An Improved Method for Prediction of Cancer Prognosis by Network Learning


Abstract

1. Introduction
2. Materials and Methods
2.1. Data Preparation
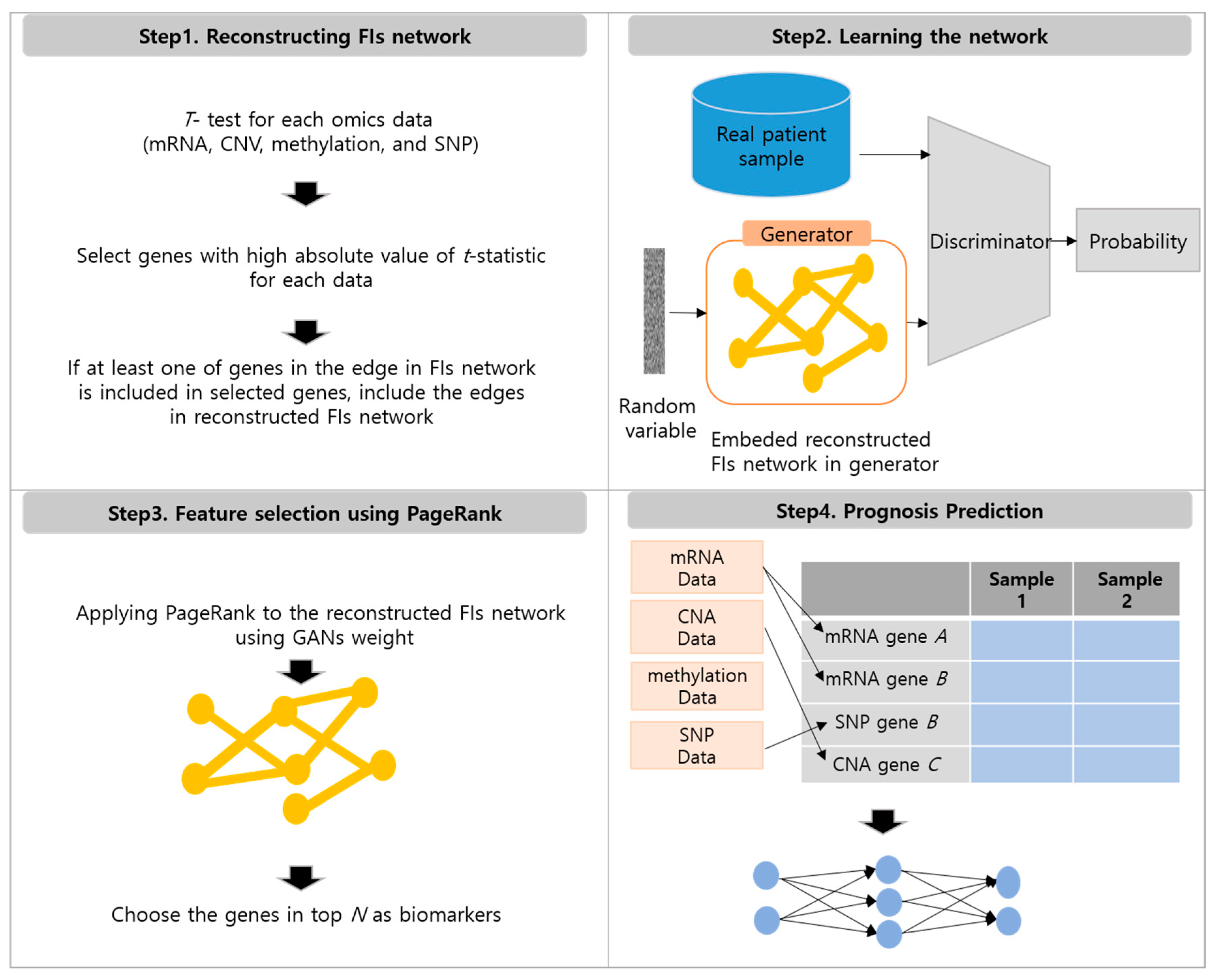
2.2. Method
2.2.1. Reconstructing FIs Network
2.2.2. Learning the Reconstructed FIs Network Using Generative Adversarial Networks
2.2.3. Feature Selection Using PageRank with Generative Adversarial Network Weights
2.2.4. Prognosis Prediction
3. Results
3.1. Parameter Selection
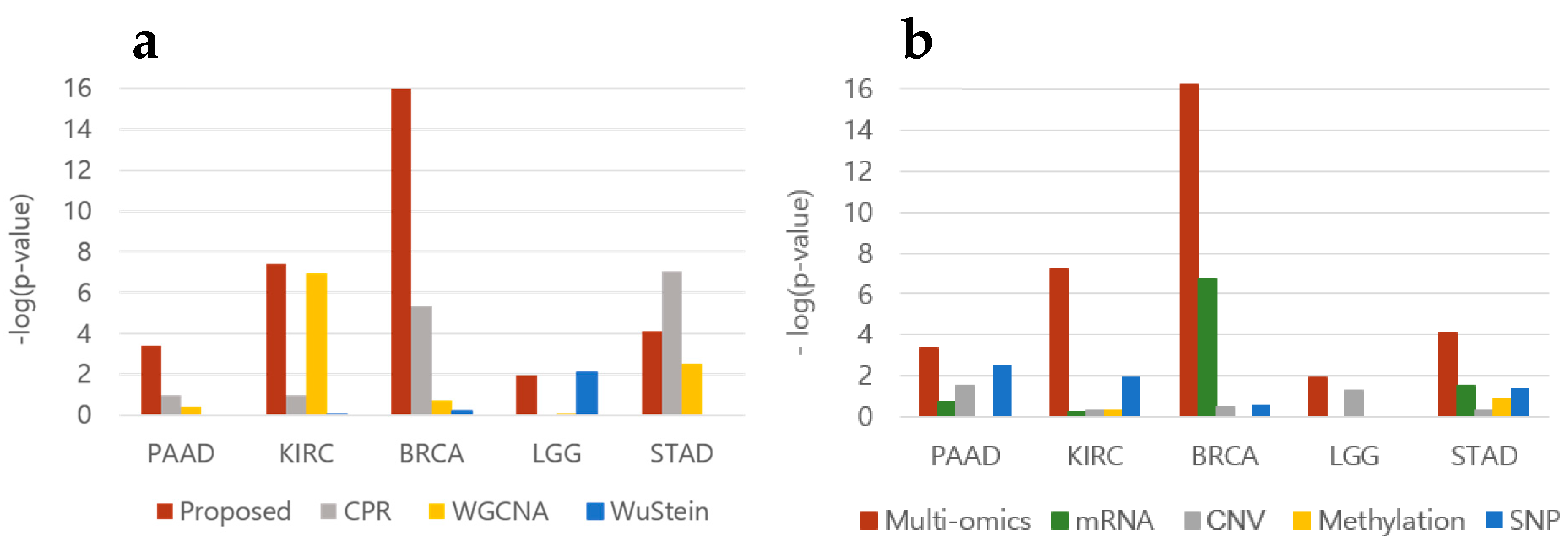
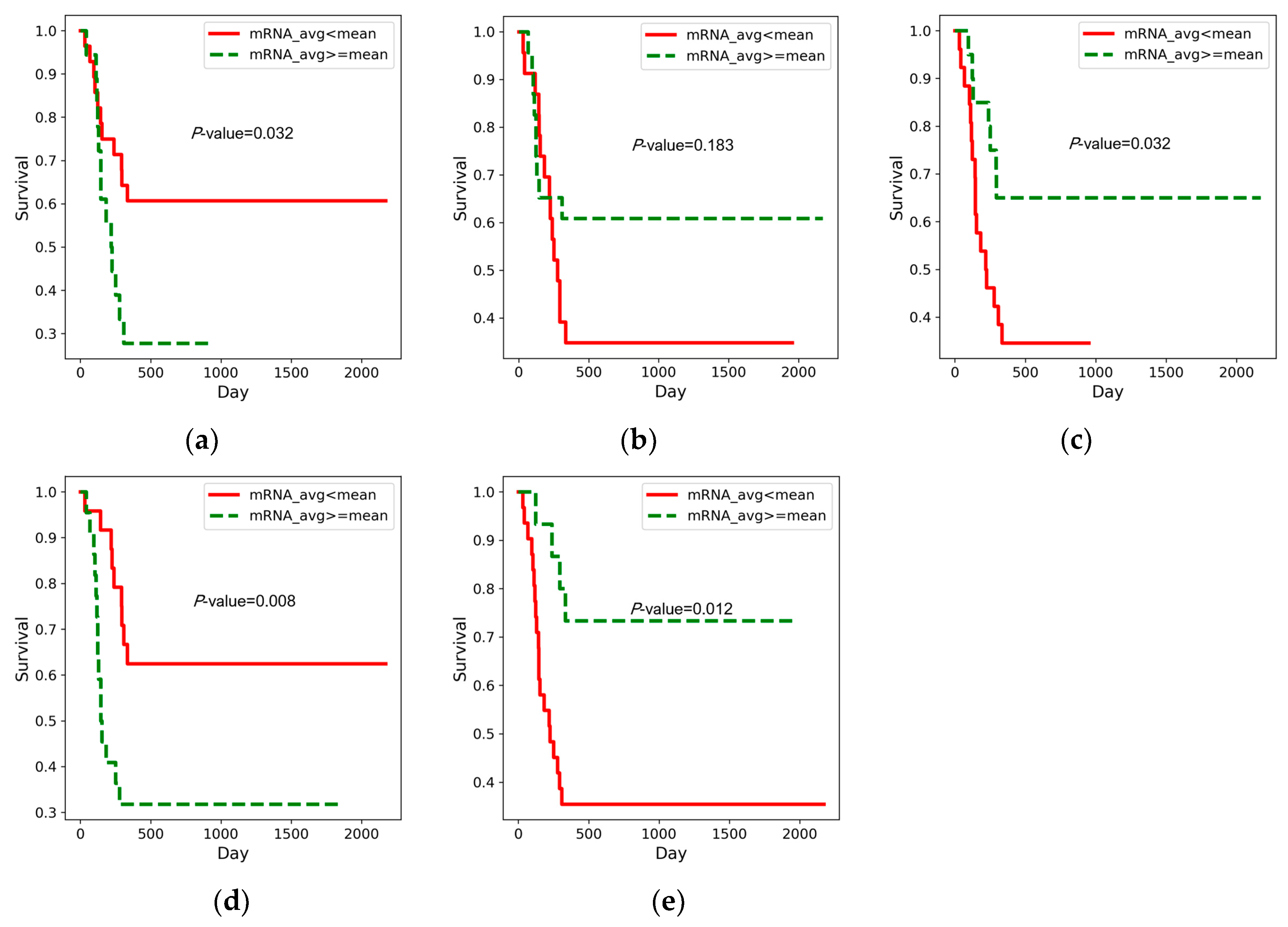
3.2. Prognostic Prediction
3.3. Oncogene Inclusion Test
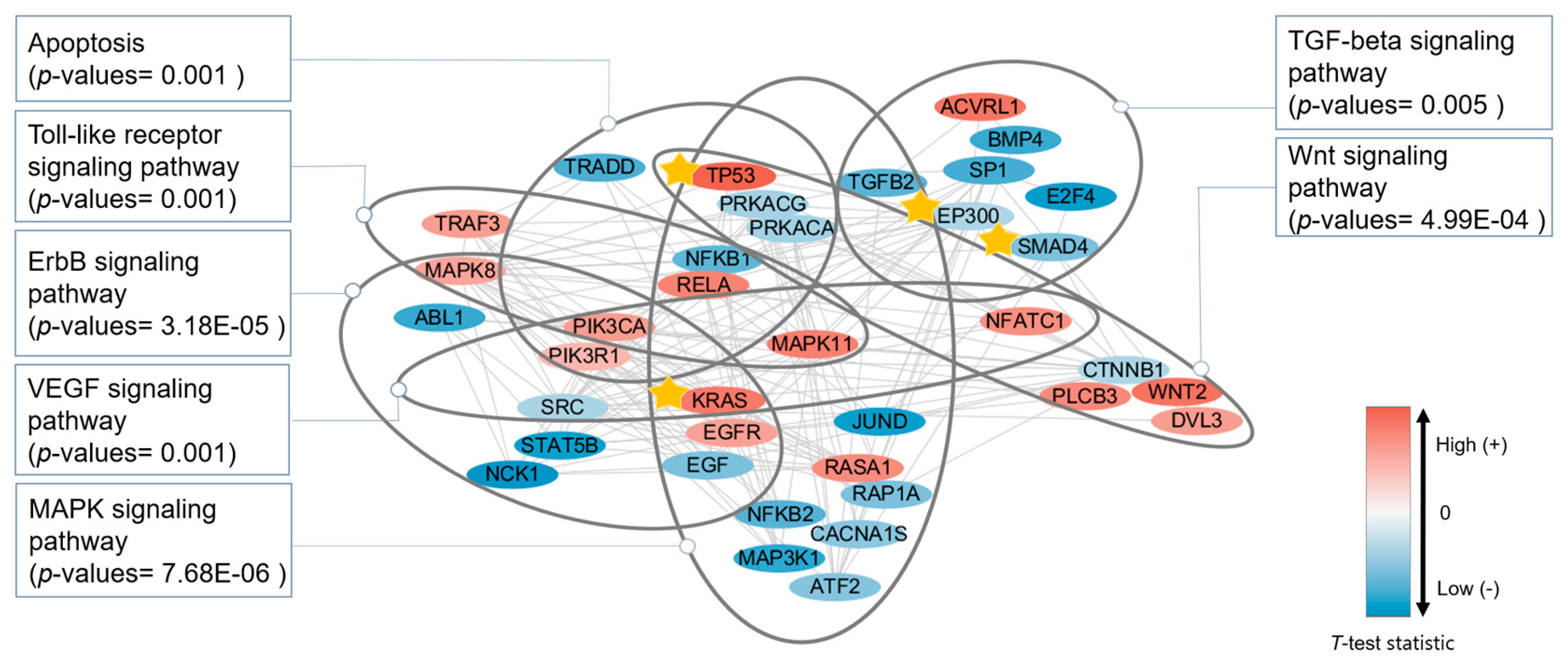
3.4. Functional Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Availability
References
- Emura, T.; Chen, Y.H. Gene selection for survival data under dependent censoring: A copula-based approach. Stat. Methods Med. Res. 2016, 25, 2840–2857. [Google Scholar] [CrossRef] [PubMed]
- Van De Vijver, M.J.; He, Y.D.; Van’t Veer, L.J.; Dai, H.; Hart, A.A.; Voskuil, D.W.; Schreiber, G.J.; Peterse, J.L.; Roberts, C.; Marton, M.J. A gene-expression signature as a predictor of survival in breast cancer. N. Engl. J. Med. 2002, 347, 1999–2009. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.Y.; Zhu, Z.H.; Li, J.; Linghu, B. Combined feature selection and cancer prognosis using support vector machine regression. IEEE/ACM Trans. Comput. Biol. Bioinform. 2011, 8, 1671–1677. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Tamayo, P.; Yang, J.-Y.; Hubbard, D.; Zhang, H.; Creighton, C.J.; Fereday, S.; Lawrence, M.; Carter, S.L.; Mermel, C.H. Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. J. Clin. Investig. 2013, 123, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. Wgcna: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Stein, L. A network module-based method for identifying cancer prognostic signatures. Genome Biol. 2012, 13, R112. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Park, S.; Yoon, Y.; Ahn, J. Improved prediction of breast cancer outcome by identifying heterogeneous biomarkers. Bioinformatics 2017, 33, 3619–3626. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K. Heterogeneity in breast cancer. J. Clin. Investig. 2011, 121, 3786–3788. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Ovaska, K.; Laakso, M.; Haapa-Paananen, S.; Louhimo, R.; Chen, P.; Aittomäki, V.; Valo, E.; Núñez-Fontarnau, J.; Rantanen, V.; Karinen, S. Large-scale data integration framework provides a comprehensive view on glioblastoma multiforme. Genome Med. 2010, 2, 65. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Chaudhary, K.; Garmire, L.X. More is better: Recent progress in multi-omics data integration methods. Front. Genet. 2017, 8, 84. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, E.; Calzone, L.; Michoel, T. Integrative multi-omics module network inference with lemon-tree. PLoS Comput. Biol. 2015, 11, e1003983. [Google Scholar] [CrossRef] [PubMed]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The cancer genome atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Jin, Z.; Yang, S.; Xu, Y.; Zhu, Y.; Ji, Y. Tcga-assembler 2: Software pipeline for retrieval and processing of TCGA/CPTAC data. Bioinformatics 2018, 34, 1615–1617. [Google Scholar] [CrossRef] [PubMed]
- Croft, D.; Mundo, A.F.; Haw, R.; Milacic, M.; Weiser, J.; Wu, G.; Caudy, M.; Garapati, P.; Gillespie, M.; Kamdar, M.R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2014, 42, D472–D477. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, A.; Sidiropoulos, K.; Garapati, P.; Gillespie, M.; Hausmann, K.; Haw, R.; Jassal, B.; Jupe, S.; Korninger, F.; McKay, S.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2016, 44, D481–D487. [Google Scholar] [CrossRef] [PubMed]
- Goodfellow, I.; Pouget-Abadie, J.; Mirza, M.; Xu, B.; Warde-Farley, D.; Ozair, S.; Courville, A.; Bengio, Y. Generative adversarial nets. In Proceedings of the Neural Information Processing Systems 27, Montreal, QC, Canada, 8–13 December 2014; pp. 2672–2680. [Google Scholar]
- Page, L.; Brin, S.; Motwani, R.; Winograd, T. The PageRank Citation Ranking: Bringing Order to the Web; Stanford InfoLab.: Stanford, CA, USA, 1999. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V. Scikit-learn: Machine learning in python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Rubio-Perez, C.; Tamborero, D.; Schroeder, M.P.; Antolín, A.A.; Deu-Pons, J.; Perez-Llamas, C.; Mestres, J.; Gonzalez-Perez, A.; Lopez-Bigas, N. In silico prescription of anticancer drugs to cohorts of 28 tumor types reveals targeting opportunities. Cancer Cell 2015, 27, 382–396. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Perez, A.; Perez-Llamas, C.; Deu-Pons, J.; Tamborero, D.; Schroeder, M.P.; Jene-Sanz, A.; Santos, A.; Lopez-Bigas, N. Intogen-mutations identifies cancer drivers across tumor types. Nat. Methods 2013, 10, 1081. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using david bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2008, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. Kegg: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2016, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T. Impacts of activation of the mitogen-activated protein kinase pathway in pancreatic cancer. Front. Oncol. 2015, 5, 23. [Google Scholar] [CrossRef] [PubMed]
- Almoguera, C.; Shibata, D.; Forrester, K.; Martin, J.; Arnheim, N.; Perucho, M. Most human carcinomas of the exocrine pancreas contain mutant ck-ras genes. Cell 1988, 53, 549–554. [Google Scholar] [CrossRef]
- Derynck, R.; Akhurst, R.J.; Balmain, A. Tgf-β signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117. [Google Scholar] [CrossRef] [PubMed]
- Friess, H.; Yamanaka, Y.; Büchler, M.; Ebert, M.; Beger, H.G.; Gold, L.I.; Korc, M. Enhanced expression of transforming growth factor β isoforms in pancreatic cancer correlates with decreased survival. Gastroenterology 1993, 105, 1846–1856. [Google Scholar] [CrossRef]
- Albo, D.; Berger, D.H.; Tuszynski, G.P. The effect of thrombospondin-1 and TGF-β1 on pancreatic cancer cell invasion. J. Surg. Res. 1998, 76, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.; Li, Y.; Tan, D.; Dong, X.; Chang, P.; Kar, S.; Li, D. Biomarkers of TGF-β signaling pathway and prognosis of pancreatic cancer. PLoS ONE 2014, 9, e85942. [Google Scholar] [CrossRef] [PubMed]
- Xue, Z.; Vis, D.J.; Bruna, A.; Sustic, T.; van Wageningen, S.; Batra, A.S.; Rueda, O.M.; Bosdriesz, E.; Caldas, C.; Wessels, L.F. Map3k1 and map2k4 mutations are associated with sensitivity to mek inhibitors in multiple cancer models. Cell Res. 2018, 28, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, L.; Mundade, R.; Korc, M.; Loehrer, P.J.; Lu, T. Critical role of NF-ΚB in pancreatic cancer. Oncotarget 2014, 5, 10969–10975. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Asano, T.; Yao, Y.; Zhang, R.; Claret, F.-X.; Korc, M.; Sabapathy, K.; Menter, D.G.; Abbruzzese, J.L.; Reddy, S.A. Activator protein-1 has an essential role in pancreatic cancer cells and is regulated by a novel akt-mediated mechanism. Mol. Cancer Res. 2009, 7, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Olayioye, M.A.; Neve, R.M.; Lane, H.A.; Hynes, N.E. The erbb signaling network: Receptor heterodimerization in development and cancer. EMBO J. 2000, 19, 3159–3167. [Google Scholar] [CrossRef] [PubMed]
- Korc, M.; Chandrasekar, B.; Yamanaka, Y.; Friess, H.; Buchier, M.; Beger, H.G. Overexpression of the epidermal growth factor receptor in human pancreatic cancer is associated with concomitant increases in the levels of epidermal growth factor and transforming growth factor alpha. J. Clin. Investig. 1992, 90, 1352–1360. [Google Scholar] [CrossRef] [PubMed]
- Durkin, A.J.; Bloomston, P.M.; Rosemurgy, A.S.; Giarelli, N.; Cojita, D.; Yeatman, T.J.; Zervos, E.E. Defining the role of the epidermal growth factor receptor in pancreatic cancer grown in vitro. Am. J. Surg. 2003, 186, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Schiemann, W.P. The TGF-β paradox in human cancer: An update. Future Oncol. 2009, 5, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Bradshaw, A.-D.; Gera, S.; Dewan, M.Z.; Xu, R. The TGF-β/smad4 signaling pathway in pancreatic carcinogenesis and its clinical significance. J. Clin. Med. 2017, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Ijichi, H. Tgf-beta signaling pathway in pancreatic cancer cells. Nihon rinsho. Jpn. J. Clin. Med. 2004, 62, 1241–1248. [Google Scholar]
- Zhu, Y.; Gu, J.; Li, Y.; Peng, C.; Shi, M.; Wang, X.; Wei, G.; Ge, O.; Wang, D.; Zhang, B. Mir-17-5p enhances pancreatic cancer proliferation by altering cell cycle profiles via disruption of rbl2/e2f4-repressing complexes. Cancer Lett. 2018, 412, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wei, Y.; Zhang, Y.; Yang, Q. Deep neural networks for high dimension, low sample size data. In Proceedings of the Twenty-Sixth International Joint Conference on Artificial Intelligence IJCAI-17, Melbourne, Australia, 19–25 August 2017; pp. 2287–2293. [Google Scholar]
- Kong, Y.; Yu, T. A graph-embedded deep feedforward network for disease outcome classification and feature selection using gene expression data. Bioinformatics 2018, 1, 11. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer | #Gene in Multi-Omics | #Gene in mRNA | #Gene in CNV | #Gene in DNA Methylation | #Gene in SNP | #Good Sample | #Poor Sample | #Criteria for Label |
|---|---|---|---|---|---|---|---|---|
| PAAD | 7371 | 11,404 | 11,637 | 11,509 | 7571 | 20 | 24 | 1 year |
| BRCA | 7908 | 11,578 | 11,093 | 9216 | 10,354 | 91 | 63 | 5 years |
| KIRC | 8801 | 11,569 | 11,643 | 11,508 | 8920 | 65 | 47 | 4 years |
| LGG | 11,419 | 11,511 | 11,642 | 11,517 | NA | 66 | 59 | 3 years |
| STAD | 5634 | 11,571 | 11,645 | 11,515 | 6836 | 29 | 16 | 1 year |
| Epoch | Batch Size | Learning Rate | Optimizer |
|---|---|---|---|
| 2 | 1 | 0.0002 | Adam optimizer |
| Data | Gene |
|---|---|
| mRNA | ABL1, E2F4, EGFR, NFKB1, NFKB2, PRKACA, RASA1, SRC, STAT5B, TGFB2, TRADD |
| CNV | DVL3, KRAS, MAPK8, NFATC1, PIK3CA, PLCB3, RELA |
| DNA methylation | ACVRL1, ATF2, BMP4, EGF, JUND, MAP3K1, MAPK11, NCK1, NFKB2, PIK3R1, PRKACG, RAP1A, SP1, TGFB2, TRAF3, WNT2 |
| SNP | CACNA1S, CTNNB1, EP300, SMAD4, TP53 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.; Oh, I.; Ahn, J. An Improved Method for Prediction of Cancer Prognosis by Network Learning. Genes 2018, 9, 478. https://doi.org/10.3390/genes9100478
Kim M, Oh I, Ahn J. An Improved Method for Prediction of Cancer Prognosis by Network Learning. Genes. 2018; 9(10):478. https://doi.org/10.3390/genes9100478
Chicago/Turabian StyleKim, Minseon, Ilhwan Oh, and Jaegyoon Ahn. 2018. "An Improved Method for Prediction of Cancer Prognosis by Network Learning" Genes 9, no. 10: 478. https://doi.org/10.3390/genes9100478
APA StyleKim, M., Oh, I., & Ahn, J. (2018). An Improved Method for Prediction of Cancer Prognosis by Network Learning. Genes, 9(10), 478. https://doi.org/10.3390/genes9100478