NF-kappaB: Two Sides of the Same Coin



{kind=link}
{kind=link}
{kind=link}
Abstract
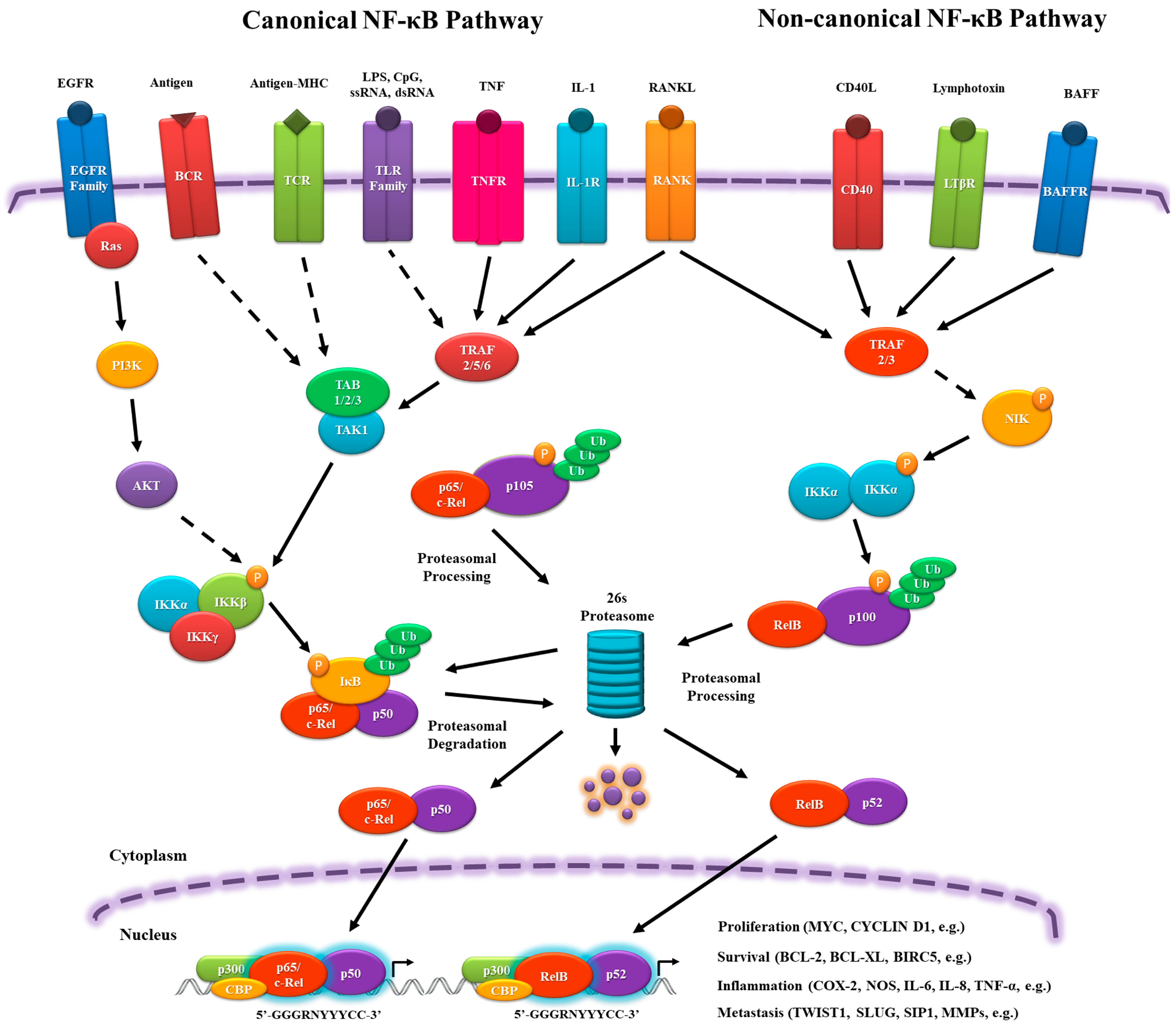
1. Evolutionary Origin of Nuclear Factor-kappa B Family and Its Signaling Pathway
2. The Role of NF-κB in Physiology, Inflammation and Cancer
Adaptive immunity
Innate Immunity
3. Role of NF-κB in Hematological Malignancies
4. Bacteria, NF-κB and Gastric Cancer
5. NF-κB and Breast Cancer
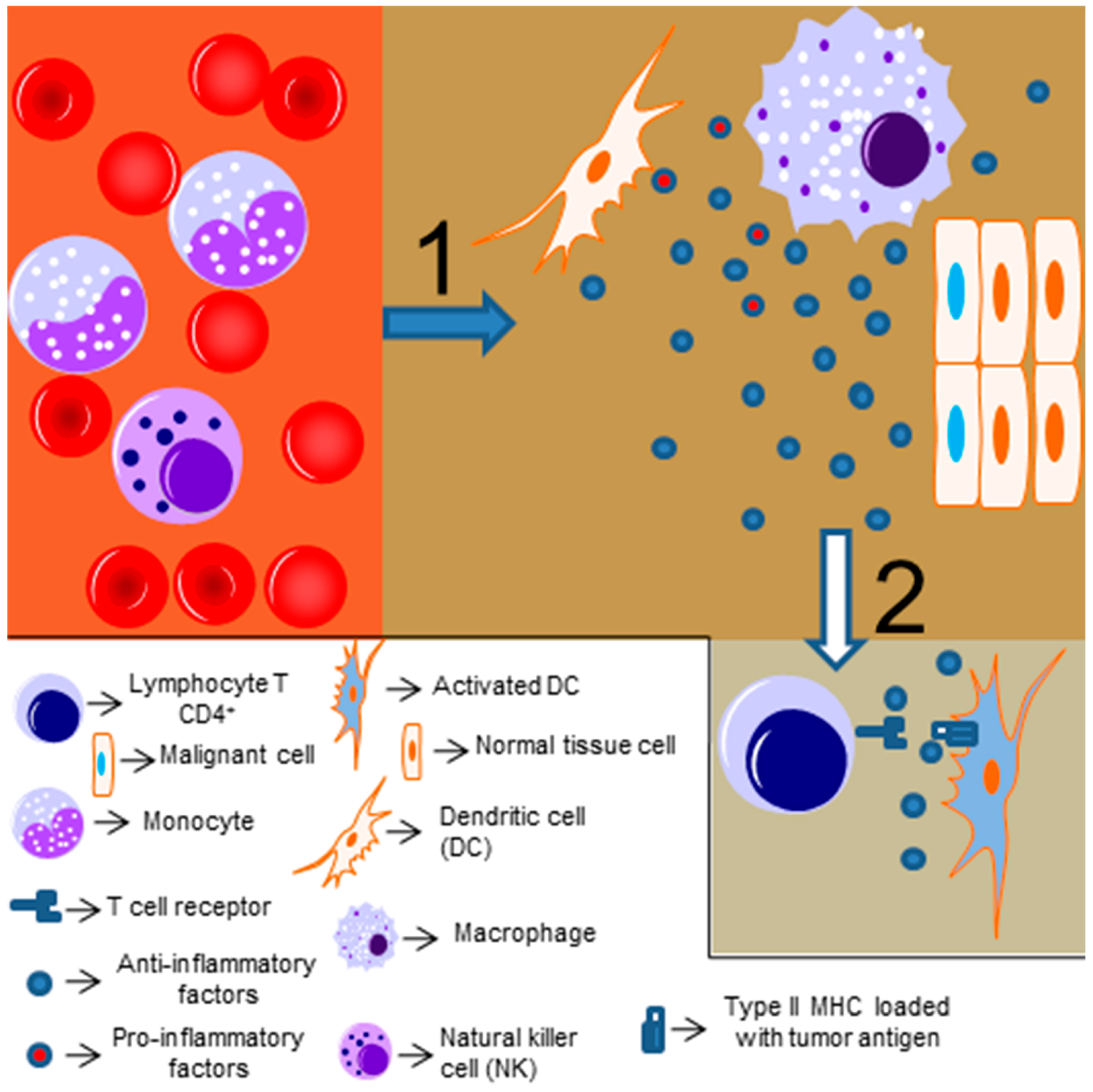
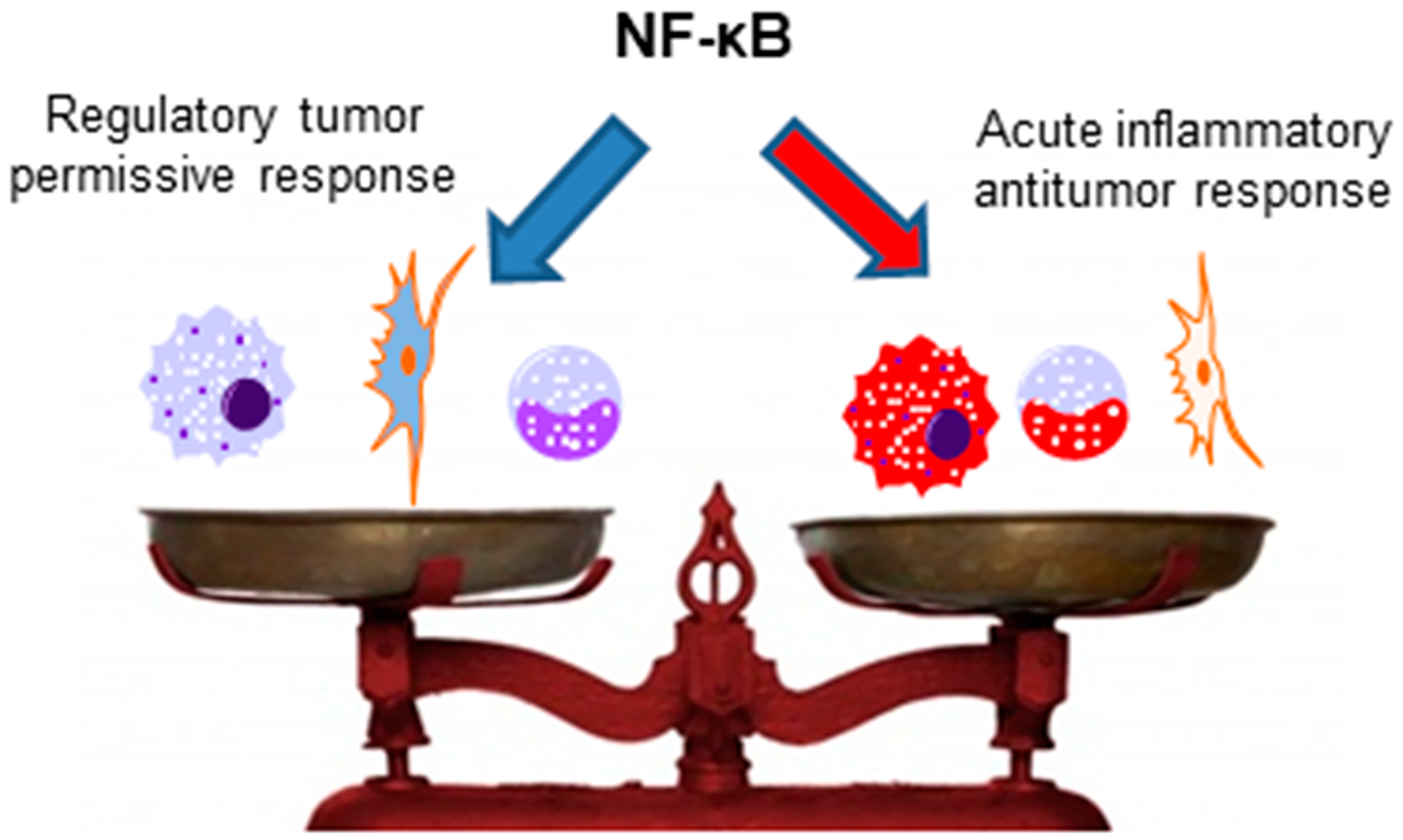
6. NF-κB and Antitumor Immune Responses
7. NF-κB and Perspectives for Therapy
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Roth, S.; Stein, D.; Nusslein-Volhard, C. A gradient of nuclear localization of the dorsal protein determines dorsoventral pattern in the drosophila embryo. Cell 1989, 59, 1189–1202. [Google Scholar] [CrossRef]
- Rushlow, C.A.; Han, K.; Manley, J.L.; Levine, M. The graded distribution of the dorsal morphogen is initiated by selective nuclear transport in drosophila. Cell 1989, 59, 1165–1177. [Google Scholar] [CrossRef]
- Steward, R. Relocalization of the dorsal protein from the cytoplasm to the nucleus correlates with its function. Cell 1989, 59, 1179–1188. [Google Scholar] [CrossRef]
- Lemaitre, B.; Meister, M.; Govind, S.; Georgel, P.; Steward, R.; Reichhart, J.M.; Hoffmann, J.A. Functional analysis and regulation of nuclear import of dorsal during the immune response in drosophila. EMBO J. 1995, 14, 536–545. [Google Scholar] [PubMed]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D.; Garbati, M.R. Inhibition of NF-κB signaling as a strategy in disease therapy. Curr. Top. Microbiol. 2011, 349, 245–263. [Google Scholar]
- Huang, D.B.; Huxford, T.; Chen, Y.Q.; Ghosh, G. The role of DNA in the mechanism of NFκB dimer formation: Crystal structures of the dimerization domains of the p50 and p65 subunits. Structure 1997, 5, 1427–1436. [Google Scholar] [CrossRef]
- Huang, D.B.; Vu, D.; Ghosh, G. NF-κB RelB forms an intertwined homodimer. Structure 2005, 13, 1365–1373. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Sundaram, C.; Reuter, S.; Aggarwal, B.B. Inhibiting NF-κB activation by small molecules as a therapeutic strategy. Biochim. Biophys. Acta 2010, 1799, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Yin, Q.; Wu, H. Structural studies of NF-κB signaling. Cell Res. 2011, 21, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, Z.J. Regulation of NF-κB by ubiquitination. Curr. Opin. Immunol. 2013, 25, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-κB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [PubMed]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C. Non-canonical NF-κB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Bandarra, D.; Biddlestone, J.; Mudie, S.; Muller, H.A.; Rocha, S. HIF-1α restricts NF-κB-dependent gene expression to control innate immunity signals. Dis. Models Mech. 2015, 8, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Dan, H.C.; Cooper, M.J.; Cogswell, P.C.; Duncan, J.A.; Ting, J.P.; Baldwin, A.S. AKT-dependent regulation of NF-κB is controlled by MTOR and raptor in association with IKK. Genes Dev. 2008, 22, 1490–1500. [Google Scholar] [CrossRef] [PubMed]
- Lam, L.T.; Wright, G.; Davis, R.E.; Lenz, G.; Farinha, P.; Dang, L.; Chan, J.W.; Rosenwald, A.; Gascoyne, R.D.; Staudt, L.M. Cooperative signaling through the signal transducer and activator of transcription 3 and nuclear factor-κB pathways in subtypes of diffuse large B-cell lymphoma. Blood 2008, 111, 3701–3713. [Google Scholar] [CrossRef] [PubMed]
- Moscat, J.; Diaz-Meco, M.T.; Rennert, P. NF-κB activation by protein kinase C isoforms and B-cell function. EMBO Rep. 2003, 4, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; Zazzeroni, F.; Bubici, C.; Jayawardena, S.; Alvarez, K.; Matsuda, S.; Nguyen, D.U.; Pham, C.G.; Nelsbach, A.H.; Melis, T.; et al. Gadd45 beta mediates the NF-kappa B suppression of JNK signalling by targeting MKK7/JNKK2. Nat. Cell Biol. 2004, 6, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Tafani, M.; Pucci, B.; Russo, A.; Schito, L.; Pellegrini, L.; Perrone, G.A.; Villanova, L.; Salvatori, L.; Ravenna, L.; Petrangeli, E.; et al. Modulators of HIF1α and NFkB in cancer treatment: Is it a rational approach for controlling malignant progression? Front. Pharmacol. 2013, 4, 13. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, C.A.; Singh, S.; Windle, B.; Sankala, H.M.; Graves, P.R.; Andrew Yeudall, W.; Deb, S.P.; Deb, S. P53 mutants induce transcription of NF-κB2 in H1299 cells through CBP and stat binding on the NF-κB2 promoter and gain of function activity. Arch. Biochem. Biophys. 2012, 518, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Shan, C.; Kong, G.; Du, Y.; Ye, L.; Zhang, X. MicroRNA-520e suppresses growth of hepatoma cells by targeting the NF-κB-inducing kinase (NIK). Oncogene 2012, 31, 3607–3620. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [PubMed]
- Castro-Alcaraz, S.; Miskolci, V.; Kalasapudi, B.; Davidson, D.; Vancurova, I. Nf-kappa B regulation in human neutrophils by nuclear I kappa B alpha: Correlation to apoptosis. J. Immunol. 2002, 169, 3947–3953. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Cheng, S.; Pear, W.S.; Liou, H.C. NF-kB inhibitor blocks B cell development at two checkpoints. Med. Immunol. 2004, 3, 1. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gerondakis, S.; Siebenlist, U. Roles of the NF-kappaB pathway in lymphocyte development and function. Cold Spring Harb. Perspect. Biol. 2010, 2, a000182. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.; Chilvers, E.R.; Lawson, M.F.; Pryde, J.G.; Fujihara, S.; Farrow, S.N.; Haslett, C.; Rossi, A.G. NF-kappaB activation is a critical regulator of human granulocyte apoptosis in vitro. J. Biol. Chem. 1999, 274, 4309–4318. [Google Scholar] [CrossRef] [PubMed]
- Wright, H.L.; Chikura, B.; Bucknall, R.C.; Moots, R.J.; Edwards, S.W. Changes in expression of membrane TNF, NF-{kappa}B activation and neutrophil apoptosis during active and resolved inflammation. Ann. Rheum. Dis. 2011, 70, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Wang, X.; Jameson, S.C.; Hogquist, K.A. Late stages of T cell maturation in the thymus involve NF-κB and tonic type I interferon signaling. Nat. Immunol. 2016, 17, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.F.; Mitchell, A.; Li, G.; Ding, S.; Fitzmaurice, A.M.; Ryan, K.; Crowe, S.; Goldberg, J.B. Toll-like receptor (TLR) 2 and TLR5, but not TLR4, are required for Helicobacter pylori-induced NF-κB activation and chemokine expression by epithelial cells. J. Biol. Chem. 2003, 278, 32552–32560. [Google Scholar] [CrossRef] [PubMed]
- Bouchery, T.; Kyle, R.; Ronchese, F.; Le Gros, G. The differentiation of CD4(+) T-helper cell subsets in the context of helminth parasite infection. Front. Immunol. 2014, 5, 487. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Lanzavecchia, A. The instructive role of dendritic cells on T-cell responses. Arthritis Res. 2002, 4, S127–S132. [Google Scholar] [CrossRef] [PubMed]
- Luckheeram, R.V.; Zhou, R.; Verma, A.D.; Xia, B. CD4+ T cells: Differentiation and functions. Clin. Dev. Immunol. 2012, 2012, 925135. [Google Scholar] [CrossRef] [PubMed]
- Corn, R.A.; Aronica, M.A.; Zhang, F.; Tong, Y.; Stanley, S.A.; Kim, S.R.; Stephenson, L.; Enerson, B.; McCarthy, S.; Mora, A.; et al. T cell-intrinsic requirement for NF-kappa B induction in postdifferentiation IFN-gamma production and clonal expansion in a Th1 response. J. Immunol. 2003, 171, 1816–1824. [Google Scholar] [CrossRef] [PubMed]
- Das, J.; Chen, C.H.; Yang, L.; Cohn, L.; Ray, P.; Ray, A. A critical role for NF-kappa B in Gata3 expression and Th2 differentiation in allergic airway inflammation. Nat. Immunol. 2001, 2, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Early, S.B.; Huyett, P.; Brown-Steinke, K.; Borish, L.; Steinke, J.W. Functional analysis of −351 interleukin-9 promoter polymorphism reveals an activator controlled by NF-κB. Genes Immun. 2009, 10, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Li-Weber, M.; Giaisi, M.; Baumann, S.; Palfi, K.; Krammer, P.H. NF-kappa B synergizes with NF-AT and NF-IL6 in activation of the IL-4 gene in T cells. Eur. J. Immunol. 2004, 34, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Q.; Kameswaran, V.; Tone, Y.; Li, L.; Liou, H.C.; Greene, M.I.; Tone, M.; Chen, Y.H. Development of Foxp3(+) regulatory T cells is driven by the c-Rel enhanceosome. Immunity 2009, 31, 932–940. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Q.; Kameswaran, V.; Zhang, Y.; Zheng, S.; Sun, J.; Wang, J.; DeVirgiliis, J.; Liou, H.C.; Beg, A.A.; Chen, Y.H. The Th17 immune response is controlled by the Rel-RORγ-RORγ T transcriptional axis. J. Exp. Med. 2011, 208, 2321–2333. [Google Scholar] [CrossRef] [PubMed]
- Gerondakis, S.; Fulford, T.S.; Messina, N.L.; Grumont, R.J. NF-κB control of T cell development. Nat. Immunol. 2014, 15, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Alcamo, E.; Hacohen, N.; Schulte, L.C.; Rennert, P.D.; Hynes, R.O.; Baltimore, D. Requirement for the NF-κB family member rela in the development of secondary lymphoid organs. J. Exp. Med. 2002, 195, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. NF-κB in immunobiology. Cell Res. 2011, 21, 223–244. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Ghosh, S. NF-κB: Roles and regulation in different CD4(+) T-cell subsets. Immunol. Rev. 2013, 252, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Mondor, I.; Schmitt-Verhulst, A.M.; Guerder, S. Rela regulates the survival of activated effector CD8 T cells. Cell Death Differ. 2005, 12, 1398–1406. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.L.; Krappmann, D. Controlling NF-κB activation in T cells by costimulatory receptors. Cell Death Differ. 2006, 13, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Ebner, K.; Bandion, A.; Binder, B.R.; de Martin, R.; Schmid, J.A. GMCSF activates NF-κB via direct interaction of the GMCSF receptor with IkappaB kinase beta. Blood 2003, 102, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Gerondakis, S.; Grossmann, M.; Nakamura, Y.; Pohl, T.; Grumont, R. Genetic approaches in mice to understand Rel/NF-κB and ikappab function: Transgenics and knockouts. Oncogene 1999, 18, 6888–6895. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [PubMed]
- Bouffi, C.; Rochman, M.; Zust, C.B.; Stucke, E.M.; Kartashov, A.; Fulkerson, P.C.; Barski, A.; Rothenberg, M.E. IL-33 markedly activates murine eosinophils by an NF-κB-dependent mechanism differentially dependent upon an IL-4-driven autoinflammatory loop. J. Immunol. 2013, 191, 4317–4325. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.K.; Wang, C.B.; Ip, W.K.; Tian, Y.P.; Lam, C.W. Role of p38 MAPK and NF-kB for chemokine release in coculture of human eosinophils and bronchial epithelial cells. Clin. Exp. Immunol. 2005, 139, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Tato, C.M.; Mason, N.; Artis, D.; Shapira, S.; Caamano, J.C.; Bream, J.H.; Liou, H.C.; Hunter, C.A. Opposing roles of NF-κB family members in the regulation of NK cell proliferation and production of IFN-gamma. Int. Immunol. 2006, 18, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Yatim, N.; Jusforgues-Saklani, H.; Orozco, S.; Schulz, O.; Barreira da Silva, R.; Reis e Sousa, C.; Green, D.R.; Oberst, A.; Albert, M.L. Ripk1 and NF-κB signaling in dying cells determines cross-priming of CD8(+) T cells. Science 2015, 350, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Vakkila, J.; Demarco, R.A.; Lotze, M.T. Coordinate NF-κB and STAT1 activation promotes development of myeloid type 1 dendritic cells. Scand. J. Immunol. 2008, 67, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Saccani, A.; Schioppa, T.; Porta, C.; Biswas, S.K.; Nebuloni, M.; Vago, L.; Bottazzi, B.; Colombo, M.P.; Mantovani, A.; Sica, A. P50 nuclear factor-kappaB overexpression in tumor-associated macrophages inhibits M1 inflammatory responses and antitumor resistance. Cancer Res. 2006, 66, 11432–11440. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Wang, Y.; Yan, F.; Zhang, P.; Li, H.; Zhao, H.; Yan, C.; Yan, F.; Ren, X. Noncanonical NF-κB activation mediates STAT3-stimulated ido upregulation in myeloid-derived suppressor cells in breast cancer. J. Immunol. 2014, 193, 2574–2586. [Google Scholar] [CrossRef] [PubMed]
- Hallam, S.; Escorcio-Correia, M.; Soper, R.; Schultheiss, A.; Hagemann, T. Activated macrophages in the tumour microenvironment-dancing to the tune of TLR and NF-κB. J. Pathol. 2009, 219, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Langereis, J.D.; Raaijmakers, H.A.; Ulfman, L.H.; Koenderman, L. Abrogation of NF-κB signaling in human neutrophils induces neutrophil survival through sustained p38-MAPK activation. J. Leukoc. Biol. 2010, 88, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Virchow, R. Cellular pathology. As based upon physiological and pathological histology. Lecture XVI--Atheromatous affection of arteries. 1858. Nutr. Rev. 1989, 47, 23–25. [Google Scholar] [CrossRef] [PubMed]
- Budhu, S.; Wolchok, J.; Merghoub, T. The importance of animal models in tumor immunity and immunotherapy. Curr. Opin. Genet. Dev. 2014, 24, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Kienle, G.S. Fever in cancer treatment: Coley’s therapy and epidemiologic observations. Glob. Adv. Health Med. 2012, 1, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Korniluk, A.; Koper, O.; Kemona, H.; Dymicka-Piekarska, V. From inflammation to cancer. Ir. J. Med. Sci. 2017, 186, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Schetter, A.J.; Heegaard, N.H.; Harris, C.C. Inflammation and cancer: Interweaving microRNA, free radical, cytokine and p53 pathways. Carcinogenesis 2010, 31, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [PubMed]
- Beug, H.; Muller, H.; Grieser, S.; Doederlein, G.; Graf, T. Hematopoietic cells transformed in vitro by REVT avian reticuloendotheliosis virus express characteristics of very immature lymphoid cells. Virology 1981, 115, 295–309. [Google Scholar] [CrossRef]
- Wilhelmsen, K.C.; Eggleton, K.; Temin, H.M. Nucleic acid sequences of the oncogene v-rel in reticuloendotheliosis virus strain T and its cellular homolog, the proto-oncogene c-rel. J. Virol. 1984, 52, 172–182. [Google Scholar] [PubMed]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Bours, V.; Bentires-Alj, M.; Hellin, A.C.; Viatour, P.; Robe, P.; Delhalle, S.; Benoit, V.; Merville, M.P. Nuclear factor-kappa B, cancer, and apoptosis. Biochem. Pharmacol. 2000, 60, 1085–1089. [Google Scholar] [CrossRef]
- Karin, M. Nuclear factor-kappaB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.T.; Kral, J.G. The NF-kappaB/IkappaB signaling system: A molecular target in breast cancer therapy. J. Surg. Res. 2005, 123, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Guzman, M.L.; Neering, S.J.; Upchurch, D.; Grimes, B.; Howard, D.S.; Rizzieri, D.A.; Luger, S.M.; Jordan, C.T. Nuclear factor-κB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood 2001, 98, 2301–2307. [Google Scholar] [CrossRef] [PubMed]
- Kordes, U.; Krappmann, D.; Heissmeyer, V.; Ludwig, W.; Scheidereit, C. Transcription factor NF-[kappa] B is constitutively activated in acute lymphoblastic leukemia cells. Leukemia 2000, 14, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Azran, I.; Schavinsky-Khrapunsky, Y.; Aboud, M. Role of tax protein in human T-cell leukemia virus type-I leukemogenicity. Retrovirology 2004, 1, 20. [Google Scholar] [CrossRef] [PubMed]
- Harhaj, E.W.; Harhaj, N.S. Mechanisms of persistent NF-κB activation by HTLV-I tax. IUBMB Life 2005, 57, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Sun, S.C. Retroviral oncoprotein tax deregulates NF-κB by activating Tak1 and mediating the physical association of Tak1–IKK. EMBO Rep. 2007, 8, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Pujari, R.; Hunte, R.; Thomas, R.; van der Weyden, L.; Rauch, D.; Ratner, L.; Nyborg, J.K.; Ramos, J.C.; Takai, Y.; Shembade, N. Human T-cell leukemia virus type 1 (HTLV-1) tax requires CADM1/TSLC1 for inactivation of the NF-κB inhibitor A20 and constitutive NF-κB signaling. PLoS Pathog. 2015, 11, e1004721. [Google Scholar] [CrossRef] [PubMed]
- Bargou, R.; Leng, C.; Krappmann, D.; Emmerich, F.; Mapara, M.; Bommert, K.; Royer, H.; Scheidereit, C.; Dorken, B. High-level nuclear NF-kappa B and Oct-2 is a common feature of cultured hodgkin/reed-sternberg cells. Blood 1996, 87, 4340–4347. [Google Scholar] [PubMed]
- Krappmann, D.; Emmerich, F.; Kordes, U.; Scharschmidt, E.; Dorken, B.; Scheidereit, C. Molecular mechanisms of constitutive NF-kB/Rel activation in Hodgkin/Reed-Sternberg cells. Oncogene 1999, 18, 943–954. [Google Scholar] [CrossRef] [PubMed]
- Cabannes, E.; Khan, G.; Aillet, F.; Jarrett, R.F.; Hay, R.T. Mutations in the IkBa gene in Hodgkin’s disease suggest a tumour suppressor role for IkappaBalpha. Oncogene 1999, 18, 3063–3070. [Google Scholar] [CrossRef] [PubMed]
- Bargou, R.C.; Emmerich, F.; Krappmann, D.; Bommert, K.; Mapara, M.Y.; Arnold, W.; Royer, H.D.; Grinstein, E.; Greiner, A.; Scheidereit, C. Constitutive nuclear factor-kappaB-RelA activation is required for proliferation and survival of Hodgkin’s disease tumor cells. J. Clin. Investig. 1997, 100, 2961–2969. [Google Scholar] [CrossRef] [PubMed]
- Barth, T.F.; Martin-Subero, J.I.; Joos, S.; Menz, C.K.; Hasel, C.; Mechtersheimer, G.; Parwaresch, R.M.; Lichter, P.; Siebert, R.; Möller, P. Gains of 2p involving the REL locus correlate with nuclear c-Rel protein accumulation in neoplastic cells of classical Hodgkin lymphoma. Blood 2003, 101, 3681–3686. [Google Scholar] [CrossRef] [PubMed]
- Mathas, S.; Jöhrens, K.; Joos, S.; Lietz, A.; Hummel, F.; Janz, M.; Jundt, F.; Anagnostopoulos, I.; Bommert, K.; Lichter, P. Elevated NF-κB p50 complex formation and Bcl-3 expression in classical hodgkin, anaplastic large-cell, and other peripheral T-cell lymphomas. Blood 2005, 106, 4287–4293. [Google Scholar] [CrossRef] [PubMed]
- Houldsworth, J.; Petlakh, M.; Chaganti, R. Identification of Genetic and Expression Markers in DLBCL Cell Lines that Influence the In Vivo Response of DLBCL to Doxorubicin; The American Society of Hematology: Washington, DC, USA, 2004. [Google Scholar]
- Eliopoulos, A.G.; Stack, M.; Dawson, C.W.; Kaye, K.M.; Hodgkin, L.; Sihota, S.; Rowe, M.; Young, L.S. Epstein-barr virus-encoded LMP1 and CD40 mediate IL-6 production in epithelial cells via an NF-kB pathway involving TNF receptor-associated factors. Oncogene 1997, 14, 2899–2916. [Google Scholar] [CrossRef] [PubMed]
- Gires, O.; Zimber-Strobl, U.; Gonnella, R.; Ueffing, M.; Marschall, G.; Zeidler, R.; Pich, D.; Hammerschmidt, W. Latent membrane protein 1 of Epstein–Barr virus mimics a constitutively active receptor molecule. EMBO J. 1997, 16, 6131–6140. [Google Scholar] [CrossRef] [PubMed]
- Luftig, M.; Yasui, T.; Soni, V.; Kang, M.-S.; Jacobson, N.; Cahir-McFarland, E.; Seed, B.; Kieff, E. Epstein–Barr virus latent infection membrane protein 1 TRAF-binding site induces NIK/IKKα-dependent noncanonical NF-κB activation. Proc. Natl. Acad. Sci. 2004, 101, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Thornburg, N.; Kulwichit, W.; Edwards, R.; Shair, K.; Bendt, K.; Raab-Traub, N. LMP1 signaling and activation of NF-κB in LMP1 transgenic mice. Oncogene 2006, 25, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Sanada, M.; Kato, I.; Sato, Y.; Takita, J.; Takeuchi, K.; Niwa, A.; Chen, Y.; Nakazaki, K.; Nomoto, J. Frequent inactivation of A20 in B-cell lymphomas. Nature 2009, 459, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.; Hansmann, M.-L.; Bohle, V.; Martin-Subero, J.I.; Hartmann, S.; Mechtersheimer, G.; Klapper, W.; Vater, I.; Giefing, M.; Gesk, S. TNFAIP3 (A20) is a tumor suppressor gene in Hodgkin lymphoma and primary mediastinal B cell lymphoma. J. Exp. Med. 2009, 206, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.E.; Brown, K.D.; Siebenlist, U.; Staudt, L.M. Constitutive nuclear factor κB activity is required for survival of activated B cell–like diffuse large B cell lymphoma cells. J. Exp. Med. 2001, 194, 1861–1874. [Google Scholar] [CrossRef] [PubMed]
- Ngo, V.N.; Davis, R.E.; Lamy, L.; Yu, X.; Zhao, H.; Lenz, G.; Lam, L.T.; Dave, S.; Yang, L.; Powell, J. A loss-of-function RNA interference screen for molecular targets in cancer. Nature 2006, 441, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Compagno, M.; Lim, W.K.; Grunn, A.; Nandula, S.V.; Brahmachary, M.; Shen, Q.; Bertoni, F.; Ponzoni, M.; Scandurra, M.; Califano, A.; et al. Mutations of multiple genes cause deregulation of NF-κB in diffuse large B-cell lymphoma. Nature 2009, 459, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Correa, P.; Piazuelo, M.B. Helicobacter pylori infection and gastric adenocarcinoma. US Gastroenterol. Hepatol. Rev. 2011, 7, 59–64. [Google Scholar] [PubMed]
- Parkin, D.M. The global health burden of infection-associated cancers in the year 2002. Int. J. Cancer 2006, 118, 3030–3044. [Google Scholar] [CrossRef] [PubMed]
- Blaser, M.J. Helicobacter pylori and the pathogenesis of gastroduodenal inflammation. J. Infect. Dis. 1990, 161, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, C.; Armstrong, J.; Marshall, B. Campylobacter pyloridis, gastritis, and peptic ulceration. J. Clin. Pathol. 1986, 39, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Aihara, M.; Tsuchimoto, D.; Takizawa, H.; Azuma, A.; Wakebe, H.; Ohmoto, Y.; Imagawa, K.; Kikuchi, M.; Mukaida, N.; Matsushima, K. Mechanisms involved in Helicobacter pylori-induced interleukin-8 production by a gastric cancer cell line, mkn45. Infect. Immun. 1997, 65, 3218–3224. [Google Scholar] [PubMed]
- Huang, J.; O’toole, P.W.; Doig, P. Stimulation of interleukin-8 production in epithelial cell lines by Helicobacter pylori. Infect. Immun. 1995, 63, 1732–1738. [Google Scholar] [PubMed]
- Keates, S.; Hitti, Y.S.; Upton, M.; Kelly, C.P. Helicobacter pylori infection activates NF-kappa B in gastric epithelial cells. Gastroenterology 1997, 113, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, J.; Farmery, S.; Lindley, I.; Figura, N.; Peichl, P.; Tompkins, D. CagA/cytotoxic strains of Helicobacter pylori and interleukin-8 in gastric epithelial cell lines. J. Clin. Pathol. 1994, 47, 945–950. [Google Scholar] [CrossRef] [PubMed]
- Covacci, A.; Censini, S.; Bugnoli, M.; Petracca, R.; Burroni, D.; Macchia, G.; Massone, A.; Papini, E.; Xiang, Z.; Figura, N. Molecular characterization of the 128-kDa immunodominant antigen of Helicobacter pylori associated with cytotoxicity and duodenal ulcer. Proc. Natl. Acad. Sci. USA 1993, 90, 5791–5795. [Google Scholar] [CrossRef] [PubMed]
- Censini, S.; Lange, C.; Xiang, Z.; Crabtree, J.E.; Ghiara, P.; Borodovsky, M.; Rappuoli, R.; Covacci, A. Cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc. Natl. Acad. Sci. USA 1996, 93, 14648–14653. [Google Scholar] [CrossRef] [PubMed]
- Fischer, W.; Püls, J.; Buhrdorf, R.; Gebert, B.; Odenbreit, S.; Haas, R. Systematic mutagenesis of the Helicobacter pylori cag pathogenicity island: Essential genes for CagA translocation in host cells and induction of interleukin-8. Mol. Microbiol. 2001, 42, 1337–1348. [Google Scholar] [CrossRef] [PubMed]
- Ogura, K.; Maeda, S.; Nakao, M.; Watanabe, T.; Tada, M.; Kyutoku, T.; Yoshida, H.; Shiratori, Y.; Omata, M. Virulence factors of Helicobacter pylori responsible for gastric diseases in mongolian gerbil. J. Exp. Med. 2000, 192, 1601–1610. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, Y. Mechanisms of disease: Helicobacter pylori virulence factors. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Brandt, S.; Kwok, T.; Hartig, R.; König, W.; Backert, S. NF-κB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc. Natl. Acad. Sci. USA 2005, 102, 9300–9305. [Google Scholar] [CrossRef] [PubMed]
- Lamb, A.; Yang, X.D.; Tsang, Y.H.N.; Li, J.D.; Higashi, H.; Hatakeyama, M.; Peek, R.M.; Blanke, S.R.; Chen, L.F. Helicobacter pylori CagA activates NF-κB by targeting TAK1 for traf6-mediated Lys 63 ubiquitination. EMBO Rep. 2009, 10, 1242–1249. [Google Scholar] [CrossRef] [PubMed]
- Glocker, E.; Lange, C.; Covacci, A.; Bereswill, S.; Kist, M.; Pahl, H.L. Proteins encoded by the cagpathogenicity island of Helicobacter pylori are required for NF-κB activation. Infect. Immun. 1998, 66, 2346–2348. [Google Scholar] [PubMed]
- Allison, C.C.; Kufer, T.A.; Kremmer, E.; Kaparakis, M.; Ferrero, R.L. Helicobacter pylori induces MAPK phosphorylation and AP-1 activation via a NOD1-dependent mechanism. J. Immunol. 2009, 183, 8099–8109. [Google Scholar] [CrossRef] [PubMed]
- Viala, J.; Chaput, C.; Boneca, I.G.; Cardona, A.; Girardin, S.E.; Moran, A.P.; Athman, R.; Mémet, S.; Huerre, M.R.; Coyle, A.J. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat. Immunol. 2004, 5, 1166–1174. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.-Q.; Xie, Z.; Chen, X.-N.; Feng, J.; Chen, J.-W.; Qin, F.-J.; Ge, L.-Y. Fas-associated factor 1 inhibits tumor growth by suppressing Helicobacter pylori-induced activation of NF-κB signaling in human gastric carcinoma. Oncotarget 2017, 8, 7999–8009. [Google Scholar] [PubMed]
- Shigematsu, Y.; Niwa, T.; Rehnberg, E.; Toyoda, T.; Yoshida, S.; Mori, A.; Wakabayashi, M.; Iwakura, Y.; Ichinose, M.; Kim, Y.-J. Interleukin-1β induced by Helicobacter pylori infection enhances mouse gastric carcinogenesis. Cancer Lett. 2013, 340, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.B.; Zuo, W.; Wang, A.J.; Lu, N.H. Helicobacter pylori infection synergistic with IL-1beta gene polymorphisms potentially contributes to the carcinogenesis of gastric cancer. Int. J. Med. Sci. 2016, 13, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Karin, M. NF-κB in mammary gland development and breast cancer. J. Mammary Gland Biol. Neoplasia 2003, 8, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Tiede, B.; Kang, Y. From milk to malignancy: The role of mammary stem cells in development, pregnancy and breast cancer. Cell Res. 2011, 21, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.R.; Hilton, H.N.; Ormandy, C.J. The alveolar switch: Coordinating the proliferative cues and cell fate decisions that drive the formation of lobuloalveoli from ductal epithelium. Breast Cancer Res. BCR 2006, 8, 207. [Google Scholar] [CrossRef] [PubMed]
- Geymayer, S.; Doppler, W. Activation of NF-κB p50/p65 is regulated in the developing mammary gland and inhibits stat5-mediated beta-casein gene expression. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2000, 14, 1159–1170. [Google Scholar]
- Liu, M.; Sakamaki, T.; Casimiro, M.C.; Willmarth, N.E.; Quong, A.A.; Ju, X.; Ojeifo, J.; Jiao, X.; Yeow, W.S.; Katiyar, S.; et al. The canonical NF-κB pathway governs mammary tumorigenesis in transgenic mice and tumor stem cell expansion. Cancer Res. 2010, 70, 10464–10473. [Google Scholar] [CrossRef] [PubMed]
- Merkhofer, E.C.; Cogswell, P.; Baldwin, A.S. Her2 activates NF-κB and induces invasion through the canonical pathway involving IKKalpha. Oncogene 2010, 29, 1238–1248. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.A.; Azoitei, N.; Baumann, B.; Grunert, S.; Sommer, A.; Pehamberger, H.; Kraut, N.; Beug, H.; Wirth, T. NF-κB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J. Clin. Investig. 2004, 114, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.A.; Beug, H.; Wirth, T. Epithelial-mesenchymal transition: NF-κB takes center stage. Cell Cycle 2004, 3, 1477–1480. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.A.; Kraut, N.; Beug, H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr. Opin. Cell Biol. 2005, 17, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Min, C.; Eddy, S.F.; Sherr, D.H.; Sonenshein, G.E. NF-κB and epithelial to mesenchymal transition of cancer. J. Cell. Biochem. 2008, 104, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Neil, J.R.; Schiemann, W.P. Altered Tab1:I kappaB kinase interaction promotes transforming growth factor beta-mediated nuclear factor-kappaB activation during breast cancer progression. Cancer Res. 2008, 68, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Pantuck, A.J.; An, J.; Liu, H.; Rettig, M.B. NF-κB-dependent plasticity of the epithelial to mesenchymal transition induced by von Hippel-Lindau inactivation in renal cell carcinomas. Cancer Res. 2010, 70, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Helfand, B.T.; Jang, T.L.; Zhu, L.J.; Chen, L.; Yang, X.J.; Kozlowski, J.; Smith, N.; Kundu, S.D.; Yang, G.; et al. Nuclear factor-kappaB-mediated transforming growth factor-beta-induced expression of vimentin is an independent predictor of biochemical recurrence after radical prostatectomy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 3557–3567. [Google Scholar] [CrossRef] [PubMed]
- Hay, E.D. An overview of epithelio-mesenchymal transformation. Acta Anat. 1995, 154, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Investig. 2009, 119, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Brickman, J.M.; Adam, M.; Ptashne, M. Interactions between an HMG-1 protein and members of the Rel family. Proc. Natl. Acad. Sci. USA 1999, 96, 10679–10683. [Google Scholar] [CrossRef] [PubMed]
- Pires, B.R.; Mencalha, A.L.; Ferreira, G.M.; de Souza, W.F.; Morgado-Diaz, J.A.; Maia, A.M.; Correa, S.; Abdelhay, E.S. NF-κB is involved in the regulation of EMT genes in breast cancer cells. PLoS ONE 2017, 12, e0169622. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.H.; Parker, J.S.; Ely, K.; Carter, J.; Yi, Y.; Murphy, B.A.; Ang, K.K.; El-Naggar, A.K.; Zanation, A.M.; Cmelak, A.J.; et al. Gene expression profiles identify epithelial-to-mesenchymal transition and activation of nuclear factor-kappaB signaling as characteristics of a high-risk head and neck squamous cell carcinoma. Cancer Res. 2006, 66, 8210–8218. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.; Cao, L.; Shen, C.; Satpathy, M.; Chelladurai, B.; Bigsby, R.M.; Nakshatri, H.; Matei, D. Epithelial-to-mesenchymal transition and ovarian tumor progression induced by tissue transglutaminase. Cancer Res. 2009, 69, 9192–9201. [Google Scholar] [CrossRef] [PubMed]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 pathways: Similarities, differences, and implications of their inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Clavijo, P.E.; Frauwirth, K.A. Anergic CD8+ T lymphocytes have impaired NF-κB activation with defects in p65 phosphorylation and acetylation. J. Immunol. 2012, 188, 1213–1221. [Google Scholar] [CrossRef] [PubMed]
- Dadi, S.; Chhangawala, S.; Whitlock, B.M.; Franklin, R.A.; Luo, C.T.; Oh, S.A.; Toure, A.; Pritykin, Y.; Huse, M.; Leslie, C.S.; et al. Cancer immunosurveillance by tissue-resident innate lymphoid cells and innate-like T cells. Cell 2016, 164, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Vallabhapurapu, S.; Powolny-Budnicka, I.; Riemann, M.; Schmid, R.M.; Paxian, S.; Pfeffer, K.; Korner, H.; Weih, F. Rel/NF-κB family member rela regulates NK1.1− to NK1.1+ transition as well as IL-15-induced expansion of NKT cells. Eur. J. Immunol. 2008, 38, 3508–3519. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, J.; Lichtenheld, M.G.; Meadows, G.G. A role for NF-kappa B activation in perforin expression of NK cells upon IL-2 receptor signaling. J. Immunol. 2002, 169, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Nenci, A.; Becker, C.; Wullaert, A.; Gareus, R.; van Loo, G.; Danese, S.; Huth, M.; Nikolaev, A.; Neufert, C.; Madison, B.; et al. Epithelial nemo links innate immunity to chronic intestinal inflammation. Nature 2007, 446, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.H.; Xiao, Y.; Hu, H.; Jin, J.; Yu, J.; Zhou, X.; Wu, X.; Johnson, H.M.; Akira, S.; Pasparakis, M.; et al. Ubc13 maintains the suppressive function of regulatory T cells and prevents their conversion into effector-like T cells. Nat. Immunol. 2012, 13, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Ha, S.Y.; Kim, H.M.; Ahn, S.M.; Kang, M.S.; Kim, K.M.; Choi, M.G.; Lee, J.H.; Sohn, T.S.; Bae, J.M.; et al. The prognostic effects of tumor infiltrating regulatory t cells and myeloid derived suppressor cells assessed by multicolor flow cytometry in gastric cancer patients. Oncotarget 2016, 7, 7940–7951. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, T.; Kambayashi, Y.; Aiba, S. Crosstalk between regulatory T cells (Tregs) and myeloid derived suppressor cells (MDSCs) during melanoma growth. Oncoimmunology 2012, 1, 1433–1434. [Google Scholar] [CrossRef] [PubMed]
- Kalathil, S.; Lugade, A.A.; Miller, A.; Iyer, R.; Thanavala, Y. Higher frequencies of GARP(+)CTLA-4(+)Foxp3(+) T regulatory cells and myeloid-derived suppressor cells in hepatocellular carcinoma patients are associated with impaired T-cell functionality. Cancer Res. 2013, 73, 2435–2444. [Google Scholar] [CrossRef] [PubMed]
- Kalathil, S.G.; Lugade, A.A.; Miller, A.; Iyer, R.; Thanavala, Y. PD-1+ and Foxp3+ T cell reduction correlates with survival of HCC patients after sorafenib therapy. JCI Insight 2016, 1. [Google Scholar] [CrossRef] [PubMed]
- Mesali, H.; Ajami, A.; Hussein-Nattaj, H.; Rafiei, A.; Rajabian, Z.; Asgarian-Omran, H.; Hosseini, V.; Taghvaei, T.; Tehrani, M. Regulatory T cells and myeloid-derived suppressor cells in patients with peptic ulcer and gastric cancer. Iran. J. Immunol. IJI 2016, 13, 167–177. [Google Scholar] [PubMed]
- Weide, B.; Martens, A.; Zelba, H.; Stutz, C.; Derhovanessian, E.; Di Giacomo, A.M.; Maio, M.; Sucker, A.; Schilling, B.; Schadendorf, D.; et al. Myeloid-derived suppressor cells predict survival of patients with advanced melanoma: Comparison with regulatory T cells and NY-ESO-1- or Melan-A-Specific T cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 1601–1609. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.M.; Subleski, J.J.; Back, T.; Chen, X.; Watkins, S.K.; Yagita, H.; Sayers, T.J.; Murphy, W.J.; Wiltrout, R.H. Regulatory T cells and myeloid-derived suppressor cells in the tumor microenvironment undergo Fas-dependent cell death during IL-2/αCD40 therapy. J. Immunol. 2014, 192, 5821–5829. [Google Scholar] [CrossRef] [PubMed]
- Baratin, M.; Foray, C.; Demaria, O.; Habbeddine, M.; Pollet, E.; Maurizio, J.; Verthuy, C.; Davanture, S.; Azukizawa, H.; Flores-Langarica, A.; et al. Homeostatic NF-κB signaling in steady-state migratory dendritic cells regulates immune homeostasis and tolerance. Immunity 2015, 42, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Overacre-Delgoffe, A.E.; Chikina, M.; Dadey, R.E.; Yano, H.; Brunazzi, E.A.; Shayan, G.; Horne, W.; Moskovitz, J.M.; Kolls, J.K.; Sander, C.; et al. Interferon-gamma drives Treg fragility to promote anti-tumor immunity. Cell 2017, 169, 1130–1141. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.Z.; Morris, G.P.; Kong, Y.C. Anti-tumor immunity and autoimmunity: A balancing act of regulatory T cells. Cancer Immunol. Immunother. CII 2004, 53, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Sopper, S.; Pircher, A.; Gastl, G.; Wolf, A.M. Treg(s) in cancer: Friends or foe? J. Cell. Physiol. 2015, 230, 2598–2605. [Google Scholar] [CrossRef] [PubMed]
- Bruey, J.M.; Bruey-Sedano, N.; Luciano, F.; Zhai, D.; Balpai, R.; Xu, C.; Kress, C.L.; Bailly-Maitre, B.; Li, X.; Osterman, A.; et al. Bcl-2 and Bcl-XL regulate proinflammatory caspase-1 activation by interaction with NALP1. Cell 2007, 129, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat. Immunol. 2011, 12, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Flint, T.R.; Janowitz, T.; Connell, C.M.; Roberts, E.W.; Denton, A.E.; Coll, A.P.; Jodrell, D.I.; Fearon, D.T. Tumor-induced IL-6 reprograms host metabolism to suppress anti-tumor immunity. Cell Metab. 2016, 24, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Zheng, L.; Jiang, J.; Zhao, Y.; Wang, X.; Shen, M.; Zhu, F.; Tian, R.; Shi, C.; Xu, M.; et al. Blocking NF-κB is essential for the immunotherapeutic effect of recombinant il18 in pancreatic cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 5939–5950. [Google Scholar] [CrossRef] [PubMed]
- Haabeth, O.A.; Lorvik, K.B.; Yagita, H.; Bogen, B.; Corthay, A. Interleukin-1 is required for cancer eradication mediated by tumor-specific Th1 cells. Oncoimmunology 2016, 5, e1039763. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, H.; Ohno, Y.; Toyoshima, Y.; Ohtake, J.; Homma, S.; Kawamura, H.; Takahashi, N.; Taketomi, A. Interleukin-6/STAT3 signaling as a promising target to improve the efficacy of cancer immunotherapy. Cancer Sci. 2017, 108, 1947–1952. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Kamata, H.; Luo, J.L.; Leffert, H.; Karin, M. Ikkbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 2005, 121, 977–990. [Google Scholar] [CrossRef] [PubMed]
- Naugler, W.E.; Sakurai, T.; Kim, S.; Maeda, S.; Kim, K.; Elsharkawy, A.M.; Karin, M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 2007, 317, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Ohno, Y.; Toyoshima, Y.; Yurino, H.; Monma, N.; Xiang, H.; Sumida, K.; Kaneumi, S.; Terada, S.; Hashimoto, S.; Ikeo, K.; et al. Lack of interleukin-6 in the tumor microenvironment augments type-1 immunity and increases the efficacy of cancer immunotherapy. Cancer Sci. 2017, 108, 1959–1966. [Google Scholar] [CrossRef] [PubMed]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-κB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lin, Y. Tumor necrosis factor and cancer, buddies or foes? Acta Pharmacol. Sin. 2008, 29, 1275–1288. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, T.; Lawrence, T.; McNeish, I.; Charles, K.A.; Kulbe, H.; Thompson, R.G.; Robinson, S.C.; Balkwill, F.R. “Re-educating” tumor-associated macrophages by targeting NF-κB. J. Exp. Med. 2008, 205, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.Q. The mechanism of the anticancer function of M1 macrophages and their use in the clinic. Chin. J. Cancer 2012, 31, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Yuan, A.; Hsiao, Y.J.; Chen, H.Y.; Chen, H.W.; Ho, C.C.; Chen, Y.Y.; Liu, Y.C.; Hong, T.H.; Yu, S.L.; Chen, J.J.; et al. Opposite effects of M1 and M2 macrophage subtypes on lung cancer progression. Sci. Rep. 2015, 5, 14273. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Hawkins, O.E.; Barham, W.; Gilchuk, P.; Boothby, M.; Ayers, G.D.; Joyce, S.; Karin, M.; Yull, F.E.; Richmond, A. Myeloid IKKbeta promotes antitumor immunity by modulating CCL11 and the innate immune response. Cancer Res. 2014, 74, 7274–7284. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Gangi, L.; Paul, S.; Schioppa, T.; Saccani, A.; Sironi, M.; Bottazzi, B.; Doni, A.; Vincenzo, B.; Pasqualini, F.; et al. A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-κB and enhanced IRF-3/STAT1 activation). Blood 2006, 107, 2112–2122. [Google Scholar] [CrossRef] [PubMed]
- Guiducci, C.; Vicari, A.P.; Sangaletti, S.; Trinchieri, G.; Colombo, M.P. Redirecting in vivo elicited tumor infiltrating macrophages and dendritic cells towards tumor rejection. Cancer Res. 2005, 65, 3437–3446. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Bilsborough, J.; George, T.C.; Norment, A.; Viney, J.L. Mucosal CD8alpha+ DC, with a plasmacytoid phenotype, induce differentiation and support function of T cells with regulatory properties. Immunology 2003, 108, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Chirdo, F.G.; Millington, O.R.; Beacock-Sharp, H.; Mowat, A.M. Immunomodulatory dendritic cells in intestinal lamina propria. Eur. J. Immunol. 2005, 35, 1831–1840. [Google Scholar] [CrossRef] [PubMed]
- Denning, T.L.; Wang, Y.C.; Patel, S.R.; Williams, I.R.; Pulendran, B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat. Immunol. 2007, 8, 1086–1094. [Google Scholar] [CrossRef] [PubMed]
- Smythies, L.E.; Shen, R.; Bimczok, D.; Novak, L.; Clements, R.H.; Eckhoff, D.E.; Bouchard, P.; George, M.D.; Hu, W.K.; Dandekar, S.; et al. Inflammation anergy in human intestinal macrophages is due to smad-induced IkappaBalpha expression and NF-κB inactivation. J. Biol. Chem. 2010, 285, 19593–19604. [Google Scholar] [CrossRef] [PubMed]
- Steinbach, E.C.; Plevy, S.E. The role of macrophages and dendritic cells in the initiation of inflammation in IBD. Inflamm. Bowel Dis. 2014, 20, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Barnes, S.E.; Wang, Y.; Chen, L.; Molinero, L.L.; Gajewski, T.F.; Evaristo, C.; Alegre, M.L. T cell-NF-κB activation is required for tumor control in vivo. J. Immunother. Cancer 2015, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.K.; Li, Y.; Oh, E.; Kim, J.; Yun, C.O. Oncolytic adenovirus expressing IL-23 and p35 elicits IFN-gamma- and TNF-alpha-co-producing T cell-mediated antitumor immunity. PLoS ONE 2013, 8, e67512. [Google Scholar] [CrossRef]
- Hopewell, E.L.; Zhao, W.; Fulp, W.J.; Bronk, C.C.; Lopez, A.S.; Massengill, M.; Antonia, S.; Celis, E.; Haura, E.B.; Enkemann, S.A.; et al. Lung tumor NF-κB signaling promotes t cell-mediated immune surveillance. J. Clin. Investig. 2013, 123, 2509–2522. [Google Scholar] [CrossRef] [PubMed]
- Gogoi, D.; Chiplunkar, S.V. Targeting gamma delta T cells for cancer immunotherapy: Bench to bedside. Indian J. Med. Res. 2013, 138, 755–761. [Google Scholar] [PubMed]
- Hadrup, S.; Donia, M.; Thor Straten, P. Effector CD4 and CD8 T cells and their role in the tumor microenvironment. Cancer Microenviron. Off. J. Int. Cancer Microenviron. Soc. 2013, 6, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Miyake, T.; Satoh, T.; Kato, H.; Matsushita, K.; Kumagai, Y.; Vandenbon, A.; Tani, T.; Muta, T.; Akira, S.; Takeuchi, O. IκBζ is essential for natural killer cell activation in response to IL-12 and IL-18. Proc. Natl. Acad. Sci. USA 2010, 107, 17680–17685. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, S.T.; Ribot, J.C.; Silva-Santos, B. Five layers of receptor signaling in gammadelta T-cell differentiation and activation. Front. Immunol. 2015, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Terabe, M.; Berzofsky, J.A. Immunoregulatory T cells in tumor immunity. Curr. Opin. Immunol. 2004, 16, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Crespo, J.; Sun, H.; Welling, T.H.; Tian, Z.; Zou, W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr. Opin. Immunol. 2013, 25, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Shrimali, R.K.; Ahmad, S.; Verma, V.; Zeng, P.; Ananth, S.; Gaur, P.; Gittelman, R.M.; Yusko, E.; Sanders, C.; Robins, H.; et al. Concurrent PD-1 blockade negates the effects of OX40 agonist antibody in combination immunotherapy through inducing T-cell apoptosis. Cancer Immunol. Res. 2017, 5, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T.; Bebien, M.; Liu, G.Y.; Nizet, V.; Karin, M. IKKalpha limits macrophage NF-κB activation and contributes to the resolution of inflammation. Nature 2005, 434, 1138–1143. [Google Scholar] [CrossRef] [PubMed]
- Hilliard, B.A.; Mason, N.; Xu, L.; Sun, J.; Lamhamedi-Cherradi, S.E.; Liou, H.C.; Hunter, C.; Chen, Y.H. Critical roles of c-Rel in autoimmune inflammation and helper T cell differentiation. J. Clin. Investig. 2002, 110, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Claudio, E.; Dambach, D.; Raventos-Suarez, C.; Ryan, C.; Bravo, R. Chronic inflammation and susceptibility to bacterial infections in mice lacking the polypeptide (p)105 precursor (NF-κB1) but expressing p50. J. Exp. Med. 1998, 187, 985–996. [Google Scholar] [CrossRef] [PubMed]
- McMillan, D.H.; Woeller, C.F.; Thatcher, T.H.; Spinelli, S.L.; Maggirwar, S.B.; Sime, P.J.; Phipps, R.P. Attenuation of inflammatory mediator production by the NF-κB member RelB is mediated by microrna-146a in lung fibroblasts. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L774–L781. [Google Scholar] [CrossRef] [PubMed]
- Yazdani, S.; Karimfar, M.H.; Imani Fooladi, A.A.; Mirbagheri, L.; Ebrahimi, M.; Ghanei, M.; Nourani, M.R. Nuclear factor kappaB1/RelA mediates the inflammation and/or survival of human airway exposed to sulfur mustard. J. Recept. Signal Transduct. Res. 2011, 31, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Dalgleish, A.G. Rationale for combining immunotherapy with chemotherapy. Immunotherapy 2015, 7, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Emens, L.A.; Middleton, G. The interplay of immunotherapy and chemotherapy: Harnessing potential synergies. Cancer Immunol. Res. 2015, 3, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Formenti, S.C.; Demaria, S. Combining radiotherapy and cancer immunotherapy: A paradigm shift. J. Natl. Cancer Inst. 2013, 105, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Roses, R.E.; Datta, J.; Czerniecki, B.J. Radiation as immunomodulator: Implications for dendritic cell-based immunotherapy. Radiat. Res. 2014, 182, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lou, X.; Jin, L.; Zhou, R.; Liu, S.; Xu, N.; Liao, D.J. Necrosis, and then stress induced necrosis-like cell death, but not apoptosis, should be the preferred cell death mode for chemotherapy: Clearance of a few misconceptions. Oncoscience 2014, 1, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, N.; Thorburn, A.N.; Macia, L.; Tan, J.; Juglair, L.; Yagita, H.; Yu, D.; Hansbro, P.M.; Mackay, C.R. Inflammation and lymphopenia trigger autoimmunity by suppression of IL-2-controlled regulatory T cell and increase of IL-21-mediated effector T cell expansion. J. Immunol. 2014, 193, 4845–4858. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chua, K.S.; Guimond, M.; Kapoor, V.; Brown, M.V.; Fleisher, T.A.; Long, L.M.; Bernstein, D.; Hill, B.J.; Douek, D.C.; et al. Lymphopenia and interleukin-2 therapy alter homeostasis of CD4+CD25+ regulatory T cells. Nat. Med. 2005, 11, 1238–1243. [Google Scholar] [CrossRef] [PubMed]
- Duarte, J.H.; Zelenay, S.; Bergman, M.L.; Martins, A.C.; Demengeot, J. Natural Treg cells spontaneously differentiate into pathogenic helper cells in lymphopenic conditions. Eur. J. Immunol. 2009, 39, 948–955. [Google Scholar] [CrossRef] [PubMed]
- Oft, M. IL-10: Master switch from tumor-promoting inflammation to antitumor immunity. Cancer Immunol. Res. 2014, 2, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Karin, M. Inflammatory cytokines in cancer: Tumour necrosis factor and interleukin 6 take the stage. Ann. Rheum. Dis. 2011, 70, i104–i108. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pires, B.R.B.; Silva, R.C.M.C.; Ferreira, G.M.; Abdelhay, E. NF-kappaB: Two Sides of the Same Coin. Genes 2018, 9, 24. https://doi.org/10.3390/genes9010024
Pires BRB, Silva RCMC, Ferreira GM, Abdelhay E. NF-kappaB: Two Sides of the Same Coin. Genes. 2018; 9(1):24. https://doi.org/10.3390/genes9010024
Chicago/Turabian StylePires, Bruno R. B., Rafael C. M. C. Silva, Gerson M. Ferreira, and Eliana Abdelhay. 2018. "NF-kappaB: Two Sides of the Same Coin" Genes 9, no. 1: 24. https://doi.org/10.3390/genes9010024
APA StylePires, B. R. B., Silva, R. C. M. C., Ferreira, G. M., & Abdelhay, E. (2018). NF-kappaB: Two Sides of the Same Coin. Genes, 9(1), 24. https://doi.org/10.3390/genes9010024