PGC1α: Friend or Foe in Cancer?

Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. The Gene and the Protein
1.2. PGC1α in Healthy Tissue and in Non-Cancer Disease
2. PGC1α in Cancer
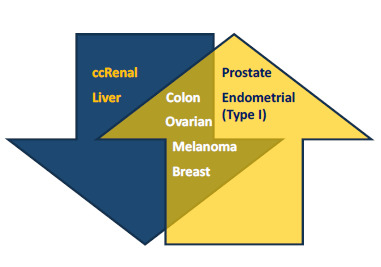
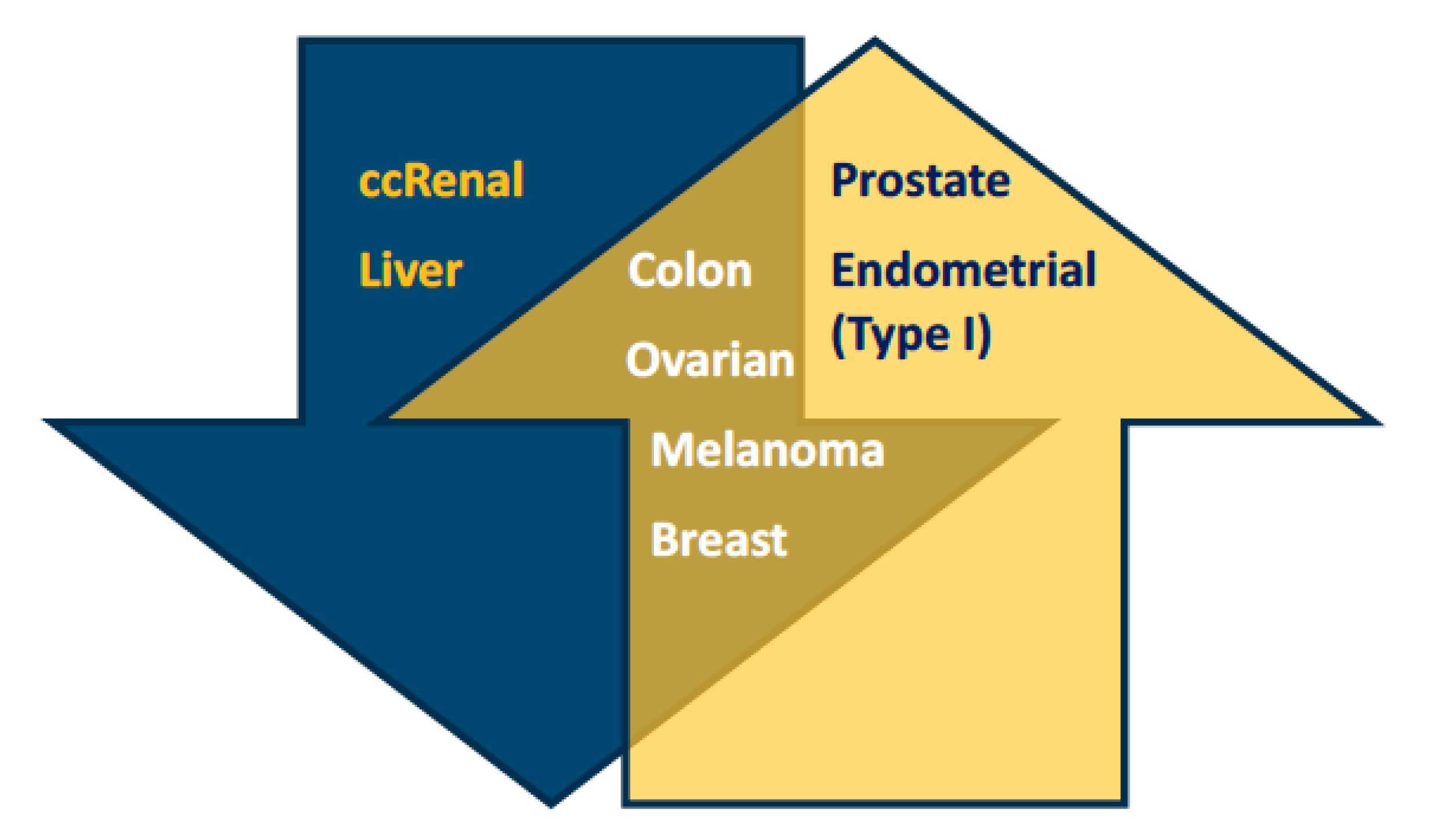
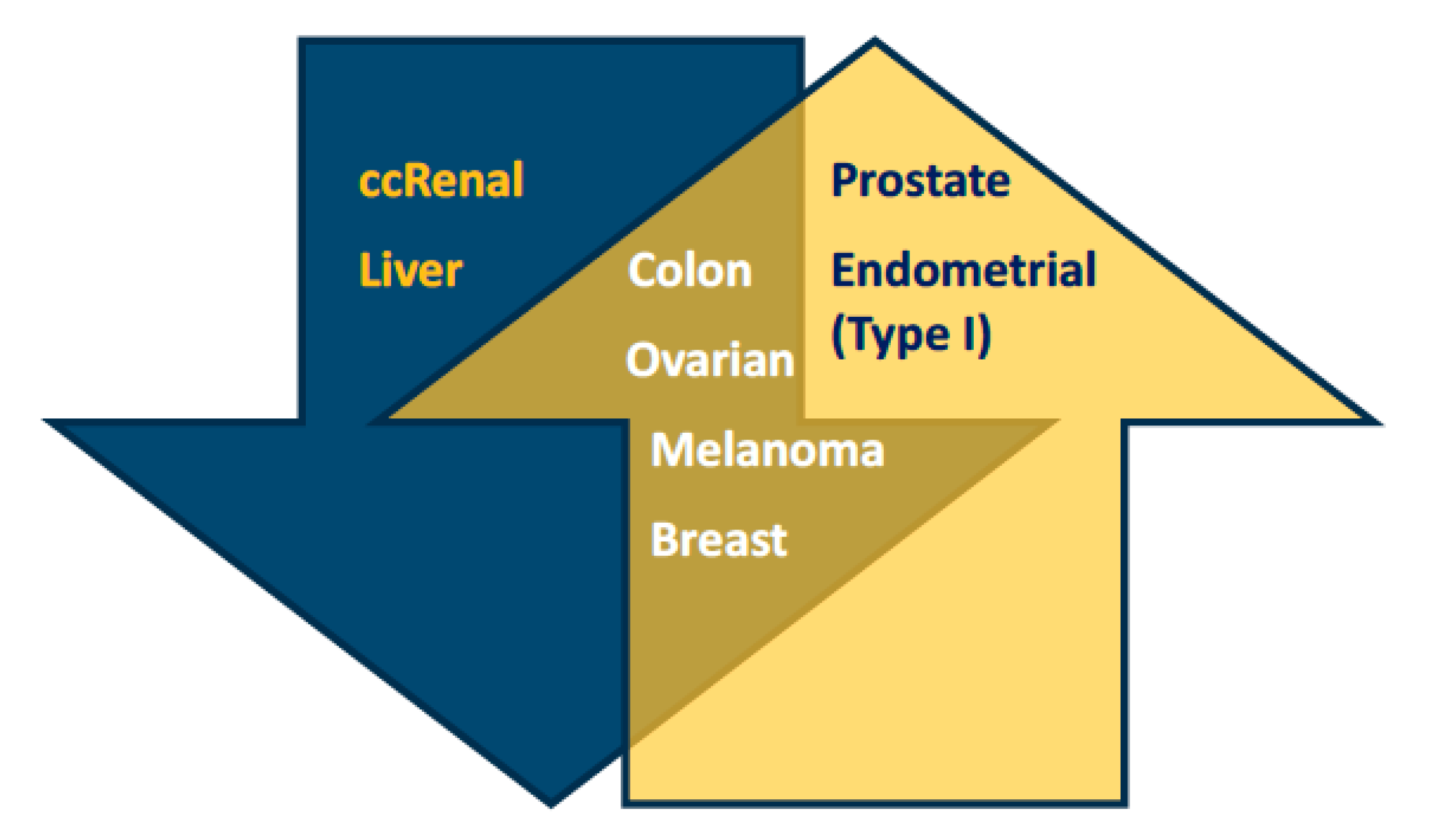
2.1. Low and High Expression of PGC1α in Cancer
2.2. PGC1α and Oncogenicity
- BRAF: Constitutively activated BRAF, commonly seen in melanomas, was shown to suppress the melanocyte lineage factor MITF, leading to loss of MITF-regulated PGC1α and upregulation of a glycolytic metabolism [39].
- p53: Experiments on cancer cell lines showed that PGC1α can bind to and potentiate p53 transactivation of cell cycle arrest and metabolic genes. Furthermore, glucose starvation and abrogation of PGC1α led to ROS overload and apoptosis [57]. It was also found that in PGC1α proficient cells, prolonged starvation led to PGC1α degradation by the ubiquitin-proteasome pathway and apoptosis [57]. However, we have not observed a similar sensitivity to starvation in an ovarian cancer cell line devoid of both p53 and PGC1α expression [37], indicating that cancer cells may develop compensating mechanisms. Conversely, in a mouse model and in samples from chronic lymphocytic leukemia patients, loss of p53 through deletion of chr17p correlated with increased expression of PGC1α and its downstream effector TFAM, and with increased mitochondrial respiratory activity [58], although no clear causality was demonstrated. Inverse correlations between p53 and PGC1α have been observed also in non-cancer contexts, for example, upregulation of p53 due to telomere dysfunction repressed both PGC1α and PGC1β [59]; such mechanisms may turn out to be of interest in the cancer field.
- MYC: An inverse relationship between MYC and PGC1α has been demonstrated in cardiac myocytes [60] as well as in pancreatic cancer stem cells [61]. The latter study also showed that PGC1α/MYC ratios represent a spectrum of tumor-promoting metabolic phenotypes ranging from OXPHOS-based to glycolytic. As MYC regulates glucose and glutamine metabolism and also mitobiogenesis in cancer cells [62], this might together with MYC-dependent PGC1β expression [63] explain why PGC1α negative tumor cells nevertheless have functioning mitochondria and metabolism. Although not formally shown, it is also conceivable that the MYC/PGC1α ratio can be regulated by levels of the transcription factor FoxO3a, since this is a direct transcriptional regulator of PGC1α [6] and is in metabolic contexts also a negative regulator of MYC [64]. Similar to PGC1α, both high and low expression of FoxO3a has been associated with cancer and worse prognosis, in line with the notion that metabolic plasticity is central to tumor progression and treatment resistance.
- ERRα Although not oncogenic as such, the role of the estrogen-related receptor (ERR) family should not be overlooked in cancer cells expressing PGC1α. Like the other members of the ERR family, ERRα does not bind estrogens and their transcriptional activities are ligand-independent. Indeed, PGC1α β act as surrogate ligands for ERRα and the resulting PGC1/ERRα axis is of importance in cancer and cancer cell metabolism [28,65]. Similar to PGC1α, ERRα is required for rapid stress responses but less so for basal energy regulation. It binds to promoters of most enzymes in glucose, glutamate and fatty acid metabolism and the TCA cycle, and is upregulated in many cancers and associated with unfavorable outcomes [65]. Interestingly, there are reports on ERRα inhibitors inhibiting the growth of PGC1α proficient cells [2,41]. In order to help clarify the roles of PGC1α and its different partners and pathways, future studies should address for instance the prognostic significance of the combined PGC1α ERRα.
2.3. Mechanisms of Regulation of PGC1α Levels in Cancer
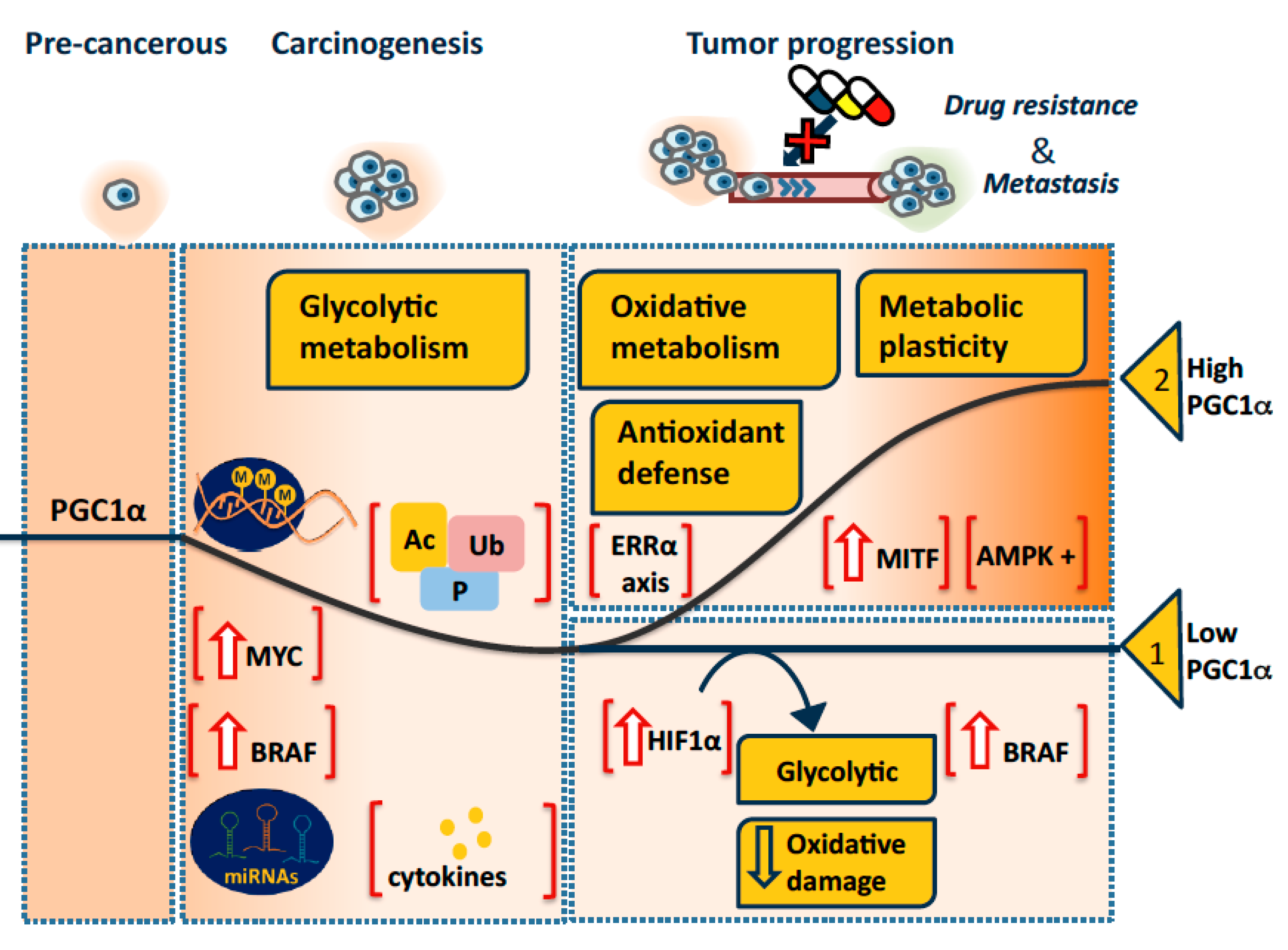
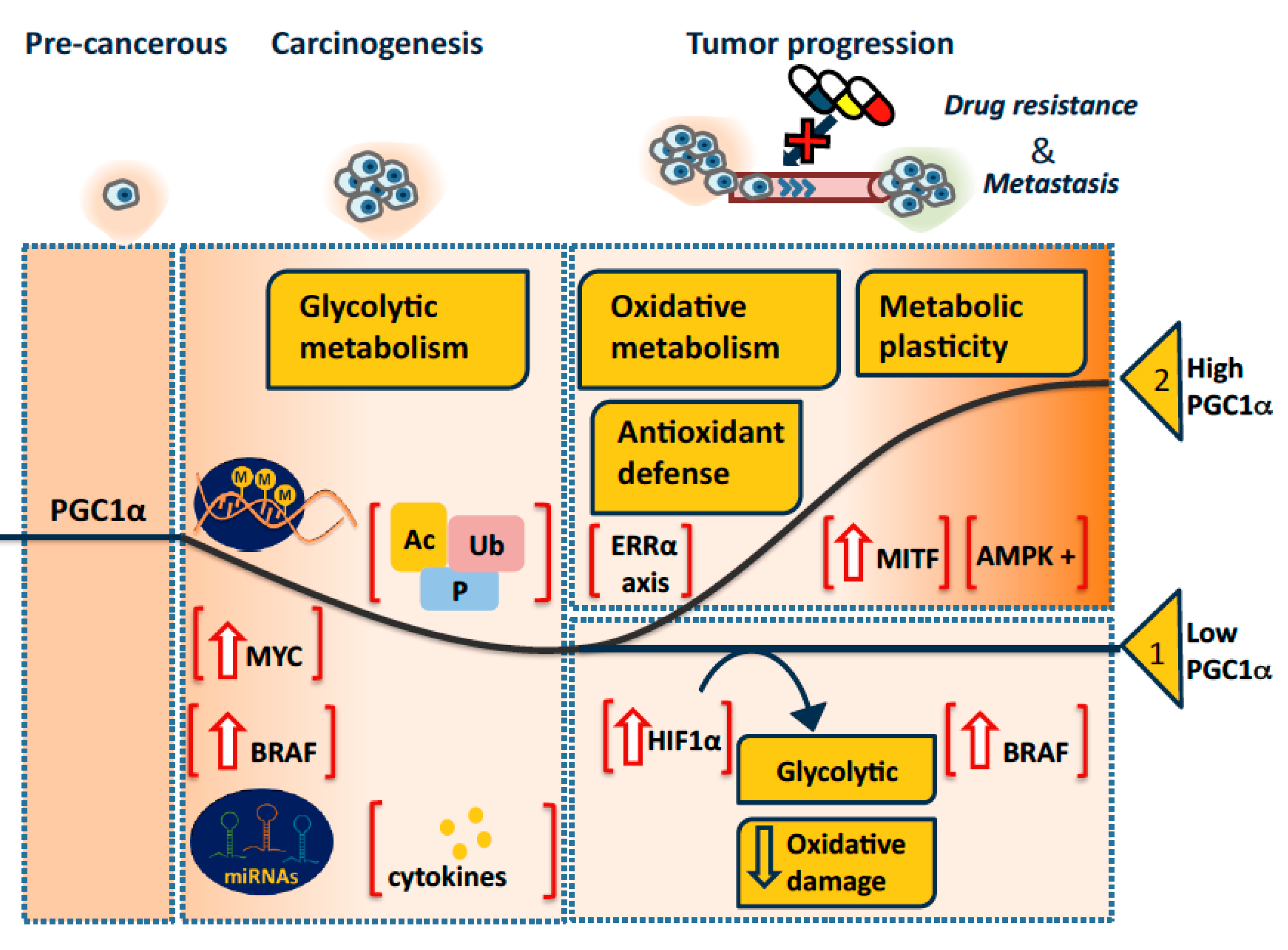
3. Pro-Oncogenic or Tumor Suppressive PGC1α in Tumorigenesis and Progression?
3.1. A Model
3.1.1. PGC1α in Tumorigenesis
3.1.2. PGC1α in Tumor Progression
3.1.3. PGC1α and Tumor-Initiating Cells
3.2. PGC1α Autophagy, Mitophagy and Mitochondrial Dynamics
4. PGC1α, Obesity and Cancer
5. PGC1β and PGC1-Related Coactivator
6. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Villena, J.A. New insights into PGC-1 coactivators: Redefining their role in the regulation of mitochondrial function and beyond. FEBS J. 2015, 282, 647–672. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Luo, X.; Xiao, L.; Tang, M.; Bode, A.M.; Dong, Z.; Cao, Y. The Role of PGC1α in Cancer Metabolism and its Therapeutic Implications. Mol. Cancer Ther. 2016, 15, 774–782. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Redondo, V.; Pettersson, A.T.; Ruas, J.L. The hitchhiker’s guide to PGC-1α isoform structure and biological functions. Diabetologia 2015, 58, 1969–1977. [Google Scholar] [CrossRef] [PubMed]
- Ranhotra, H.S. Estrogen-related receptor alpha and mitochondria: Tale of the titans. J. Recept. Signal Transduct. Res. 2015, 35, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Spiegelman, B.M. Transcriptional control of mitochondrial energy metabolism through the PGC1 coactivators. Novartis Found. Symp. 2007, 287, 60–63, discussion 3–9. [Google Scholar] [PubMed]
- Olmos, Y.; Valle, I.; Borniquel, S.; Tierrez, A.; Soria, E.; Lamas, S.; Monsalve, M. Mutual dependence of Foxo3a and PGC-1alpha in the induction of oxidative stress genes. J. Biol. Chem. 2009, 284, 14476–14484. [Google Scholar] [CrossRef] [PubMed]
- Dominy, J.E., Jr.; Lee, Y.; Gerhart-Hines, Z.; Puigserver, P. Nutrient-dependent regulation of PGC-1alph’s acetylation state and metabolic function through the enzymatic activities of Sirt1/GCN5. Biochim. Biophys. Acta 2010, 1804, 1676–1683. [Google Scholar] [CrossRef] [PubMed]
- Trausch-Azar, J.; Leone, T.C.; Kelly, D.P.; Schwartz, A.L. Ubiquitin proteasome-dependent degradation of the transcriptional coactivator PGC-1α via the N-terminal pathway. J. Biol. Chem. 2010, 285, 40192–40200. [Google Scholar] [CrossRef] [PubMed]
- Trausch-Azar, J.S.; Abed, M.; Orian, A.; Schwartz, A.L. Isoform-specific SCF(Fbw7) ubiquitination mediates differential regulation of PGC-1alpha. J. Cell. Physiol. 2015, 230, 842–852. [Google Scholar] [CrossRef] [PubMed]
- Aquilano, K.; Vigilanza, P.; Baldelli, S.; Pagliei, B.; Rotilio, G.; Ciriolo, M.R. Peroxisome proliferator-activated receptor gamma co-activator 1alpha (PGC-1α) and sirtuin 1 (SIRT1) reside in mitochondria: Possible direct function in mitochondrial biogenesis. J. Biol. Chem. 2010, 285, 21590–21599. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.L. Sirt1 and the Mitochondria. Mol. Cells 2016, 39, 87–95. [Google Scholar] [PubMed]
- Lehman, J.J.; Barger, P.M.; Kovacs, A.; Saffitz, J.E.; Medeiros, D.M.; Kelly, D.P. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J. Clin. Investig. 2000, 106, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Girnun, G.D. The diverse role of the PPARγ coactivator 1 family of transcriptional coactivators in cancer. Semin. Cell Dev. Biol. 2012, 23, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Nijland, P.G.; Witte, M.E.; van het Hof, B.; Bauer, J.; Lassmann, H.; van der Valk, P.; de Vries, H.E.; van Horssen, J. Astroglial PGC-1alpha increases mitochondrial antioxidant capacity and suppresses inflammation: Implications for multiple sclerosis. Acta Neuropathol. Commun. 2014, 2, 170. [Google Scholar] [CrossRef] [PubMed]
- Daitoku, H.; Yamagata, K.; Matsuzaki, H.; Hatta, M.; Fukamizu, A. Regulation of PGC-1 promoter activity by protein kinase B and the forkhead transcription factor FKHR. Diabetes 2003, 52, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Jiang, B.; Qiu, Y.; Guan, J.; Jain, M.; Cao, X.; Bauer, M.; Su, L.; Burkly, L.C.; Leone, T.C.; et al. PGC1α plays a critical role in TWEAK-induced cardiac dysfunction. PLoS ONE 2013, 8, e54054. [Google Scholar] [CrossRef] [PubMed]
- Onyango, I.G.; Khan, S.M.; Bennett, J.P., Jr. Mitochondria in the pathophysiology of Alzheimer’s and Parkinson’s diseases. Front. Biosci. 2017, 22, 854–872. [Google Scholar] [CrossRef]
- Su, X.; Chu, Y.; Kordower, J.H.; Li, B.; Cao, H.; Huang, L.; Nishida, M.; Song, L.; Wang, D.; Federoff, H.J. PGC-1α Promoter Methylation in Parkinson’s disease. PLoS ONE 2015, 10, e0134087. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Ko, H.S.; Kang, H.; Lee, Y.; Lee, Y.I.; Pletinkova, O.; Troconso, J.C.; Dawson, V.L.; Dawson, T.M. PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson’s disease. Cell 2011, 144, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Ciron, C.; Lengacher, S.; Dusonchet, J.; Aebischer, P.; Schneider, B.L. Sustained expression of PGC-1alpha in the rat nigrostriatal system selectively impairs dopaminergic function. Hum. Mol. Genet. 2012, 21, 1861–1876. [Google Scholar] [CrossRef] [PubMed]
- Besseiche, A.; Riveline, J.P.; Gautier, J.F.; Breant, B.; Blondeau, B. Metabolic roles of PGC-1alpha and its implications for type 2 diabetes. Diabetes Metab. 2015, 41, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, E.J.; LeRoith, D. Obesity and Diabetes: The Increased Risk of Cancer and Cancer-Related Mortality. Physiol. Rev. 2015, 95, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.M.; Tewari, S.; Benite-Ribeiro, S.A. The effect of exercise on epigenetic modifications of PGC1: The impact on type 2 diabetes. Med. Hypotheses 2014, 82, 748–753. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Barres, R.; Osler, M.E.; Yan, J.; Rune, A.; Fritz, T.; Caidahl, K.; Krook, A.; Zierath, J.R. Non-CpG methylation of the PGC-1α promoter through DNMT3B controls mitochondrial density. Cell Metab. 2009, 10, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Crunkhorn, S.; Dearie, F.; Mantzoros, C.; Gami, H.; da Silva, W.S.; Espinoza, D.; Faucette, R.; Barry, K.; Bianco, A.C.; Patti, M.E. Peroxisome proliferator activator receptor gamma coactivator-1 expression is reduced in obesity: Potential pathogenic role of saturated fatty acids and p38 mitogen-activated protein kinase activation. J. Biol. Chem. 2007, 282, 15439–15450. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Ogura, Y.; Tajrishi, M.M.; Kumar, A. Elevated levels of TWEAK in skeletal muscle promote visceral obesity, insulin resistance, and metabolic dysfunction. FASEB J. 2015, 29, 988–1002. [Google Scholar] [CrossRef] [PubMed]
- Deblois, G.; St-Pierre, J.; Giguere, V. The PGC-1/ERR signaling axis in cancer. Oncogene 2013, 32, 3483–3490. [Google Scholar] [CrossRef] [PubMed]
- LaGory, E.L.; Wu, C.; Taniguchi, C.M.; Ding, C.C.; Chi, J.T.; von Eyben, R.; Scott, D.A.; Richardson, A.D.; Giaccia, A.J. Suppression of PGC-1α Is Critical for Reprogramming Oxidative Metabolism in Renal Cell Carcinoma. Cell Rep. 2015, 12, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Zhang, H.; Zhang, Y.; Li, S.; Wang, X.; Wang, X.; Wang, C.; Liu, B.; Zen, K.; Zhang, C.Y.; et al. Peroxisome proliferator-activated receptor gamma coactivator-1 alpha acts as a tumor suppressor in hepatocellular carcinoma. Tumour Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Iakova, P.; Jiang, Y.; Lewis, K.; Sullivan, E.; Jawanmardi, N.; Donehower, L.; Timchenko, L.; Timchenko, N.A. Transcriptional and translational regulation of C/EBPβ-HDAC1 protein complexes controls different levels of p53, SIRT1, and PGC1α proteins at the early and late stages of liver cancer. J. Biol. Chem. 2013, 288, 14451–14462. [Google Scholar] [CrossRef] [PubMed]
- Tennakoon, J.B.; Shi, Y.; Han, J.J.; Tsouko, E.; White, M.A.; Burns, A.R.; Zhang, A.; Xia, X.; Ilkayeva, O.R.; Xin, L.; et al. Androgens regulate prostate cancer cell growth via an AMPK-PGC-1α-mediated metabolic switch. Oncogene 2014, 33, 5251–5261. [Google Scholar] [CrossRef] [PubMed]
- Cormio, A.; Guerra, F.; Cormio, G.; Pesce, V.; Fracasso, F.; Loizzi, V.; Cantatore, P.; Selvaggi, L.; Gadaleta, M.N. The PGC-1alpha-dependent pathway of mitochondrial biogenesis is upregulated in type I endometrial cancer. Biochem. Biophys. Res. Commun. 2009, 390, 1182–1185. [Google Scholar] [CrossRef] [PubMed]
- D’Errico, I.; Salvatore, L.; Murzilli, S.; Lo Sasso, G.; Latorre, D.; Martelli, N.; Egorova, A.V.; Polishuck, R.; Madeyski-Bengtson, K.; Lelliott, C.; et al. Peroxisome proliferator-activated receptor-gamma coactivator 1-alpha (PGC1alpha) is a metabolic regulator of intestinal epithelial cell fate. Proc. Natl. Acad. Sci. USA 2011, 108, 6603–6608. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, K.; Hwang, B.J.; Dewi, R.E.; Ou, L.; Twaddel, W.; Fang, H.B.; Vafai, S.B.; Vazquez, F.; Puigserver, P.; Boros, L. PGC1α promotes tumor growth by inducing gene expression programs supporting lipogenesis. Cancer Res. 2011, 71, 6888–6898. [Google Scholar] [CrossRef] [PubMed]
- Dar, S.; Chhina, J.; Mert, I.; Chitale, D.; Buekers, T.; Kaur, H.; Giri, S.; Munkarah, A.; Rattan, R. Bioenergetic Adaptations in Chemoresistant Ovarian Cancer Cells. Sci. Rep. 2017, 7, 8760. [Google Scholar] [CrossRef] [PubMed]
- Gabrielson, M.; Bjorklund, M.; Carlson, J.; Shoshan, M. Expression of mitochondrial regulators PGC1α and TFAM as putative markers of subtype and chemoresistance in epithelial ovarian carcinoma. PLoS ONE 2014, 9, e107109. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Jung, J.W.; Jung, J.; Han, Y.; Suh, D.H.; Kim, H.S.; Dhanasekaran, D.N.; Song, Y.S. PGC1α induced by reactive oxygen species contributes to chemoresistance of ovarian cancer cells. Oncotarget 2017, 8, 60299–60311. [Google Scholar] [PubMed]
- Haq, R.; Shoag, J.; Andreu-Perez, P.; Yokoyama, S.; Edelman, H.; Rowe, G.C.; Frederick, D.T.; Hurley, A.D.; Nellore, A.; Kung, A.L.; et al. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell 2013, 23, 302–315. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, F.; Lim, J.H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1α expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Lim, J.H.; Lee, Y.; Granter, S.R.; Thomas, A.; Vazquez, F.; Widlund, H.R.; Puigserver, P. A PGC1α-mediated transcriptional axis suppresses melanoma metastasis. Nature 2016, 537, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Watkins, G.; Douglas-Jones, A.; Mansel, R.E.; Jiang, W.G. The localisation and reduction of nuclear staining of PPARgamma and PGC-1 in human breast cancer. Oncol. Rep. 2004, 12, 483–488. [Google Scholar] [PubMed]
- Jiang, W.G.; Douglas-Jones, A.; Mansel, R.E. Expression of peroxisome-proliferator activated receptor-gamma (PPARgamma) and the PPARgamma co-activator, PGC-1, in human breast cancer correlates with clinical outcomes. Int. J. Cancer 2003, 106, 752–757. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003. [Google Scholar] [CrossRef] [PubMed]
- Andrzejewski, S.; Klimcakova, E.; Johnson, R.M.; Tabariès, S.; Annis, M.G.; McGuirk, S.; Northey, J.J.; Chénard, V.; Sriram, U.; Papadopoli, D.J.; et al. PGC-1α Promotes Breast Cancer Metastasis and Confers Bioenergetic Flexibility against Metabolic Drugs. Cell Metab. 2017, 26, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Cai, F.F.; Xu, C.; Pan, X.; Cai, L.; Lin, X.Y.; Chen, S.; Biskup, E. Prognostic value of plasma levels of HIF-1a and PGC-1a in breast cancer. Oncotarget 2016, 7, 77793–77806. [Google Scholar] [CrossRef] [PubMed]
- Savagner, F.; Mirebeau, D.; Jacques, C.; Guyetant, S.; Morgan, C.; Franc, B.; Reynier, P.; Malthièry, Y. PGC-1-related coactivator and targets are upregulated in thyroid oncocytoma. Biochem. Biophys. Res. Commun. 2003, 310, 779–784. [Google Scholar] [CrossRef] [PubMed]
- De Luise, M.; Girolimetti, G.; Okere, B.; Porcelli, A.M.; Kurelac, I.; Gasparre, G. Molecular and metabolic features of oncocytomas: Seeking the blueprints of indolent cancers. Biochim. Biophys. Acta 2017, 1858, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Cormio, A.; Guerra, F.; Cormio, G.; Pesce, V.; Fracasso, F.; Loizzi, V.; Resta, L.; Putignano, G.; Cantatore, P.; Selvaggi, L.E.; et al. Mitochondrial DNA content and mass increase in progression from normal to hyperplastic to cancer endometrium. BMC Res. Notes 2012, 5, 279. [Google Scholar] [CrossRef] [PubMed]
- Guerra, F.; Kurelac, I.; Cormio, A.; Zuntini, R.; Amato, L.B.; Ceccarelli, C.; Santini, D.; Cormio, G.; Fracasso, F.; Selvaggi, L.; et al. Placing mitochondrial DNA mutations within the progression model of type I endometrial carcinoma. Hum. Mol. Genet. 2011, 20, 2394–2405. [Google Scholar] [CrossRef] [PubMed]
- Gasparre, G.; Romeo, G.; Rugolo, M.; Porcelli, A.M. Learning from oncocytic tumors: Why choose inefficient mitochondria? Biochim. Biophys. Acta 2011, 1807, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Permuth-Wey, J.; Chen, Y.A.; Tsai, Y.Y.; Chen, Z.; Qu, X.; Lancaster, J.M.; Stockwell, H.; Dagne, G.; Iversen, E.; Risch, H.; et al. Inherited variants in mitochondrial biogenesis genes may influence epithelial ovarian cancer risk. Cancer Epidemiol. Biomark. Prev. 2011, 20, 1131–1145. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.A.; Lee, J.; Oh, J.H.; Chang, H.J.; Sohn, D.K.; Shin, A.; Kim, J. Genetic variation in PPARGC1A may affect the role of diet-associated inflammation in colorectal carcinogenesis. Oncotarget 2017, 8, 8550–8558. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017, 45, D777–D783. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed]
- Iommarini, L.; Ghelli, A.; Gasparre, G.; Porcelli, A.M. Mitochondrial metabolism and energy sensing in tumor progression. Biochim. Biophys. Acta 2017, 1858, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.; Satija, Y.K.; Das, S. PGC-1alpha, a key modulator of p53, promotes cell survival upon metabolic stress. Mol. Cell 2011, 44, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, M.A.; Liu, J.; Pelicano, H.; Hammoudi, N.; Croce, C.M.; Keating, M.J.; Huang, P.; et al. Alterations of mitochondrial biogenesis in chronic lymphocytic leukemia cells with loss of p53. Mitochondrion 2016, 31, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Sahin, E.; Colla, S.; Liesa, M.; Moslehi, J.; Müller, F.L.; Guo, M.; Cooper, M.; Kotton, D.; Fabian, A.J.; Walkey, C.; Maser, R.S.; et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 2011, 470, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, P.; Zhao, P.; Angelis, E.; Ruan, H.; Korge, P.; Olson, A.; Wang, Y.; Jin, E.S.; Jeffrey, F.M.; Portman, M.; et al. Myc controls transcriptional regulation of cardiac metabolism and mitochondrial biogenesis in response to pathological stress in mice. J. Clin. Investig. 2010, 120, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Bou Kheir, T.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Graña, O.; et al. MYC/PGC-1α Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a014217. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gao, P.; Fukuda, R.; Kumar, G.; Krishnamachary, B.; Zeller, K.I.; Dang, C.V.; Semenza, G.L. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell 2007, 11, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Peck, B.; Ferber, E.C.; Schulze, A. Antagonism between FOXO and MYC Regulates Cellular Powerhouse. Front. Oncol. 2013, 3, 96. [Google Scholar] [CrossRef] [PubMed]
- Tam, I.S.; Giguere, V. There and back again: The journey of the estrogen-related receptors in the cancer realm. J. Steroid Biochem. Mol. Biol. 2016, 157, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Jager, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranhotra, H.S. The orphan estrogen-related receptor alpha and metabolic regulation: New frontiers. J. Recept. Signal Transduct. Res. 2015, 35, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Perfilyev, A.; Dahlman, I.; Gillberg, L.; Rosqvist, F.; Iggman, D.; Volkov, P.; Nilsson, E.; Risérus, U.; Ling, C. Impact of polyunsaturated and saturated fat overfeeding on the DNA-methylation pattern in human adipose tissue: A randomized controlled trial. Am. J. Clin. Nutr. 2017, 105, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xu, J. DNA methyltransferases and their roles in tumorigenesis. Biomark. Res. 2017, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.M.; Barger, J.L.; Edwards, M.G.; Braun, K.H.; O'Connor, C.E.; Prolla, T.A.; Weindruch, R. Dynamic regulation of PGC-1alpha localization and turnover implicates mitochondrial adaptation in calorie restriction and the stress response. Aging Cell 2008, 7, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, K.; Yasuda, T.; Shiraishi, C.; Fujiwara, K.; Przedborski, S.; Mochizuki, H.; Yoshikawa, K. Promotion of mitochondrial biogenesis by necdin protects neurons against mitochondrial insults. Nat. Commun. 2016, 7, 10943. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Beggs, S.M.; Gildea, D.; Bupp, S.; Lichtenberg, J.; Trivedi, N.S.; NISC Comparative Sequencing Program; Hu, Y.; Bodine, D.M.; Crawford, N.P. Necdin is a breast cancer metastasis suppressor that regulates the transcription of c-Myc. Oncotarget 2015, 6, 31557–31568. [Google Scholar] [CrossRef] [PubMed]
- De Faveri, L.E.; Hurst, C.D.; Platt, F.M.; Taylor, C.F.; Roulson, J.A.; Sanchez-Carbayo, M.; Knowles, M.A.; Chapman, E.J. Putative tumour suppressor gene necdin is hypermethylated and mutated in human cancer. Br. J. Cancer 2013, 108, 1368–1377. [Google Scholar] [CrossRef] [PubMed]
- Haviland, R.; Eschrich, S.; Bloom, G.; Ma, Y.; Minton, S.; Jove, R.; Cress, W.D. Necdin, a negative growth regulator, is a novel STAT3 target gene down-regulated in human cancer. PLoS ONE 2011, 6, e24923. [Google Scholar] [CrossRef] [PubMed]
- Bohm, A.; Hoffmann, C.; Irmler, M.; Schneeweiss, P.; Schnauder, G.; Sailer, C.; Schmid, V.; Hudemann, J.; Machann, J.; Schick, F.; et al. TGF-β Contributes to Impaired Exercise Response by Suppression of Mitochondrial Key Regulators in Skeletal Muscle. Diabetes 2016, 65, 2849–2861. [Google Scholar] [CrossRef] [PubMed]
- Sohn, E.J.; Kim, J.; Hwang, Y.; Im, S.; Moon, Y.; Kang, D.M. TGF-β suppresses the expression of genes related to mitochondrial function in lung A549 cells. Cell. Mol. Biol. 2012, 58, OL1763–OL1767. [Google Scholar] [PubMed]
- Li, J.; Ke, W.; Zhou, Q.; Wu, Y.; Lou, H.; Yang, N.; Guo, Y.; Zheng, Q.; Zhang, Y. Tumour necrosis factor-α promotes liver ischaemia-reperfusion injury through the PGC-1alpha/Mfn2 pathway. J. Cell. Mol. Med. 2014, 18, 1863–1873. [Google Scholar] [CrossRef] [PubMed]
- Barroso, W.A.; Victorino, V.J.; Jeremias, I.C.; Petroni, R.C.; Ariga, S.K.K.; Salles, T.A.; Barbeiro, D.F.; de Lima, T.M.; de Souza, H.P. High-fat diet inhibits PGC-1α suppressive effect on NFκB signaling in hepatocytes. Eur. J. Nutr. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Andres, O.; Suarez-Alvarez, B.; Sanchez-Ramos, C.; Monsalve, M.; Sanchez-Niño, M.D.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A.; Sanz, A.B. The inflammatory cytokine TWEAK decreases PGC-1alpha expression and mitochondrial function in acute kidney injury. Kidney Int. 2016, 89, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, J.P.; Lazarou, M.; Dewson, G. Parkin and mitophagy in cancer. Oncogene 2017, 36, 1315–1327. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Li, Y.; Wang, X.Y.; Zhang, D.; Zhang, H.; Wu, Q.; He, Y.Q.; Wang, J.Y.; Zhang, L.; Xia, H.; et al. Circulating miR-130b mediates metabolic crosstalk between fat and muscle in overweight/obesity. Diabetologia 2013, 56, 2275–2285. [Google Scholar] [CrossRef] [PubMed]
- Lou, C.; Xiao, M.; Cheng, S.; Lu, X.; Jia, S.; Ren, Y.; Li, Z. MiR-485-3p and miR-485-5p suppress breast cancer cell metastasis by inhibiting PGC-1α expression. Cell Death Dis. 2016, 7, e2159. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Hsu, S.H.; Frankel, W.; Ghoshal, K.; Jacob, S.T. Stat3-mediated activation of microRNA-23a suppresses gluconeogenesis in hepatocellular carcinoma by down-regulating glucose-6-phosphatase and peroxisome proliferator-activated receptor gamma, coactivator 1 alpha. Hepatology 2012, 56, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Roufayel, R.; Kadry, S. Expression of miR-23a by apoptotic regulators in human cancer: A review. Cancer Biol. Ther. 2017, 18, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, X.; Liu, J.; Guo, H.; Xu, H.; Zhang, G. PGC-1 alpha interacts with microRNA-217 to functionally regulate breast cancer cell proliferation. Biomed. Pharmacother. 2017, 85, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.J.; Man, N.; Tan, Y.; Nimer, S.D.; Wang, L. The Role of Histone Acetyltransferases in Normal and Malignant Hematopoiesis. Front. Oncol. 2015, 5, 108. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.W.; Jin, H.J.; Zhao, W.; Gao, B.; Fang, J.; Wei, J.; Zhang, D.D.; Zhang, J.; Fang, D. The Histone Acetyltransferase GCN5 Expression Is Elevated and Regulated by c-Myc and E2F1 Transcription Factors in Human Colon Cancer. Gene Exp. 2015, 16, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Poulose, N.; Raju, R. Aging and injury: Alterations in cellular energetics and organ function. Aging Dis. 2014, 5, 101–108. [Google Scholar] [PubMed]
- Sczelecki, S.; Besse-Patin, A.; Abboud, A.; Kleiner, S.; Laznik-Bogoslavski, D.; Wrann, C.D.; Ruas, J.L.; Haibe-Kains, B.; Estall, J.L. Loss of Pgc-1α expression in aging mouse muscle potentiates glucose intolerance and systemic inflammation. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E157–E167. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lluch, G.; Irusta, P.M.; Navas, P.; de Cabo, R. Mitochondrial biogenesis and healthy aging. Exp. Gerontol. 2008, 43, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Luo, C.; Vazquez, F.; Puigserver, P. Targeting mitochondrial oxidative metabolism in melanoma causes metabolic compensation through glucose and glutamine utilization. Cancer Res. 2014, 74, 3535–3545. [Google Scholar] [CrossRef] [PubMed]
- Morandi, A.; Giannoni, E.; Chiarugi, P. Nutrient Exploitation within the Tumor-Stroma Metabolic Crosstalk. Trends Cancer 2016, 2, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Payen, V.L.; Baselet, B.; Sonveaux, P. Metabolic changes associated with tumor metastasis, part 2: Mitochondria, lipid and amino acid metabolism. Cell Mol. Life Sci. 2016, 73, 1349–1363. [Google Scholar] [CrossRef] [PubMed]
- Alix-Panabieres, C.; Cayrefourcq, L.; Mazard, T.; Maudelonde, T.; Assenat, E.; Assou, S. Molecular Portrait of Metastasis-Competent Circulating Tumor Cells in Colon Cancer Reveals the Crucial Role of Genes Regulating Energy Metabolism and DNA Repair. Clin. Chem. 2017, 63, 700–713. [Google Scholar] [CrossRef] [PubMed]
- Ertel, A.; Tsirigos, A.; Whitaker-Menezes, D.; Birbe, R.C.; Pavlides, S.; Martinez-Outschoorn, U.E.; Pestell, R.G.; Howell, A.; Sotgia, F.; Lisanti, M.P. Is cancer a metabolic rebellion against host aging? In the quest for immortality, tumor cells try to save themselves by boosting mitochondrial metabolism. Cell Cycle 2012, 11, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Payen, V.L.; Perez-Escuredo, J.; De Saedeleer, C.J.; Danhier, P.; Copetti, T.; Dhup, S.; Tardy, M.; Vazeille, T.; Bouzin, C.; et al. A mitochondrial switch promotes tumor metastasis. Cell Rep. 2014, 8, 754–766. [Google Scholar] [CrossRef] [PubMed]
- Feron, O. Pyruvate into lactate and back: From the Warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother. Oncol. 2009, 92, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.; Sotgia, F.; Lisanti, M.P. Tumor microenvironment and metabolic synergy in breast cancers: Critical importance of mitochondrial fuels and function. Semin. Oncol. 2014, 41, 195–216. [Google Scholar] [CrossRef] [PubMed]
- Wanet, A.; Arnould, T.; Najimi, M.; Renard, P. Connecting Mitochondria, Metabolism, and Stem Cell Fate. Stem Cells Dev. 2015, 24, 1957–1971. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Fiorillo, M.; Peiris-Pages, M.; Ozsvari, B.; Smith, D.L.; Sanchez-Alvarez, R.; Martinez-Outschoorn, U.E.; Cappello, A.R.; Pezzi, V.; Lisanti, M.P.; et al. Mitochondrial biogenesis is required for the anchorage-independent survival and propagation of stem-like cancer cells. Oncotarget 2015, 6, 14777–14795. [Google Scholar] [CrossRef] [PubMed]
- Pasto, A.; Bellio, C.; Pilotto, G.; Ciminale, V.; Silic-Benussi, M.; Guzzo, G.; Rasola, A.; Frasson, C.; Nardo, G.; Zulato, E.; et al. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget 2014, 5, 4305–4319. [Google Scholar] [CrossRef] [PubMed]
- Wintzell, M.; Lofstedt, L.; Johansson, J.; Pedersen, A.B.; Fuxe, J.; Shoshan, M. Repeated cisplatin treatment can lead to a multiresistant tumor cell population with stem cell features and sensitivity to 3-bromopyruvate. Cancer Biol. Ther. 2012, 13, 1454–1462. [Google Scholar] [CrossRef] [PubMed]
- Kulikov, A.V.; Luchkina, E.A.; Gogvadze, V.; Zhivotovsky, B. Mitophagy: Link to cancer development and therapy. Biochem. Biophys. Res. Commun. 2017, 482, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Drake, L.E.; Springer, M.Z.; Poole, L.P.; Kim, C.J.; Macleod, K.F. Expanding perspectives on the significance of mitophagy in cancer. Semin. Cancer Biol. 2017, 47, 110–124. [Google Scholar] [CrossRef] [PubMed]
- Neill, T.; Torres, A.; Buraschi, S.; Owens, R.T.; Hoek, J.B.; Baffa, R.; Iozzo, R.V. Decorin induces mitophagy in breast carcinoma cells via peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) and mitostatin. J. Biol. Chem. 2014, 289, 4952–4968. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wei, H.; Sehgal, S.A.; Liu, L.; Chen, Q. Mitophagy receptors sense stress signals and couple mitochondrial dynamic machinery for mitochondrial quality control. Free Radic. Biol. Med. 2016, 100, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhang, J.; Yu, M.; Xie, Y.; Huang, Y.; Wolff, D.W.; Abel, P.W.; Tu, Y. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 2013, 32, 4814–4824. [Google Scholar] [CrossRef] [PubMed]
- Soriano, F.X.; Liesa, M.; Bach, D.; Chan, D.C.; Palacin, M.; Zorzano, A. Evidence for a mitochondrial regulatory pathway defined by peroxisome proliferator-activated receptor-gamma coactivator-1 alpha, estrogen-related receptor-alpha, and mitofusin 2. Diabetes 2006, 55, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Martin, O.J.; Lai, L.; Soundarapandian, M.M.; Leone, T.C.; Zorzano, A.; Keller, M.P.; Attie, A.D.; Muoio, D.M.; Kelly, D.P. A role for peroxisome proliferator-activated receptor γ coactivator-1 in the control of mitochondrial dynamics during postnatal cardiac growth. Circ. Res. 2014, 114, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.P.; Schragenheim, J.; Cao, J.; Falck, J.R.; Abraham, N.G.; Bellner, L. PGC-1 alpha regulates HO-1 expression, mitochondrial dynamics and biogenesis: Role of epoxyeicosatrienoic acid. Prostaglandins Other Lipid Mediat. 2016, 125, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Doerstling, S.S.; O’Flanagan, C.H.; Hursting, S.D. Obesity and Cancer Metabolism: A Perspective on Interacting Tumor-Intrinsic and Extrinsic Factors. Front. Oncol. 2017, 7, 216. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; Spiegelman, B.M. What we talk about when we talk about fat. Cell 2014, 156, 20–44. [Google Scholar] [CrossRef] [PubMed]
- Nieman, K.M.; Romero, I.L.; Van Houten, B.; Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta 2013, 1831, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.A.; Xing, X.; Harris, J.W.; Zaytseva, Y.Y.; Mitov, M.I.; Napier, D.L.; Weiss, H.L.; Mark Evers, B.; Gao, T. Adipocytes activate mitochondrial fatty acid oxidation and autophagy to promote tumor growth in colon cancer. Cell Death Dis. 2017, 8, e2593. [Google Scholar] [CrossRef] [PubMed]
- Barres, R.; Kirchner, H.; Rasmussen, M.; Yan, J.; Kantor, F.R.; Krook, A.; Näslund, E.; Zierath, J.R. Weight loss after gastric bypass surgery in human obesity remodels promoter methylation. Cell Rep. 2013, 3, 1020–1027. [Google Scholar] [CrossRef] [PubMed]
- May-Wilson, S.; Sud, A.; Law, P.J.; Palin, K.; Tuupanen, S.; Gylfe, A.; Hänninen, U.A.; Cajuso, T.; Tanskanen, T.; Kondelin, J.; et al. Pro-inflammatory fatty acid profile and colorectal cancer risk: A Mendelian randomisation analysis. Eur. J. Cancer 2017, 84, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Iwabu, M.; Yamauchi, T.; Okada-Iwabu, M.; Sato, K.; Nakagawa, T.; Funata, M.; Yamaguchi, M.; Namiki, S.; Nakayama, R.; Tabata, M.; et al. Adiponectin and AdipoR1 regulate PGC-1alpha and mitochondria by Ca(2+) and AMPK/SIRT1. Nature 2010, 464, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- Blanquer-Rossello Mdel, M.; Oliver, J.; Sastre-Serra, J.; Valle, A.; Roca, P. Leptin regulates energy metabolism in MCF-7 breast cancer cells. Int. J. Biochem. Cell Biol. 2016, 72, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Diedrich, J.; Gusky, H.C.; Podgorski, I. Adipose tissue dysfunction and its effects on tumor metabolism. Horm. Mol. Biol. Clin. Investig. 2015, 21, 17–41. [Google Scholar]
- Lezi, E.; Swerdlow, R.H. Lactate’s effect on human neuroblastoma cell bioenergetic fluxes. Biochem. Pharmacol. 2016, 99, 88–100. [Google Scholar]
- Bartoletti-Stella, A.; Mariani, E.; Kurelac, I.; Maresca, A.; Caratozzolo, M.F.; Iommarini, L.; Carelli, V.; Eusebi, L.H.; Guido, A.; Cenacchi, G.; et al. Gamma rays induce a p53-independent mitochondrial biogenesis that is counter-regulated by HIF1α. Cell Death Dis. 2013, 4, e663. [Google Scholar] [CrossRef] [PubMed]
- Lyons, A.; Coleman, M.; Riis, S.; Favre, C.; O'Flanagan, C.H.; Zhdanov, A.V.; Papkovsky, D.B.; Hursting, S.D.; O'Connor, R. Insulin-like growth factor 1 signaling is essential for mitochondrial biogenesis and mitophagy in cancer cells. J. Biol. Chem. 2017, 292, 16983–16998. [Google Scholar] [CrossRef] [PubMed]
- Gleyzer, N.; Scarpulla, R.C. Activation of a PGC-1-related coactivator (PRC)-dependent inflammatory stress program linked to apoptosis and premature senescence. J. Biol. Chem. 2013, 288, 8004–8015. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mastropasqua, F.; Girolimetti, G.; Shoshan, M. PGC1α: Friend or Foe in Cancer? Genes 2018, 9, 48. https://doi.org/10.3390/genes9010048
Mastropasqua F, Girolimetti G, Shoshan M. PGC1α: Friend or Foe in Cancer? Genes. 2018; 9(1):48. https://doi.org/10.3390/genes9010048
Chicago/Turabian StyleMastropasqua, Francesca, Giulia Girolimetti, and Maria Shoshan. 2018. "PGC1α: Friend or Foe in Cancer?" Genes 9, no. 1: 48. https://doi.org/10.3390/genes9010048
APA StyleMastropasqua, F., Girolimetti, G., & Shoshan, M. (2018). PGC1α: Friend or Foe in Cancer? Genes, 9(1), 48. https://doi.org/10.3390/genes9010048