Molecular Network-Based Identification of Competing Endogenous RNAs in Thyroid Carcinoma

Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection and Pre-Processing
2.2. Differential Gene Expression Analysis
2.3. Construction of Gene Regulatory Network
2.4. Construction of Gene Co-Expression Network
2.5. Survival Analysis
2.6. Function Enrichment
3. Results
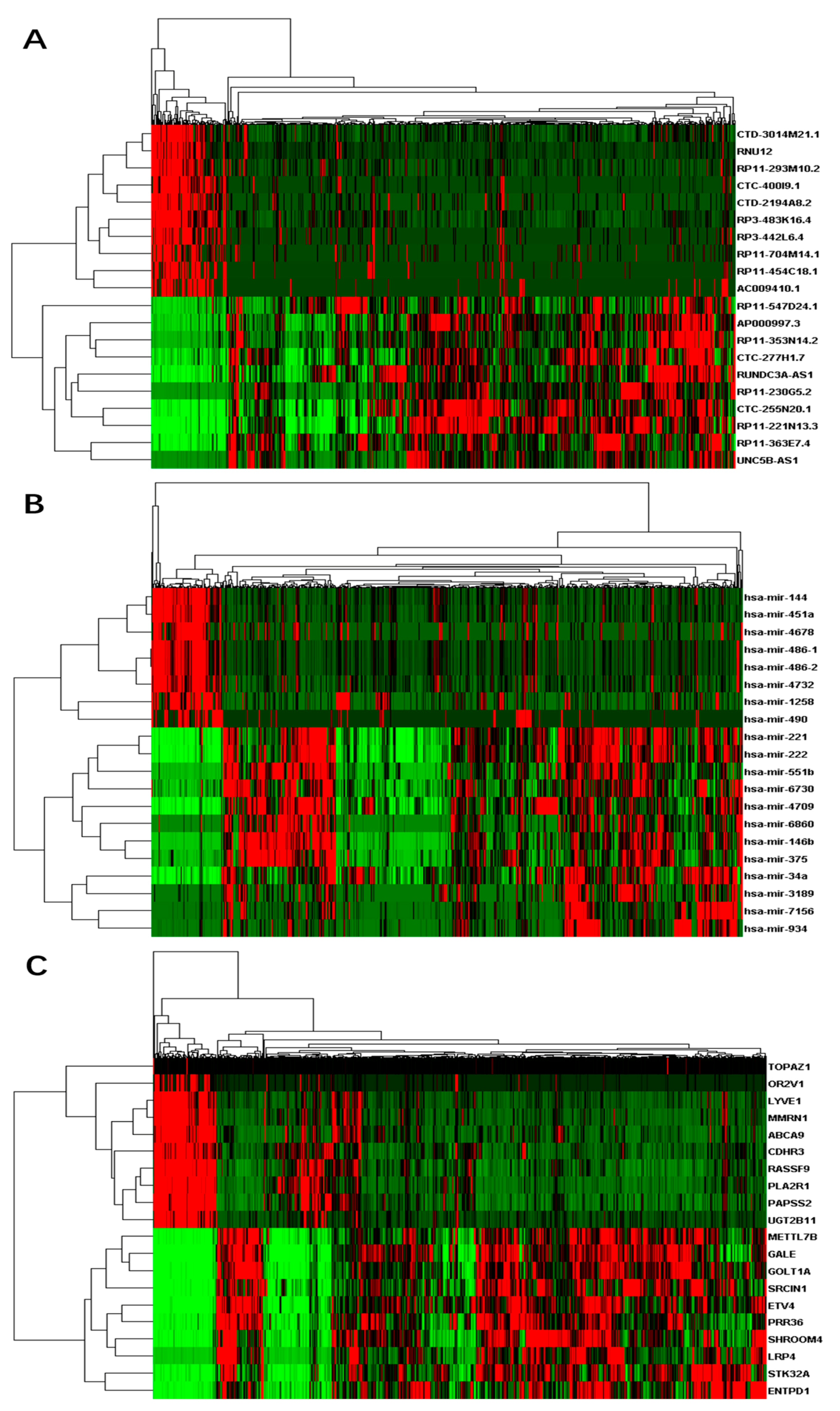
3.1. Differentially Expressed RNAs between Primary Tumor and Control Samples
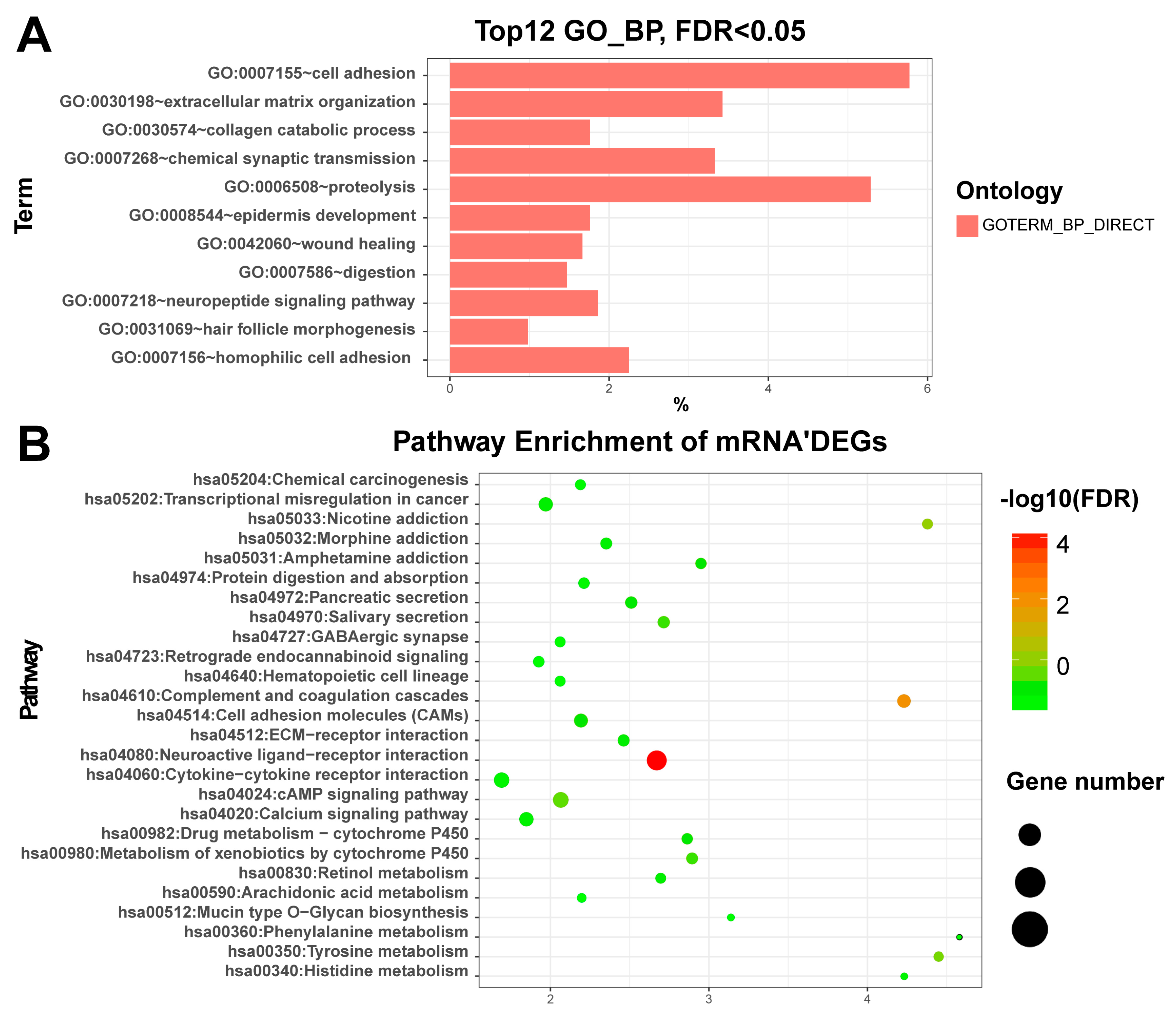
3.2. Enriched Functions of Differentially Expressed RNAs
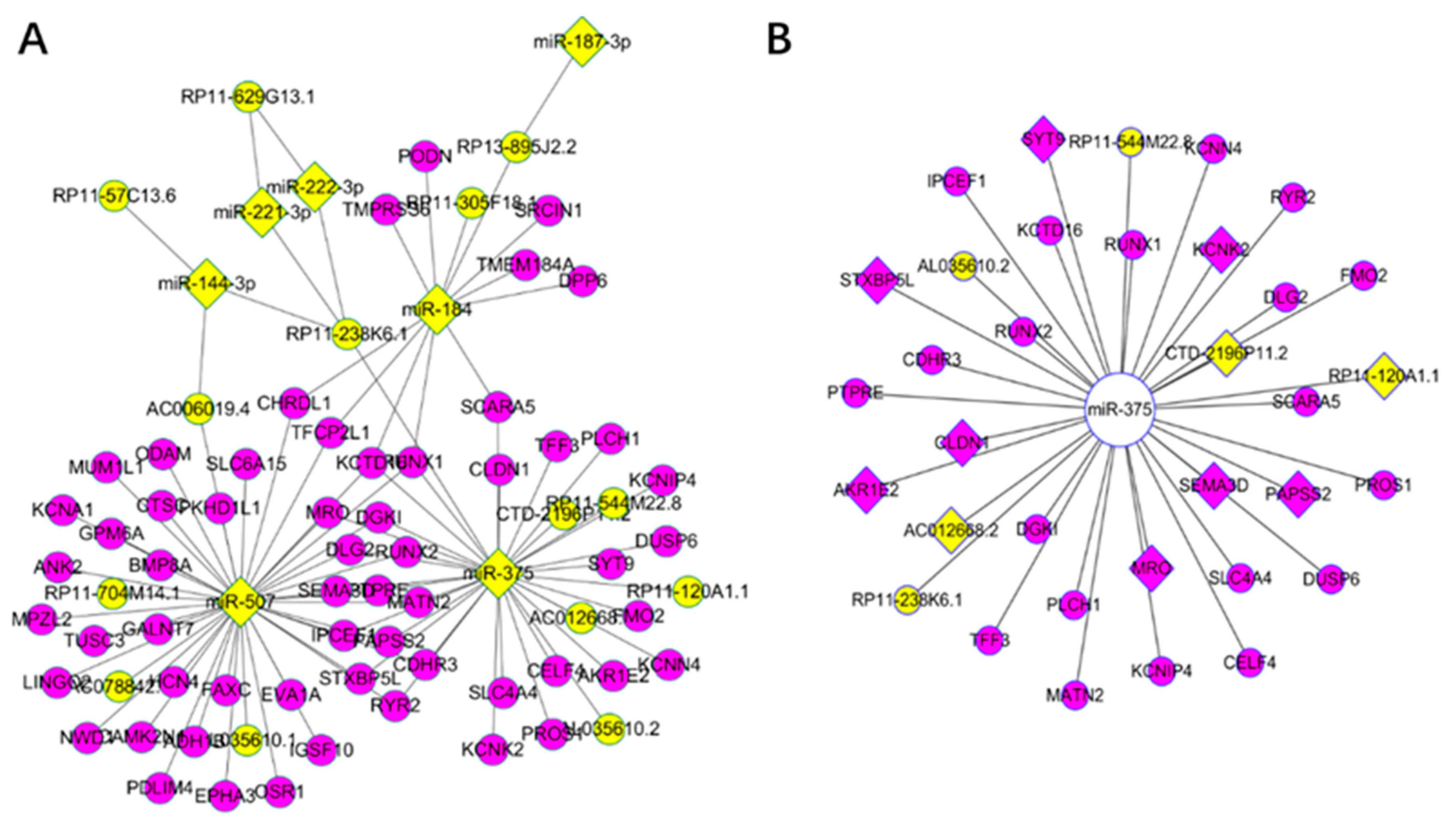
3.3. Key Driver Analysis
3.4. Competing Endogenous RNA Network Reveals Competing Endogenous Mechanisms of Long Non-Coding RNAs and Messenger RNAs
3.5. Survival Analysis of Key Driver Genes
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nikiforov, Y.E.; Nikiforova, M.N. Molecular genetics and diagnosis of thyroid cancer. Nat. Rev. Endocrinol. 2011, 7, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Livolsi, V.A. Papillary thyroid carcinoma: An update. Mod. Pathol. 2011, 24 (Suppl. 2), S1–S9. [Google Scholar] [CrossRef] [PubMed]
- Xing, M. BRAF mutation in thyroid cancer. Endocr. Relat. Cancer 2005, 12, 245–262. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Akbani, R.; Aksoy, B.A.; Ally, A.; Arachchi, H.; Asa, S.L.; Auman, J.T.; Balasundaram, M.; Balu, S.; Baylin, S.B. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Zhang, W.; Xia, Q.; Liu, F.; Li, L.; Zhao, S.; Gao, X.; Zang, C.; Ge, R.; Sun, Y. RNA sequencing identifies crucial genes in papillary thyroid carcinoma (PTC) progression. Exp. Mol. Pathol. 2016, 100, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Yapa, S.; Mulla, O.; Green, V.; England, J.; Greenman, J. The Role of Chemokines in Thyroid Carcinoma. Thyroid 2017, 27, 1347–1359. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. SEER Cancer Statistics Review 1975–2009. National Cancer Institute: Bethesda, MD, USA. Available online: http://seer.cancer.gov/archive/csr/1975_2009_pops09 (accessed on 12 December 2017).
- Udelsman, R.; Zhang, Y. The epidemic of thyroid cancer in the United States: The role of endocrinologists and ultrasounds. Thyroid 2014, 24, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Matson, D.R.; Hardin, H.; Buehler, D.; Lloyd, R.V. AKT activity is elevated in aggressive thyroid neoplasms where it promotes proliferation and invasion. Exp. Mol. Pathol. 2017, 103, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigó, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; Thurman, R.E. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799–816. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tao, Y.; Liao, Q. Long non-coding RNA: A crosslink in biological regulatory network. Brief. Bioinform. 2017. [Google Scholar] [CrossRef]
- Wang, Q.; Yang, H.; Wu, L.; Yao, J.; Meng, X.; Jiang, H.; Xiao, C.; Wu, F. Identification of Specific Long Non-Coding RNA Expression: Profile and Analysis of Association with Clinicopathologic Characteristics and BRAF Mutation in Papillary Thyroid Cancer. Thyroid 2016, 26, 1719–1732. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Hardin, H.; Chen, J.; Guo, Z.; Lloyd, R.V. Non-Coding RNAs in Thyroid Cancer. Endocr. Pathol. 2016, 27, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Nagano, T.; Fraser, P. No-nonsense functions for long non-coding RNAs. Cell 2011, 145, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Poliseno, L.; Salmena, L.; Zhang, J.; Carver, B.; Haveman, W.J.; Pandolfi, P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010, 465, 1033–1038. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H. Redefining MicroRNA Targets. Curr. Biol. 2009, 19, 870–873. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; Liao, Q.; Jiang, X.; Shao, Y.; Xiao, B.; Xi, Y.; Guo, J. Long non-coding RNA associated-competing endogenous RNAs in gastric cancer. Sci. Rep. 2014, 4, 6088. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The rosetta stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Tay, Y.; Rinn, J.; Pandolfi, P.P. The multilayered complexity of ceRNA crosstalk and competition. Nature 2014, 505, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Jens, M.; Rajewsky, N. Competition between target sites of regulators shapes post-transcriptional gene regulation. Nat. Rev. Genet. 2015, 16, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Tay, Y.; Kats, L.; Salmena, L.; Weiss, D.; Shen, M.T.; Ala, U.; Karreth, F.; Poliseno, L.; Provero, P.; Cunto, F.D. Coding-independent regulation of the tumor suppressor PTEN by competing endogenous mRNAs. Cell 2011, 147, 344–357. [Google Scholar] [CrossRef] [PubMed]
- Cesana, M.; Cacchiarelli, D.; Legnini, I.; Santini, T.; Sthandier, O.; Chinappi, M.; Tramontano, A.; Bozzoni, I. A long non-coding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 2011, 147, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Martirosyan, A.; Figliuzzi, M.; Marinari, E.; Martino, A.D. Probing the limits to microRNA-mediated control of gene expression. PLoS Comput. Biol. 2016, 12, e1004715. [Google Scholar] [CrossRef] [PubMed]
- Geng, C.; Wang, Z.; Wang, D.; Qiu, C.; Liu, M.; Xing, C.; Zhang, Q.; Yan, G.; Cui, Q. LncRNADisease: A database for long-non-coding RNA-associated diseases. Nucleic Acids Res. 2013, 41, D983. [Google Scholar]
- Li, Y.; Qiu, C.; Tu, J.; Geng, B.; Yang, J.; Jiang, T.; Cui, Q. HMDD v2.0: A database for experimentally supported human microRNA and disease associations. Nucleic Acids Res. 2014, 42, 1070–1074. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Ren, F.; Liu, C.; He, S.; Sun, G.; Gao, Q.; Yao, L.; Zhang, Y.; Miao, R.; Cao, Y. dbDEMC: A database of differentially expressed miRNAs in human cancers. BMC Genom. 2010, 11 (Suppl. 4), S5. [Google Scholar] [CrossRef] [PubMed]
- Ashwini, J.; Marks, D.S.; Erik, L. miRcode: A map of putative microRNA target sites in the long non-coding transcriptome. Bioinformatics 2012, 28, 2062–2063. [Google Scholar]
- Betel, D.; Koppal, A.; Agius, P.; Sander, C.; Leslie, C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010, 11, R90. [Google Scholar] [CrossRef] [PubMed]
- Betel, D.; Wilson, M.; Gabow, A.; Marks, D.S.; Sander, C. The microRNA.org resource: Targets and expression. Nucleic Acids Res. 2008, 36, D149. [Google Scholar] [CrossRef] [PubMed]
- Enright, A.J.; John, B.; Gaul, U.; Tuschl, T.; Sander, C.; Marks, D.S. MicroRNA targets in Drosophila. Genome Biol. 2003, 5, R1. [Google Scholar] [CrossRef] [PubMed]
- Hofacker, I.L.; Fontana, W.; Stadler, P.F.; Bonhoeffer, L.S.; Tacker, M.; Schuster, P. Fast folding and comparison of RNA secondary structures. Monatshefte für Chemie 1994, 125, 167–188. [Google Scholar] [CrossRef]
- John, B.; Enright, A.J.; Aravin, A.; Tuschl, T.; Sander, C.; Marks, D.S. Human MicroRNA targets. PLoS Biol. 2004, 2, e363. [Google Scholar] [CrossRef] [PubMed]
- Landgraf, P.; Rusu, M.; Sheridan, R.; Sewer, A.; Iovino, N.; Aravin, A.; Pfeffer, S.; Rice, A.; Kamphorst, A.O.; Landthaler, M. A Mammalian microRNA Expression Atlas Based on Small RNA Library Sequencing. Cell 2007, 129, 1401–1414. [Google Scholar] [CrossRef] [PubMed]
- Mccaskill, J.S. The equilibrium partition function and base pair binding probabilities for RNA secondary structure. Biopolymers 1990, 29, 1105–1119. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M.; Stiegler, P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res. 1981, 9, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Ghosal, S.; Sen, R.; Chakrabarti, J. lnCeDB: Database of human long non-coding RNA acting as competing endogenous RNA. PLoS ONE 2014, 9, e98965. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; Mccarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted gene co-expression network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- GDC Data Portal. Available online: http://portal.gdc.cancer.gov/projects/TCGA-THCA (accessed on 21 December 2016).
- The Cancer Genome Atlas. Available online: http://cancergenome.nih.gov/ (accessed on 12 December 2017).
- Fang, S.M.; Hu, B.L.; Zhou, Q.Z.; Yu, Q.Y.; Zhang, Z. Comparative analysis of the silk gland transcriptomes between the domestic and wild silkworms. BMC Genom. 2015, 16, 60. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling The False Discovery Rate—A Practical And Powerful Approach to Multiple Testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar]
- Zhang, B.; Zhu, J. Identification of key causal regulators in gene networks. Lect. Notes Eng. Comput. Sci. 2013, 2205, 1309–1312. [Google Scholar]
- Yang, J.; Qiu, J.; Wang, K.; Zhu, L.; Fan, J.; Zheng, D.; Meng, X.; Yang, J.; Peng, L.; Fu, Y. Using molecular functional networks to manifest connections between obesity and obesity-related diseases. Oncotarget 2017, 8, 85136–85149. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2004, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Zou, A.E.; Zheng, H.; Saad, M.A.; Rahimy, M.; Ku, J.; Kuo, S.Z.; Honda, T.K.; Wangrodriguez, J.; Xuan, Y.; Korrapati, A. The non-coding landscape of head and neck squamous cell carcinoma. Oncotarget 2016, 7, 51211–51222. [Google Scholar] [CrossRef] [PubMed]
- De Hoon, M.J.; Imoto, S.; Nolan, J.; Miyano, S. Open Source Clustering Software. Bioinformatics 2004, 20, 1453–1454. [Google Scholar] [CrossRef] [PubMed]
- Page, R.D. TREEVIEW: An application to display phylogenetic trees on personal computers. Comput. Appl. Biosci. 1996, 12, 357–358. [Google Scholar] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef] [PubMed]
- Murugan, A.K.; Munirajan, A.K.; Alzahrani, A.S. Long non-coding RNAs: Emerging players in thyroid cancer pathogenesis. Endocr. Relat. Cancer 2017. [Google Scholar] [CrossRef]
- Wu, D.; Wang, B.; Shang, J.; Song, J.; Zhang, H. miR-31 Reduces Cell Growth of Papillary Thyroid Carcinoma by RNA-Binding Protein HuR. Clin. Lab. 2015, 61, 1625–1634. [Google Scholar] [CrossRef] [PubMed]
- Amaral, P.P.; Dinger, M.E.; Mercer, T.R.; Mattick, J.S. The Eukaryotic genome as an RNA machine. Science 2008, 319, 1787–1789. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, T.; Suresh, P.S.; Tsutsumi, R. Non-coding RNAs: Functions and applications in endocrine-related cancer. Mol. Cell. Endocrinol. 2015, 416, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Vester, B.; Wengel, J. LNA (locked nucleic acid): High-affinity targeting of complementary RNA and DNA. Biochemistry 2004, 43, 13233–13244. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Hardin, H.; Montemayor-Garcia, C.; Asioli, S.; Righi, A.; Maletta, F.; Sapino, A.; Lloyd, R.V. In Situ Hybridization Analysis of miR-146b-5p and miR-21 in Thyroid Nodules: Diagnostic Implications. Endocr. Pathol. 2015, 26, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Fuziwara, C.S.; Kimura, E.T. MicroRNA deregulation in anaplastic thyroid cancer biology. Int. J. Endocrinol. 2014, 2014, 743450. [Google Scholar] [CrossRef] [PubMed]
- Visone, R.; Russo, L.; Pallante, P.; De, M.I.; Ferraro, A.; Leone, V.; Borbone, E.; Petrocca, F.; Alder, H.; Croce, C.M. MicroRNAs (miR)-221 and miR-222, both overexpressed in human thyroid papillary carcinomas, regulate p27Kip1 protein levels and cell cycle. Endocr. Relat. Cancer 2007, 14, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Pallante, P.; Battista, S.; Pierantoni, G.M.; Fusco, A. Deregulation of microRNA expression in thyroid neoplasias. Nat. Rev. Endocrinol. 2014, 10, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Chruścik, A.; Lam, A.K. Clinical pathological impacts of microRNAs in papillary thyroid carcinoma: A crucial review. Exp. Mol. Pathol. 2015, 99, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 27, 29–34. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, Y.; Chen, M.; Sun, L.; Han, J.; Elena, V.K.; Qiao, H. CXCL12 methylation-mediated epigenetic regulation of gene expression in papillary thyroid carcinoma. Sci. Rep. 2017, 7, 44033. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.K.; Chung, K.W.; Yang, S.K.; Park, M.J.; Min, H.S.; Kim, S.W.; Kang, H.S. DNA methylation of MAPK signal-inhibiting genes in papillary thyroid carcinoma. Anticancer Res. 2013, 33, 4833–4839. [Google Scholar] [PubMed]
- Du, Y.; Xia, W.; Zhang, J.; Wan, D.; Yang, Z.; Li, X. Comprehensive analysis of long non-coding RNA-mRNA co-expression patterns in thyroid cancer. Mol. Biosyst. 2017, 13, 2107–2115. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Song, M.; Song, M.K.; Kim, Y.J.; Ryu, J.C. Analysis of differentially expressed genes by Mirex ‘persistent organic pollutant’ in HepG2 cells. Toxicol. Environ. Health Sci. 2011, 3, 245–253. [Google Scholar] [CrossRef]
- Blasi, F.; Sidenius, N. The urokinase receptor: Focused cell surface proteolysis, cell adhesion and signaling. FEBS Lett. 2010, 584, 1923–1930. [Google Scholar] [CrossRef] [PubMed]
- Casey, T.M.E.J.; Crocker, A.; White, J.; Tessitore, J.; Stanley, M.; Harlow, S.; Bunn, J.Y.; Weaver, D.; Muss, H.; Plaut, K. Cancer associated fibroblasts stimulated by transforming growth factor beta1 (TGF-β1) increase invasion rate of tumor cells: A population study. Breast Cancer Res. Treat. 2008, 110, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Delys, L.; Detours, V.; Franc, B.; Thomas, G.; Bogdanova, T.; Tronko, M.; Libert, F.; Dumont, J.E.; Maenhaut, C. Gene expression and the biological phenotype of papillary thyroid carcinomas. Oncogene 2007, 26, 7894–7903. [Google Scholar] [CrossRef] [PubMed]
- Unoki, M.; Nakamura, Y. Growth-suppressive effects of BPOZ and EGR2, two genes involved in the PTEN signaling pathway. Oncogene 2001, 20, 4457–4465. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Jin, H.; Yang, Z.; Luo, G.; Lu, Y.; Li, K.; Ren, G.; Su, T.; Pan, Y.; Feng, B. MiR-150 promotes gastric cancer proliferation by negatively regulating the pro-apoptotic gene EGR2. Biochem. Biophys. Res. Commun. 2010, 392, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Meunier, D.; Patra, K.; Smits, R.; Hägebarth, A.; Lüttges, A.; Jaussi, R.; Wieduwilt, M.J.; Quintanilla-Fend, L.; Himmelbauer, H.; Fodde, R. Expression analysis of proline rich 15 (Prr15) in mouse and human gastrointestinal tumors. Mol. Carcinog. 2011, 50, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.L.L.; He, R.; Wen, D.; Deng, G.; Yang, H.; He, Y.; Ma, W.; Cai, X.; Chen, J.; Chen, G. RNA-sequencing investigation identifies an effective risk score generated by three novel lncRNAs for the survival of papillary thyroid cancer patients. Oncotarget 2017, 8, 74139–74158. [Google Scholar] [CrossRef] [PubMed]
- Huarte, M. The emerging role of lncRNAs in cancer. Nat. Med. 2015, 21, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Li, H.M.; Yang, H.; Wen, D.Y.; Luo, Y.H.; Liang, C.Y.; Pan, D.H.; Ma, W.; Chen, G.; He, Y.; Chen, J.Q. Overexpression of LncRNA HOTAIR is Associated with Poor Prognosis in Thyroid Carcinoma: A Study Based on TCGA and GEO Data. Horm. Metab. Res. 2017, 49, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Hudson, J.; Duncavage, E.; Tamburrino, A.; Salerno, P.; Xi, L.; Raffeld, M.; Moley, J.; Chernock, R.D. Overexpression of mir-10a and mir-375 and downregulation of yap1 in medullary thyroid carcinoma. Exp. Mol. Pathol. 2013, 95, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Mian, C.; Pennelli, G.; Fassan, M.; Balistreri, M.; Barollo, S.; Cavedon, E.; Galuppini, F.; Pizzi, M.; Vianello, F.; Pelizzo, M.R. MicroRNA profiles in familial and sporadic medullary thyroid carcinoma: Preliminary relationships with ret status and outcome. Thyroid 2012, 22, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Gundara, J.S.; Zhao, J.T.; Gill, A.J.; Clifton-Bligh, R.; Robinson, B.G.; Delbridge, L.; Sidhu, S.B. Nodal metastasis microRNA expression correlates with the primary tumour in MTC. ANZ J. Surg. 2012, 84, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Lassalle, S.; Zangari, J.; Popa, A.; Ilie, M.; Hofman, V.; Long, E.; Patey, M.; Tissier, F.; Belléannée, G.; Trouette, H. MicroRNA-375/SEC23A as biomarkers of the in vitro efficacy of vandetanib. Oncotarget 2016, 7, 30461–30478. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Zhao, S.M.; Luo, Y.; Zhang, A.W.; Wei, L.H.; Xie, Z.Y.; Li, Y.Y.; Ma, W. Mir-375: A prospective regulator in medullary thyroid cancer based on microarray data and bioinformatics analyses. Pathol. Res. Pract. 2017, 213, 1344–1354. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Z.; Hang, Y.K.; Liu, J.B.; Hou, Y.Q.; Wang, N.; Wang, M.J. Over-expression of microRNA-375 inhibits papillary thyroid carcinoma cell proliferation and induces cell apoptosis by targeting ERBB2. J. Pharmacol. Sci. 2016, 130, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Dettmer, M.; Perren, A.; Moch, H.; Komminoth, P.; Nikiforov, Y.E.; Nikiforova, M.N. Comprehensive microRNA expression profiling identifies novel markers in follicular variant of papillary thyroid carcinoma. Thyroid 2013, 23, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- Sabrina, L.B.; Sophie, D.; Arnaud, C.; Chloé, B.; Franck, A.; Gilles, T.; Gilles, L.; Sepideh, S.; Isabelle, B.; Pond, A.L. Microarray analysis reveals complex remodeling of cardiac ion channel expression with altered thyroid status: Relation to cellular and integrated electrophysiology. Circ. Res. 2003, 92, 234–242. [Google Scholar]
- Zhu, J.; Zhang, Y.; Zhang, W.; Zhang, W.; Fan, L.; Wang, L.; Liu, Y.; Liu, S.; Guo, Y.; Wang, Y. MicroRNA-142-5p contributes to Hashimoto’s thyroiditis by targeting CLDN1. J. Transl. Med. 2016, 14, 166. [Google Scholar] [CrossRef] [PubMed]
- Gomezrueda, H.; Palacioscorona, R.; Gutiérrezhermosillo, H.; Trevino, V. A robust biomarker of differential correlations improves the diagnosis of cytologically indeterminate thyroid cancers. Int. J. Mol. Med. 2016, 37, 1355–1362. [Google Scholar] [CrossRef] [PubMed]
- Sass, S.; Dietmann, S.; Burk, U.; Brabletz, S.; Lutter, D.; Kowarsch, A.; Mayer, K.F.; Brabletz, T.; Ruepp, A.; Theis, F. MicroRNAs coordinately regulate protein complexes. BMC Syst. Biol. 2011, 5, 136. [Google Scholar] [CrossRef] [PubMed]
- Cockburn, J.G.; Richardson, D.S.; Gujral, T.S.; Mulligan, L.M. Ret-mediated cell adhesion and migration require multiple integrin subunits. J. Clin. Endocrinol. Metab. 2010, 95, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Rath, G.M.; Schneider, C.; Dedieu, S.; Rothhut, B.; Soula-Rothhut, M.; Ghoneim, C.; Sid, B.; Morjani, H.; Btaouri, H.E.; Martiny, L. The C-terminal CD47/IAP-binding domain of thrombospondin-1 prevents camptothecin- and doxorubicin-induced apoptosis in human thyroid carcinoma cells. Biochim. Biophys. Acta Mol. Cell Res. 2006, 1763, 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Figliuzzi, M.; Marinari, E.; De, M.A. MicroRNAs as a selective channel of communication between competing RNAs: A steady-state theory. Biophys. J. 2013, 104, 1203–1213. [Google Scholar] [CrossRef] [PubMed]
- Bosson, A.D.; Zamudio, J.R.; Sharp, P.A. Endogenous miRNA and target concentrations determine susceptibility to potential ceRNA competition. Mol. Cell 2014, 56, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Denzler, R.; Agarwal, V.; Stefano, J.; Bartel, D.P.; Stoffel, M. Assessing the ceRNA hypothesis with quantitative measurements of miRNA and target abundance. Mol. Cell 2014, 54, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Figliuzzi, M.; De, M.A.; Marinari, E. RNA-based regulation: Dynamics and response to perturbations of competing RNAs. Biophys. J. 2014, 107, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Sumazin, P.; Yang, X.; Chiu, H.S.; Chung, W.J.; Iyer, A.; Llobet-Navas, D.; Rajbhandari, P.; Bansal, M.; Guarnieri, P.; Silva, J.; et al. An extensive microRNA-mediated network of RNA-RNA interactions regulates established oncogenic pathways in glioblastoma. Cell 2011, 147, 370–381. [Google Scholar] [CrossRef] [PubMed]
- Bosia, C.; Pagnani, A.; Zecchina, R. Modelling competing endogenous RNA networks. PLoS ONE 2013, 8, e66609. [Google Scholar] [CrossRef] [PubMed]
- Martirosyan, A.; Marsili, M.; De, M.A. Translating ceRNA susceptibilities into correlation functions. Biophys. J. 2017, 113, 206–213. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Numbers | |
|---|---|---|
| Sample type | Primary tumor | 501 |
| Solid tissue normal | 58 | |
| Age | Median | 47 |
| Range [years] | 15~89 | |
| Sex | Male | 152 |
| Female | 407 | |
| Vital status | Alive | 539 |
| Dead | 20 | |
| Stage | I | 315 |
| II | 59 | |
| III | 124 | |
| IV | 2 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, M.; Xu, X.; Xi, B.; Dai, Q.; Li, C.; Su, L.; Zhou, X.; Tang, M.; Yao, Y.; Yang, J. Molecular Network-Based Identification of Competing Endogenous RNAs in Thyroid Carcinoma. Genes 2018, 9, 44. https://doi.org/10.3390/genes9010044
Lu M, Xu X, Xi B, Dai Q, Li C, Su L, Zhou X, Tang M, Yao Y, Yang J. Molecular Network-Based Identification of Competing Endogenous RNAs in Thyroid Carcinoma. Genes. 2018; 9(1):44. https://doi.org/10.3390/genes9010044
Chicago/Turabian StyleLu, Minjia, Xingyu Xu, Baohang Xi, Qi Dai, Chenli Li, Li Su, Xiaonan Zhou, Min Tang, Yuhua Yao, and Jialiang Yang. 2018. "Molecular Network-Based Identification of Competing Endogenous RNAs in Thyroid Carcinoma" Genes 9, no. 1: 44. https://doi.org/10.3390/genes9010044
APA StyleLu, M., Xu, X., Xi, B., Dai, Q., Li, C., Su, L., Zhou, X., Tang, M., Yao, Y., & Yang, J. (2018). Molecular Network-Based Identification of Competing Endogenous RNAs in Thyroid Carcinoma. Genes, 9(1), 44. https://doi.org/10.3390/genes9010044