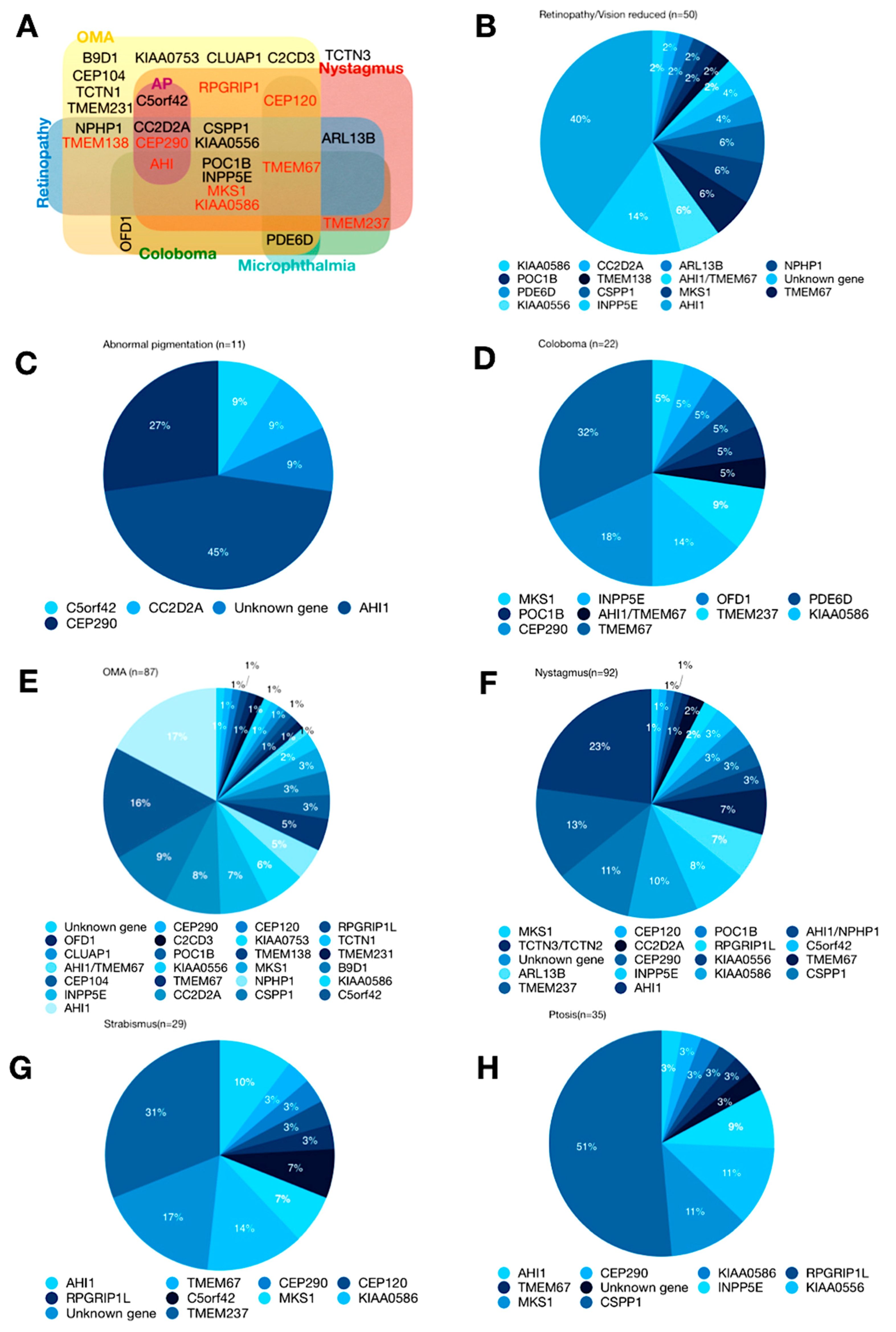
4. Genetics
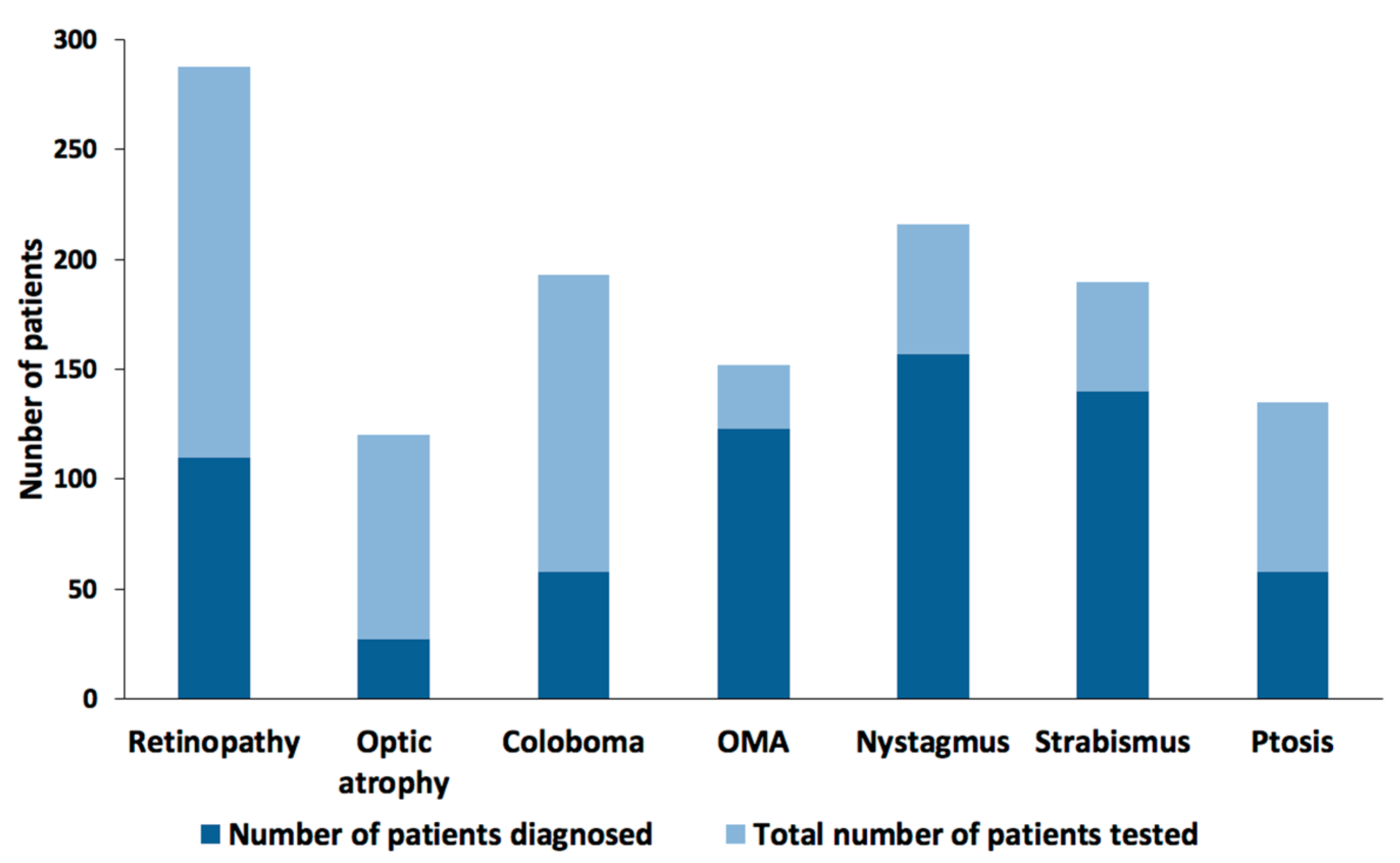
5. Ophthalmic Phenotypes/Presentation
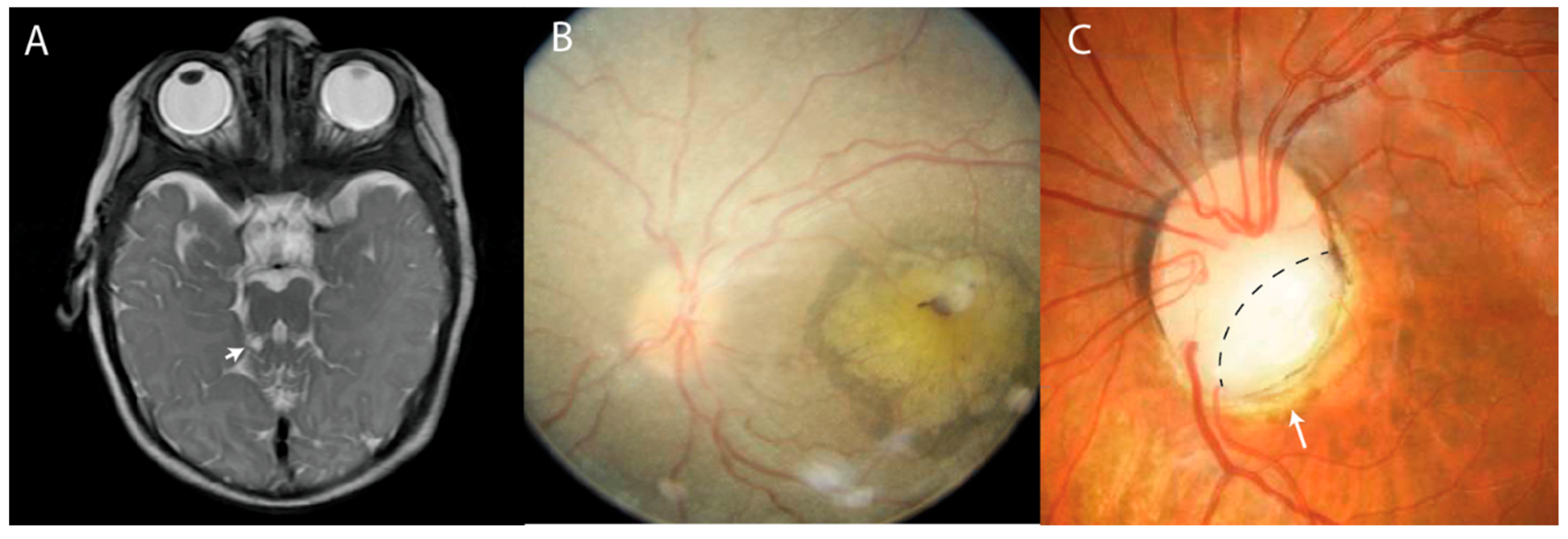
6. Retinal Dystrophy
6.1. Retinitis Pigmentosa
6.2. Leber Congenital Amaurosis
6.3. Abnormal Pigmentation
7. Ocular Colobomas
7.1. Oculomotor Defects
7.1.1. Oculomotor Apraxia
7.1.2. Nystagmus and Strabismus
7.2. Ptosis
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Joubert, M.; Eisenring, J.J.; Robb, J.P.; Andermann, F. Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology 1969, 19, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Maria, B.L.; Boltshauser, E.; Palmer, S.C.; Tran, T.X. Clinical features and revised diagnostic criteria in Joubert syndrome. J. Child Neurol. 1999, 14, 583–590, discussion 590-591. [Google Scholar] [CrossRef] [PubMed]
- Boltshauser, E.; Isler, W. Joubert syndrome: episodic hyperpnea, abnormal eye movements, retardation and ataxia, associated with dysplasia of the cerebellar vermis. Neuropadiatrie 1977, 8, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Parisi, M.; Glass, I. Joubert Syndrome. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Brancati, F.; Dallapiccola, B.; Valente, E.M. Joubert Syndrome and related disorders. Orphanet J. Rare Dis. 2010, 5, 20. [Google Scholar] [CrossRef]
- Romani, M.; Micalizzi, A.; Valente, E.M. Joubert syndrome: Congenital cerebellar ataxia with the molar tooth. Lancet Neurol. 2013, 12, 894–905. [Google Scholar] [CrossRef]
- Hildebrandt, F.; Benzing, T.; Katsanis, N. Ciliopathies. N. Engl. J. Med. 2011, 364, 1533–1543. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.R.; Kriss, A.; Gresty, M.; Benton, S.; Taylor, D. Joubert syndrome. Arch. Ophthalmol. 1989, 107, 709–713. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.O.; Oystreck, D.T.; Seidahmed, M.Z.; AlDrees, A.; Elmalik, S.A.; Alorainy, I.A.; Salih, M.A. Ophthalmic features of Joubert syndrome. Ophthalmology 2008, 115, 2286–2289. [Google Scholar] [CrossRef] [PubMed]
- Maria, B.L.; Hoang, K.B.; Tusa, R.J.; Mancuso, A.A.; Hamed, L.M.; Quisling, R.G.; Hove, M.T.; Fennell, E.B.; Booth-Jones, M.; Ringdahl, D.M.; et al. “Joubert syndrome” revisited: key ocular motor signs with magnetic resonance imaging correlation. J. Child Neurol. 1997, 12, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Sturm, V.; Leiba, H.; Menke, M.N.; Valente, E.M.; Poretti, A.; Landau, K.; Boltshauser, E. Ophthalmological findings in Joubert syndrome. Eye (Lond) 2010, 24, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Hodgkins, P.R.; Harris, C.M.; Shawkat, F.S.; Thompson, D.A.; Chong, K.; Timms, C.; Russell-Eggitt, I.; Taylor, D.S.; Kriss, A. Joubert syndrome: Long-term follow-up. Dev. Med. Child Neurol. 2004, 46, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Gill, H.; Muthusamy, B.; Atan, D.; Williams, C.; Ellis, M. Joubert syndrome presenting with motor delay and oculomotor apraxia. Case Rep. Pediatr. 2011, 2011, 262641. [Google Scholar] [CrossRef] [PubMed]
- Kroes, H.Y.; Monroe, G.R.; van der Zwaag, B.; Duran, K.J.; de Kovel, C.G.; van Roosmalen, M.J.; Harakalova, M.; Nijman, I.J.; Kloosterman, W.P.; Giles, R.H.; et al. Joubert syndrome: Genotyping a Northern European patient cohort. Eur. J. Hum. Genet. 2016, 24, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Parisi, M.A.; Doherty, D.; Chance, P.F.; Glass, I.A. Joubert syndrome (and related disorders) (OMIM 213300). Eur. J. Hum. Genet. 2007, 15, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Srour, M.; Schwartzentruber, J.; Hamdan, F.F.; Ospina, L.H.; Patry, L.; Labuda, D.; Massicotte, C.; Dobrzeniecka, S.; Capo-Chichi, J.-M.; Papillon-Cavanagh, S.; et al. Mutations in C5ORF42 cause Joubert syndrome in the French Canadian population. Am. J. Hum. Genet. 2012, 90, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Srour, M.; Hamdan, F.F.; McKnight, D.; Davis, E.; Mandel, H.; Schwartzentruber, J.; Martin, B.; Patry, L.; Nassif, C.; Dionne-Laporte, A.; et al. Joubert Syndrome in French Canadians and Identification of Mutations in CEP104. Am. J. Hum. Genet. 2015, 97, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Srour, M.; Hamdan, F.F.; Schwartzentruber, J.A.; Patry, L.; Ospina, L.H.; Shevell, M.I.; Désilets, V.; Dobrzeniecka, S.; Mathonnet, G.; Lemyre, E.; et al. Mutations in TMEM231 cause Joubert syndrome in French Canadians. J. Med. Genet. 2012, 49, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Edvardson, S.; Shaag, A.; Zenvirt, S.; Erlich, Y.; Hannon, G.J.; Shanske, A.L.; Gomori, J.M.; Ekstein, J.; Elpeleg, O. Joubert syndrome 2 (JBTS2) in Ashkenazi Jews is associated with a TMEM216 mutation. Am. J. Hum. Genet. 2010, 86, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Szymanska, K.; Jensen, V.L.; Janecke, A.R.; Innes, A.M.; Davis, E.E.; Frosk, P.; Li, C.; Willer, J.R.; Chodirker, B.N.; et al. TMEM237 is mutated in individuals with a Joubert syndrome related disorder and expands the role of the TMEM family at the ciliary transition zone. Am. J. Hum. Genet. 2011, 89, 713–730. [Google Scholar] [CrossRef]
- Shaheen, R.; Shamseldin, H.E.; Loucks, C.M.; Seidahmed, M.Z.; Ansari, S.; Ibrahim Khalil, M.; Al-Yacoub, N.; Davis, E.E.; Mola, N.A.; Szymanska, K.; et al. Mutations in CSPP1, encoding a core centrosomal protein, cause a range of ciliopathy phenotypes in humans. Am. J. Hum. Genet. 2014, 94, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Miyake, N.; Tsurusaki, Y.; Okamoto, N.; Alkindy, A.; Inaba, A.; Sato, M.; Ito, S.; Muramatsu, K.; Kimura, S.; et al. Molecular genetic analysis of 30 families with Joubert syndrome. Clin. Genet. 2016, 90, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Doherty, D.; Parisi, M.A.; Finn, L.S.; Gunay-Aygun, M.; Al-Mateen, M.; Bates, D.; Clericuzio, C.; Demir, H.; Dorschner, M.; van Essen, A.J.; et al. Mutations in 3 genes (MKS3, CC2D2A and RPGRIP1L) cause COACH syndrome (Joubert syndrome with congenital hepatic fibrosis). J. Med. Genet. 2010, 47, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Iannicelli, M.; Brancati, F.; Mougou-Zerelli, S.; Mazzotta, A.; Thomas, S.; Elkhartoufi, N.; Travaglini, L.; Gomes, C.; Ardissino, G.L.; Bertini, E.; et al. Novel TMEM67 mutations and genotype-phenotype correlates in meckelin-related ciliopathies. Hum. Mutat. 2010, 31, E1319–E1331. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Marsh, S.E.; Castori, M.; Dixon-Salazar, T.; Bertini, E.; Al-Gazali, L.; Messer, J.; Barbot, C.; Woods, C.G.; Boltshauser, E.; et al. Distinguishing the four genetic causes of Jouberts syndrome-related disorders. Ann. Neurol. 2005, 57, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Senocak, E.U.; Oğuz, K.K.; Haliloğlu, G.; Topçu, M.; Cila, A. Structural abnormalities of the brain other than molar tooth sign in Joubert syndrome-related disorders. Diagn Interv. Radiol. 2010, 16, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Bachmann-Gagescu, R.; Dempsey, J.C.; Phelps, I.G.; O’Roak, B.J.; Knutzen, D.M.; Rue, T.C.; Ishak, G.E.; Isabella, C.R.; Gorden, N.; Adkins, J.; et al. Joubert syndrome: A model for untangling recessive disorders with extreme genetic heterogeneity. J. Med. Genet. 2015, 52, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Lehman, A.M.; Eydoux, P.; Doherty, D.; Glass, I.A.; Chitayat, D.; Chung, B.Y.H.; Langlois, S.; Yong, S.L.; Lowry, R.B.; Hildebrandt, F.; et al. Co-occurrence of Joubert syndrome and Jeune asphyxiating thoracic dystrophy. Am. J. Med. Genet. A 2010, 152A, 1411–1419. [Google Scholar] [CrossRef]
- Shaheen, R.; Schmidts, M.; Faqeih, E.; Hashem, A.; Lausch, E.; Holder, I.; Superti-Furga, A.; UK10K Consortium; Mitchison, H.M.; Almoisheer, A.; et al. A founder CEP120 mutation in Jeune asphyxiating thoracic dystrophy expands the role of centriolar proteins in skeletal ciliopathies. Hum. Mol. Genet. 2015, 24, 1410–1419. [Google Scholar] [CrossRef]
- Adams, N.A.; Awadein, A.; Toma, H.S. The retinal ciliopathies. Ophthalmic Genet. 2007, 28, 113–125. [Google Scholar] [CrossRef]
- Vilboux, T.; Doherty, D.A.; Glass, I.A.; Parisi, M.A.; Phelps, I.G.; Cullinane, A.R.; Zein, W.; Brooks, B.P.; Heller, T.; Soldatos, A.; et al. Molecular genetic findings and clinical correlations in 100 patients with Joubert syndrome and related disorders prospectively evaluated at a single center. Genet. Med. 2017, 19, 875–882. [Google Scholar] [CrossRef]
- Parisi, M.A.; Doherty, D.; Eckert, M.L.; Shaw, D.W.W.; Ozyurek, H.; Aysun, S.; Giray, O.; Al Swaid, A.; Al Shahwan, S.; Dohayan, N.; et al. AHI1 mutations cause both retinal dystrophy and renal cystic disease in Joubert syndrome. J. Med. Genet. 2006, 43, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Westfall, J.E.; Hoyt, C.; Liu, Q.; Hsiao, Y.-C.; Pierce, E.A.; Page-McCaw, P.S.; Ferland, R.J. Retinal degeneration and failure of photoreceptor outer segment formation in mice with targeted deletion of the Joubert syndrome gene, Ahi1. J. Neurosci. 2010, 30, 8759–8768. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, J.M.; Baraitser, M. Joubert syndrome: a review. Am. J. Med. Genet. 1992, 43, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.P.; Zein, W.M.; Thompson, A.H.; Mokhtarzadeh, M.; Doherty, D.A.; Parisi, M.; Glass, I.A.; Malicdan, M.C.; Vilboux, T.; Vemulapalli, M.; et al. Joubert Syndrome: Ophthalmological Findings in Correlation with Genotype and Hepatorenal Disease in 99 Patients Prospectively Evaluated at a Single Center. Ophthalmology 2018. [Google Scholar] [CrossRef] [PubMed]
- Baba, S.; Takeshita, E.; Yamazaki, H.; Tarashima, M.; Sasaki, M. Disruption of the Photoreceptor Inner Segment-Outer Segment Junction in a 6-Year-Old Girl with Joubert Syndrome. Neuroophthalmology 2017, 41, 19–23. [Google Scholar] [CrossRef]
- Dixon-Salazar, T.; Silhavy, J.L.; Marsh, S.E.; Louie, C.M.; Scott, L.C.; Gururaj, A.; Al-Gazali, L.; Al-Tawari, A.A.; Kayserili, H.; Sztriha, L.; et al. Mutations in the AHI1 gene, encoding jouberin, cause Joubert syndrome with cortical polymicrogyria. Am. J. Hum. Genet. 2004, 75, 979–987. [Google Scholar] [CrossRef]
- Eley, L.; Gabrielides, C.; Adams, M.; Johnson, C.A.; Hildebrandt, F.; Sayer, J.A. Jouberin localizes to collecting ducts and interacts with nephrocystin-1. Kidney Int. 2008, 74, 1139–1149. [Google Scholar] [CrossRef]
- Romani, S.; Illi, B.; De Mori, R.; Savino, M.; Gleeson, J.G.; Valente, E.M. The ciliary proteins Meckelin and Jouberin are required for retinoic acid-dependent neural differentiation of mouse embryonic stem cells. Differentiation 2014, 87, 134–146. [Google Scholar] [CrossRef]
- Louie, C.M.; Caridi, G.; Lopes, V.S.; Brancati, F.; Kispert, A.; Lancaster, M.A.; Schlossman, A.M.; Otto, E.A.; Leitges, M.; Gröne, H.-J.; et al. AHI1 is required for photoreceptor outer segment development and is a modifier for retinal degeneration in nephronophthisis. Nat. Genet. 2010, 42, 175–180. [Google Scholar] [CrossRef]
- Jacoby, M.; Cox, J.J.; Gayral, S.; Hampshire, D.J.; Ayub, M.; Blockmans, M.; Pernot, E.; Kisseleva, M.V.; Compère, P.; Schiffmann, S.N.; et al. INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat. Genet. 2009, 41, 1027–1031. [Google Scholar] [CrossRef]
- Chawla, R.; Bypareddy, R.; Vekaria, L.; Venkatesh, P.; Ananthashayana, V.H. Fundus findings in a case of Joubert syndrome. Indian J. Ophthalmol. 2017, 65, 329–330. [Google Scholar] [CrossRef]
- Abouammoh, M.A.; Al-Shibani, S.K.; Alhawwas, A.; Bosley, T.M. Coats-like retinopathy in Joubert syndrome. J. AAPOS 2016, 20, 372–374. [Google Scholar] [CrossRef] [PubMed]
- Yzer, S.; den Hollander, A.I.; Lopez, I.; Pott, J.-W.R.; de Faber, J.T.H.N.; Cremers, F.P.M.; Koenekoop, R.K.; van den Born, L.I. Ocular and extra-ocular features of patients with Leber congenital amaurosis and mutations in CEP290. Mol. Vis. 2012, 18, 412–425. [Google Scholar]
- Valente, E.M.; Silhavy, J.L.; Brancati, F.; Barrano, G.; Krishnaswami, S.R.; Castori, M.; Lancaster, M.A.; Boltshauser, E.; Boccone, L.; Al-Gazali, L.; et al. Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat. Genet. 2006, 38, 623–625. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, S.; Biler, E.D.; Solmaz, A.E.; Serdaroglu, G.; Tekin, H.G.; Gokben, S. Optic disc drusen mimicking papilledema in an infant with Joubert syndrome. Genet. Couns. 2015, 26, 35–39. [Google Scholar]
- Erkkilä, H. Clinical appearance of optic disc drusen in childhood. Albrecht Von Graefes Arch. Klin. Exp. Ophthalmol. 1975, 193, 1–18. [Google Scholar]
- Wilkins, J.M.; Pomeranz, H.D. Visual manifestations of visible and buried optic disc drusen. J. Neuroophthalmol. 2004, 24, 125–129. [Google Scholar] [CrossRef]
- Chung, D.C.; Traboulsi, E.I. Leber congenital amaurosis: clinical correlations with genotypes, gene therapy trials update, and future directions. J. AAPOS 2009, 13, 587–592. [Google Scholar] [CrossRef]
- Waters, A.M.; Beales, P.L. Ciliopathies: an expanding disease spectrum. Pediatr. Nephrol. 2011, 26, 1039–1056. [Google Scholar] [CrossRef]
- Heher, K.L.; Traboulsi, E.I.; Maumenee, I.H. The natural history of Leber’s congenital amaurosis. Age-related findings in 35 patients. Ophthalmology 1992, 99, 241–245. [Google Scholar] [CrossRef]
- Koenekoop, R.K. An overview of Leber congenital amaurosis: a model to understand human retinal development. Surv. Ophthalmol. 2004, 49, 379–398. [Google Scholar] [CrossRef] [PubMed]
- Sohocki, M.M.; Sullivan, L.S.; Mintz-Hittner, H.A.; Birch, D.; Heckenlively, J.R.; Freund, C.L.; McInnes, R.R.; Daiger, S.P. A range of clinical phenotypes associated with mutations in CRX, a photoreceptor transcription-factor gene. Am. J. Hum. Genet. 1998, 63, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Tzekov, R.T.; Liu, Y.; Sohocki, M.M.; Zack, D.J.; Daiger, S.P.; Heckenlively, J.R.; Birch, D.G. Autosomal dominant retinal degeneration and bone loss in patients with a 12-bp deletion in the CRX gene. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1319–1327. [Google Scholar]
- Nichols, L.L.; Alur, R.P.; Boobalan, E.; Sergeev, Y.V.; Caruso, R.C.; Stone, E.M.; Swaroop, A.; Johnson, M.A.; Brooks, B.P. Two novel CRX mutant proteins causing autosomal dominant Leber congenital amaurosis interact differently with NRL. Hum. Mutat. 2010, 31, E1472–E1483. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Yao, F.; Liang, X.; Xu, F.; Li, H.; Sui, R.; Dong, F. De novo mutations in the cone-rod homeobox gene associated with leber congenital amaurosis in Chinese patients. Ophthalmic Genet. 2015, 36, 21–26. [Google Scholar] [CrossRef]
- Beck, B.B.; Phillips, J.B.; Bartram, M.P.; Wegner, J.; Thoenes, M.; Pannes, A.; Sampson, J.; Heller, R.; Göbel, H.; Koerber, F.; et al. Mutation of POC1B in a severe syndromic retinal ciliopathy. Hum. Mutat. 2014, 35, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Chacon-Camacho, O.F.; Zenteno, J.C. Review and update on the molecular basis of Leber congenital amaurosis. World J. Clin. Cases 2015, 3, 112–124. [Google Scholar] [CrossRef]
- Gregory-Evans, C.Y.; Williams, M.J.; Halford, S.; Gregory-Evans, K. Ocular coloboma: A reassessment in the age of molecular neuroscience. J. Med. Genet. 2004, 41, 881–891. [Google Scholar] [CrossRef]
- Arrigoni, F.; Romaniello, R.; Peruzzo, D.; De Luca, A.; Parazzini, C.; Valente, E.M.; Borgatti, R.; Triulzi, F. Anterior mesencephalic cap dysplasia: Novel brain stem malformative features associated with Joubert Syndrome. AJNR Am. J. Neuroradiol. 2017, 38, 2385–2390. [Google Scholar] [CrossRef]
- Zhang, K.; Meng, C.; Ma, J.; Gao, M.; Lv, Y.; Liu, Y.; Gai, Z. Novel OFD1 frameshift mutation in a Chinese boy with Joubert syndrome: A case report and literature review. Clin. Dysmorphol. 2017, 26, 135–141. [Google Scholar] [CrossRef]
- Enokizono, M.; Aida, N.; Niwa, T.; Osaka, H.; Naruto, T.; Kurosawa, K.; Ohba, C.; Suzuki, T.; Saitsu, H.; Goto, T.; et al. Neuroimaging findings in Joubert syndrome with C5orf42 gene mutations: A milder form of molar tooth sign and vermian hypoplasia. J. Neurol. Sci. 2017, 376, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Malicdan, M.C.V.; Vilboux, T.; Stephen, J.; Maglic, D.; Mian, L.; Konzman, D.; Guo, J.; Yildirimli, D.; Bryant, J.; Fischer, R.; et al. Mutations in human homologue of chicken talpid3 gene (KIAA0586) cause a hybrid ciliopathy with overlapping features of Jeune and Joubert syndromes. J. Med. Genet. 2015, 52, 830–839. [Google Scholar] [CrossRef]
- Bachmann-Gagescu, R.; Phelps, I.G.; Dempsey, J.C.; Sharma, V.A.; Ishak, G.E.; Boyle, E.A.; Wilson, M.; Marques Lourenço, C.; Arslan, M.; University of Washington Center for Mendelian Genomics; et al. KIAA0586 is Mutated in Joubert Syndrome. Hum. Mutat. 2015, 36, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Makino, S.; Tampo, H. Ocular findings in two siblings with Joubert syndrome. Clin. Ophthalmol. 2014, 8, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Wright, K.J.; Le Corre, S.; Micalizzi, A.; Romani, M.; Abhyankar, A.; Saada, J.; Perrault, I.; Amiel, J.; Litzler, J.; et al. A homozygous PDE6D mutation in Joubert syndrome impairs targeting of farnesylated INPP5E protein to the primary cilium. Hum. Mutat. 2014, 35, 137–146. [Google Scholar] [CrossRef] [PubMed]
- İncecik, F.; Hergüner, M.Ö.; Altunbaşak, Ş.; Gleeson, J.G. Joubert syndrome: Report of 11 cases. Turk. J. Pediatr. 2012, 54, 605–611. [Google Scholar] [PubMed]
- Satran, D.; Pierpont, M.E.; Dobyns, W.B. Cerebello-oculo-renal syndromes including Arima, Senior-Löken and COACH syndromes: More than just variants of Joubert syndrome. Am. J. Med. Genet. 1999, 86, 459–469. [Google Scholar] [CrossRef]
- Brancati, F.; Iannicelli, M.; Travaglini, L.; Mazzotta, A.; Bertini, E.; Boltshauser, E.; D’Arrigo, S.; Emma, F.; Fazzi, E.; Gallizzi, R.; et al. MKS3/TMEM67 mutations are a major cause of COACH Syndrome, a Joubert Syndrome related disorder with liver involvement. Hum. Mutat. 2009, 30, E432–E442. [Google Scholar] [CrossRef] [PubMed]
- Doherty, D. Joubert syndrome: Insights into brain development, cilium biology, and complex disease. Semin. Pediatr. Neurol. 2009, 16, 143–154. [Google Scholar] [CrossRef]
- Apostolou, T.; Nikolopoulou, N.; Theodoridis, M.; Koumoustiotis, V.; Pavlopoulou, E.; Chondros, D.; Billis, A. Late onset of renal disease in nephronophthisis with features of Joubert syndrome type B. Nephrol. Dial. Transplant. 2001, 16, 2412–2415. [Google Scholar] [CrossRef]
- Couch, S.M.; Brodie, S.E.; Leavitt, J.A.; Brodsky, M.C. Something to sink your teeth into. Surv. Ophthalmol. 2011, 56, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Papanagnu, E.; Klaehn, L.D.; Bang, G.M.; Ghadban, R.; Mohney, B.G.; Brodsky, M.C. Congenital ocular motor apraxia with wheel-rolling ocular torsion-a neurodiagnostic phenotype of Joubert syndrome. J. AAPOS 2014, 18, 404–407. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.J.; Lee, C.; Wentzensen, I.M.; Parisi, M.A.; Crenshaw, M.M.; Sapp, J.C.; Gross, J.M.; Wallingford, J.B.; Biesecker, L.G. Compound heterozygous alterations in intraflagellar transport protein CLUAP1 in a child with a novel Joubert and oral-facial-digital overlap syndrome. Cold Spring Harb. Mol. Case Stud. 2017, 3. [Google Scholar] [CrossRef] [PubMed]
- Stephen, J.; Vilboux, T.; Mian, L.; Kuptanon, C.; Sinclair, C.M.; Yildirimli, D.; Maynard, D.M.; Bryant, J.; Fischer, R.; Vemulapalli, M.; et al. Mutations in KIAA0753 cause Joubert syndrome associated with growth hormone deficiency. Hum. Genet. 2017, 136, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Wente, S.; Schröder, S.; Buckard, J.; Büttel, H.-M.; von Deimling, F.; Diener, W.; Häussler, M.; Hübschle, S.; Kinder, S.; Kurlemann, G.; et al. Nosological delineation of congenital ocular motor apraxia type Cogan: An observational study. Orphanet. J. Rare Dis. 2016, 11, 104. [Google Scholar] [CrossRef] [PubMed]
- Roosing, S.; Rosti, R.O.; Rosti, B.; de Vrieze, E.; Silhavy, J.L.; van Wijk, E.; Wakeling, E.; Gleeson, J.G. Identification of a homozygous nonsense mutation in KIAA0556 in a consanguineous family displaying Joubert syndrome. Hum. Genet. 2016, 135, 919–921. [Google Scholar] [CrossRef]
- Slaats, G.G.; Isabella, C.R.; Kroes, H.Y.; Dempsey, J.C.; Gremmels, H.; Monroe, G.R.; Phelps, I.G.; Duran, K.J.; Adkins, J.; Kumar, S.A.; et al. MKS1 regulates ciliary INPP5E levels in Joubert syndrome. J. Med. Genet. 2016, 53, 62–72. [Google Scholar] [CrossRef]
- Roosing, S.; Romani, M.; Isrie, M.; Rosti, R.O.; Micalizzi, A.; Musaev, D.; Mazza, T.; Al-Gazali, L.; Altunoglu, U.; Boltshauser, E.; et al. Mutations in CEP120 cause Joubert syndrome as well as complex ciliopathy phenotypes. J. Med. Genet. 2016, 53, 608–615. [Google Scholar] [CrossRef]
- Gagliardi, C.; Brenna, V.; Romaniello, R.; Arrigoni, F.; Tavano, A.; Romani, M.; Valente, E.M.; Borgatti, R. Cognitive rehabilitation in a child with Joubert Syndrome: Developmental trends and adaptive changes in a single case report. Res. Dev. Disabil. 2015, 47, 375–384. [Google Scholar] [CrossRef]
- Hsu, C.C.-T.; Kwan, G.N.C.; Bhuta, S. High-Resolution Diffusion Tensor Imaging and Tractography in Joubert Syndrome: Beyond Molar Tooth Sign. Pediatr. Neurol. 2015, 53, 47–52. [Google Scholar] [CrossRef]
- Thomas, S.; Cantagrel, V.; Mariani, L.; Serre, V.; Lee, J.-E.; Elkhartoufi, N.; de Lonlay, P.; Desguerre, I.; Munnich, A.; Boddaert, N.; et al. Identification of a novel ARL13B variant in a Joubert syndrome-affected patient with retinal impairment and obesity. Eur. J. Hum. Genet. 2015, 23, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Dekair, L.H.; Kamel, H.; El-Bashir, H.O. Joubert syndrome labeled as hypotonic cerebral palsy. Neurosciences (Riyadh) 2014, 19, 233–235. [Google Scholar] [PubMed]
- Poretti, A.; Christen, H.-J.; Elton, L.E.; Baumgartner, M.; Korenke, G.C.; Sukhudyan, B.; Hethey, S.; Cross, E.; Steinlin, M.; Boltshauser, E. Horizontal head titubation in infants with Joubert syndrome: a new finding. Dev. Med. Child Neurol. 2014, 56, 1016–1020. [Google Scholar] [CrossRef] [PubMed]
- Tuz, K.; Bachmann-Gagescu, R.; O’Day, D.R.; Hua, K.; Isabella, C.R.; Phelps, I.G.; Stolarski, A.E.; O’Roak, B.J.; Dempsey, J.C.; Lourenco, C.; et al. Mutations in CSPP1 cause primary cilia abnormalities and Joubert syndrome with or without Jeune asphyxiating thoracic dystrophy. Am. J. Hum. Genet. 2014, 94, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Akizu, N.; Silhavy, J.L.; Rosti, R.O.; Scott, E.; Fenstermaker, A.G.; Schroth, J.; Zaki, M.S.; Sanchez, H.; Gupta, N.; Kabra, M.; et al. Mutations in CSPP1 lead to classical Joubert syndrome. Am. J. Hum. Genet. 2014, 94, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Nag, C.; Ghosh, M.; Das, K.; Ghosh, T. Joubert syndrome: The molar tooth sign of the mid-brain. Ann. Med. Health Sci. Res. 2013, 3, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Théoret, H.; Gleeson, J.; Pascual-Leone, A. Neurophysiologic characterization of motor and sensory projections in Joubert syndrome. Clin. Neurophysiol. 2013, 124, 2283–2284. [Google Scholar] [CrossRef]
- Deacon, B.S.; Lowery, R.S.; Phillips, P.H.; Schaefer, G.B. Congenital ocular motor apraxia, the NPHP1 gene, and surveillance for nephronophthisis. J. AAPOS 2013, 17, 332–333. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, J.-L.; Tang, B.-S.; Sun, Z.-F.; Shi, Y.-T.; Shen, L.; Lei, L.-F.; Wei, X.-M.; Xiao, J.-J.; Hu, Z.-M.; et al. Using next-generation sequencing as a genetic diagnostic tool in rare autosomal recessive neurologic Mendelian disorders. Neurobiol. Aging 2013, 34, 2442.e11–2442.e17. [Google Scholar] [CrossRef]
- Brodsky, M.C. Marshall M. Parks Memorial Lecture: Ocular Motor Misbehavior in Children: Where Neuro-Ophthalmology Meets Strabismus. Ophthalmology 2017, 124, 835–842. [Google Scholar] [CrossRef]
- Steinlin, M.; Schmid, M.; Landau, K.; Boltshauser, E. Follow-up in children with Joubert syndrome. Neuropediatrics 1997, 28, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.H.; Doherty, D.; Parisi, M.; Shaw, D.; Glass, I.; Phillips, J.O. Eye movement abnormalities in Joubert syndrome. Invest. Ophthalmol. Vis. Sci. 2009, 50, 4669–4677. [Google Scholar] [CrossRef] [PubMed]
- Lamônica, D.A.C.; Ribeiro, C.d.C.; Richieri-Costa, A.; Giacheti, C.M. Language, behavior and neurodevelopment in Joubert syndrome: A case report. Codas 2016, 28, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Lv, C.; Zhang, Z.; Wu, M.; Zheng, X.; Yang, L.; Li, X.; Wu, G.; Chen, J. Novel CC2D2A compound heterozygous mutations cause Joubert syndrome. Mol. Med. Rep. 2017, 15, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Singh, D. Joubert Syndrome Mimicking Hypotonic Cerebral Palsy. Indian J. Pediatr. 2016, 83, 1505. [Google Scholar] [CrossRef]
- Bader, I.; Decker, E.; Mayr, J.A.; Lunzer, V.; Koch, J.; Boltshauser, E.; Sperl, W.; Pietsch, P.; Ertl-Wagner, B.; Bolz, H.; et al. MKS1 mutations cause Joubert syndrome with agenesis of the corpus callosum. Eur. J. Med. Genet. 2016, 59, 386–391. [Google Scholar] [CrossRef] [PubMed]
- Salva, I.; Albuquerque, C.; Moreira, A.; Dâmaso, C. Nystagmus in a newborn: A manifestation of Joubert syndrome in the neonatal period. BMJ Case Rep. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Sanders, A.A.W.M.; de Vrieze, E.; Alazami, A.M.; Alzahrani, F.; Malarkey, E.B.; Sorusch, N.; Tebbe, L.; Kuhns, S.; van Dam, T.J.P.; Alhashem, A.; et al. KIAA0556 is a novel ciliary basal body component mutated in Joubert syndrome. Genome Biol. 2015, 16, 293. [Google Scholar] [CrossRef] [PubMed]
- Chafai-Elalaoui, S.; Chalon, M.; Elkhartoufi, N.; Kriouele, Y.; Mansouri, M.; Attié-Bitach, T.; Sefiani, A.; Baala, L. A homozygous AHI1 gene mutation (p.Thr304AsnfsX6) in a consanguineous Moroccan family with Joubert syndrome: a case report. J. Med. Case Rep. 2015, 9, 254. [Google Scholar] [CrossRef]
- Sönmez, F.; Güzünler-Şen, M.; Yılmaz, D.; Cömertpay, G.; Heise, M.; Çırak, S.; Uyanık, G. Development of end-stage renal disease at a young age in two cases with Joubert syndrome. Turk. J. Pediatr. 2014, 56, 458–461. [Google Scholar]
- Purkait, R.; Basu, R.; Das, R.; Chatterjee, U. Association of Joubert syndrome and Hirschsprung disease. Indian Pediatr. 2015, 52, 61–62. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Brancati, F.; Silhavy, J.L.; Castori, M.; Marsh, S.E.; Barrano, G.; Bertini, E.; Boltshauser, E.; Zaki, M.S.; Abdel-Aleem, A.; et al. AHI1 gene mutations cause specific forms of Joubert syndrome-related disorders. Ann. Neurol. 2006, 59, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Huppke, P.; Wegener, E.; Böhrer-Rabel, H.; Bolz, H.J.; Zoll, B.; Gärtner, J.; Bergmann, C. Tectonic gene mutations in patients with Joubert syndrome. Eur. J. Hum. Genet. 2015, 23, 616–620. [Google Scholar] [CrossRef] [PubMed]
- Kendall, B.; Kingsley, D.; Lambert, S.R.; Taylor, D.; Finn, P. Joubert syndrome: A clinico-radiological study. Neuroradiology 1990, 31, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, K.; Kaneko, C.R.; Fuchs, A.F. The neuronal substrate of integration in the oculomotor system. Prog. Neurobiol. 1992, 39, 609–639. [Google Scholar] [CrossRef]
- Helmchen, C.; Rambold, H.; Fuhry, L.; Büttner, U. Deficits in vertical and torsional eye movements after uni- and bilateral muscimol inactivation of the interstitial nucleus of Cajal of the alert monkey. Exp. Brain Res. 1998, 119, 436–452. [Google Scholar] [CrossRef] [PubMed]
- Crawford, J.D.; Cadera, W.; Vilis, T. Generation of torsional and vertical eye position signals by the interstitial nucleus of Cajal. Science 1991, 252, 1551–1553. [Google Scholar] [CrossRef] [PubMed]
- Yachnis, A.T.; Rorke, L.B. Neuropathology of Joubert syndrome. J. Child Neurol. 1999, 14, 655–659, discussion 669–672. [Google Scholar] [CrossRef] [PubMed]
- Parisi, M.A.; Pinter, J.D.; Glass, I.A.; Field, K.; Maria, B.L.; Chance, P.F.; Mahurin, R.K.; Cramer, S.C. Cerebral and cerebellar motor activation abnormalities in a subject with Joubert syndrome: Functional magnetic resonance imaging (MRI) study. J. Child Neurol. 2004, 19, 214–218. [Google Scholar] [PubMed]
- Poretti, A.; Boltshauser, E.; Loenneker, T.; Valente, E.M.; Brancati, F.; Il’yasov, K.; Huisman, T.a.G.M. Diffusion tensor imaging in Joubert syndrome. AJNR Am. J. Neuroradiol. 2007, 28, 1929–1933. [Google Scholar] [CrossRef] [PubMed]
- Widjaja, E.; Blaser, S.; Raybaud, C. Diffusion tensor imaging of midline posterior fossa malformations. Pediatr. Radiol. 2006, 36, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Salman, M.S.; Chodirker, B.N. Neuro-Ophthalmological Findings in Children and Adolescents with Chronic Ataxia. Neuroophthalmology 2015, 39, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Usta, M.; Urganci, N.; Özçelik, G.; Çetinçelik, Ü.; Kafadar, I.; Özgüven, B.Y. Joubert syndrome and related disorders: A rare cause of intrahepatic portal hypertension in childhood. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 2297–2300. [Google Scholar] [PubMed]
- Lee, N.; Nam, S.-O.; Kim, Y.M.; Lee, Y.-J. A neonate with Joubert syndrome presenting with symptoms of Horner syndrome. Korean J. Pediatr. 2016, 59, S32–S36. [Google Scholar] [CrossRef] [PubMed]
- Stephen, L.A.; Tawamie, H.; Davis, G.M.; Tebbe, L.; Nürnberg, P.; Nürnberg, G.; Thiele, H.; Thoenes, M.; Boltshauser, E.; Uebe, S.; et al. TALPID3 controls centrosome and cell polarity and the human ortholog KIAA0586 is mutated in Joubert syndrome (JBTS23). eLife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Burt, B.; Levine, J.; Le, K. Adult-onset bulbar ptosis in Joubert syndrome. Clin. Ophthalmol. 2012, 6, 151–153. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| JS-Related Gene | Protein | No. of pts | Eye Phenotypes | References (PMID) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OMA | Nystagmus | Coloboma | Retinopathy (Vision Reduced) | Microphthalmia (Dysplasia) | Abnormal Pigmentation | Myopia/Hyperopia | Strabismus | Ptosis | ||||
| AHI1 | Abelson helper integration site 1 | 36 | + | + | + | + | + | + | + | + | 28431631; 25920555; 26759440; 26541515; 16453322; 19461662; 19443711; 29987673; 21458016 | |
| ARL13B | ADP-ribosylation factor-like 13B | 6 | + | + | 25138100; 29255182 | |||||||
| B9D1 | B9 domain containing 1 | 3 | + | 26477546; 24886560 | ||||||||
| C2CD3 | C2 calcium dependent domain containing 3 | 2 | + | 26477546 | ||||||||
| C5orf42 | CPLANE1, ciliogenesis and planar polarity effector 1 | 31 | + | + | + | 28431631; 26477546; 25920555; 25173907; 27866068; 24178751 | ||||||
| CC2D2A | Coiled-coil and C2 domain containing 2A | 8 | + | + | + | + | 26477546; 25920555; 27959436; 25173907; 29987673 | |||||
| CEP104 | Centrosomal protein 104 | 3 | + | 26477546 | ||||||||
| CEP120 | Centrosomal protein 120 | 6 | + | + | + | + | 27208211 | |||||
| CEP290 | Centrosomal protein 290 | 7 | + | + | + | + | + | + | 26477546; 25920555; 19461662; 25818971; 24850569 | |||
| CLUAP1 | Clusterin associated protein 1 | 1 | + | 28679688 | ||||||||
| CSPP1 | Centrosome and spindle pole associated protein 1 | 25 | + | + | + | + | 24360808; 24360807; | |||||
| INPP5E | Inositol polyphosphate-5-phosphatase E | 11 | + | + | + | + | + | + | 25920555; 25818971; 29052317; 20446224; 29987673 | |||
| KIAA0556 | KIAA0556 | 5 | + | + | + | + | 27245168; 26714646 | |||||
| KIAA0586 | KIAA0586 | 26 | + | + | + | + | + | + | + | 26386247; 26386044; 26096313 | ||
| KIAA0753 | KIAA0753 | 2 | + | 28220259 | ||||||||
| MKS1 | Meckel syndrome, type 1 | 12 | + | + | + | + | + | + | 27377014; 26490104; 24886560 | |||
| NPHP1 | Nephrocystin 1 | 5 | + | + | 26477546; 28347285; 23683649 | |||||||
| OFD1 | OFD1, centriole and centriolar satellite protein | 3 | + | + | 26477546; 25920555; 28505061 | |||||||
| PDE6D | Phosphodiesterase 6D | 3 | + | + | + | 24166846 | ||||||
| POC1B | POC1 centriolar protein B | 1 | + | + | + | + | 25044745 | |||||
| RPGRIP1L | RPGR interacting protein 1 | 5 | + | + | + | + | 25920555; 19461662; 18565097 | |||||
| TCTN1 | Tectonic family member 1 | 2 | + | 26477546; 26489806 | ||||||||
| TCTN3 | Tectonic family member 3 | 1 | 25118024 | |||||||||
| TMEM138 | Transmembrane protein 138 | 1 | + | + | + | 28102635 | ||||||
| TMEM231 | Transmembrane protein 231 | 1 | + | 25920555 | ||||||||
| TMEM237 | Transmembrane protein 237 | 19 | + | + | + | 22152675 | ||||||
| TMEM67 | Transmembrane protein 67 | 9 | + | + | + | + | + | + | + | 28838911; 28431631; 26477546; 26166658; 25920555 | ||
| AHI1/NPHP1 | 1 | + | 19443711 | |||||||||
| AHI1/TMEM67 | 1 | + | + | + | + | 25920555 | ||||||
| TCTN3/TCTN2 | 1 | + | + | 25118024 | ||||||||
| Unknown gene | - | 8 | + | + | + | + | + | + | 28838911; 19461662; 28018441 | |||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.F.; Kowal, T.J.; Ning, K.; Koo, E.B.; Wu, A.Y.; Mahajan, V.B.; Sun, Y. Review of Ocular Manifestations of Joubert Syndrome. Genes 2018, 9, 605. https://doi.org/10.3390/genes9120605
Wang SF, Kowal TJ, Ning K, Koo EB, Wu AY, Mahajan VB, Sun Y. Review of Ocular Manifestations of Joubert Syndrome. Genes. 2018; 9(12):605. https://doi.org/10.3390/genes9120605
Chicago/Turabian StyleWang, Stephanie F., Tia J. Kowal, Ke Ning, Euna B. Koo, Albert Y. Wu, Vinit B. Mahajan, and Yang Sun. 2018. "Review of Ocular Manifestations of Joubert Syndrome" Genes 9, no. 12: 605. https://doi.org/10.3390/genes9120605
APA StyleWang, S. F., Kowal, T. J., Ning, K., Koo, E. B., Wu, A. Y., Mahajan, V. B., & Sun, Y. (2018). Review of Ocular Manifestations of Joubert Syndrome. Genes, 9(12), 605. https://doi.org/10.3390/genes9120605