Transcription Factor and lncRNA Regulatory Networks Identify Key Elements in Lung Adenocarcinoma

Abstract
1. Introduction
2. Materials and Methods
2.1. Multidimensional Genomics Datasets
2.2. Differential Analysis
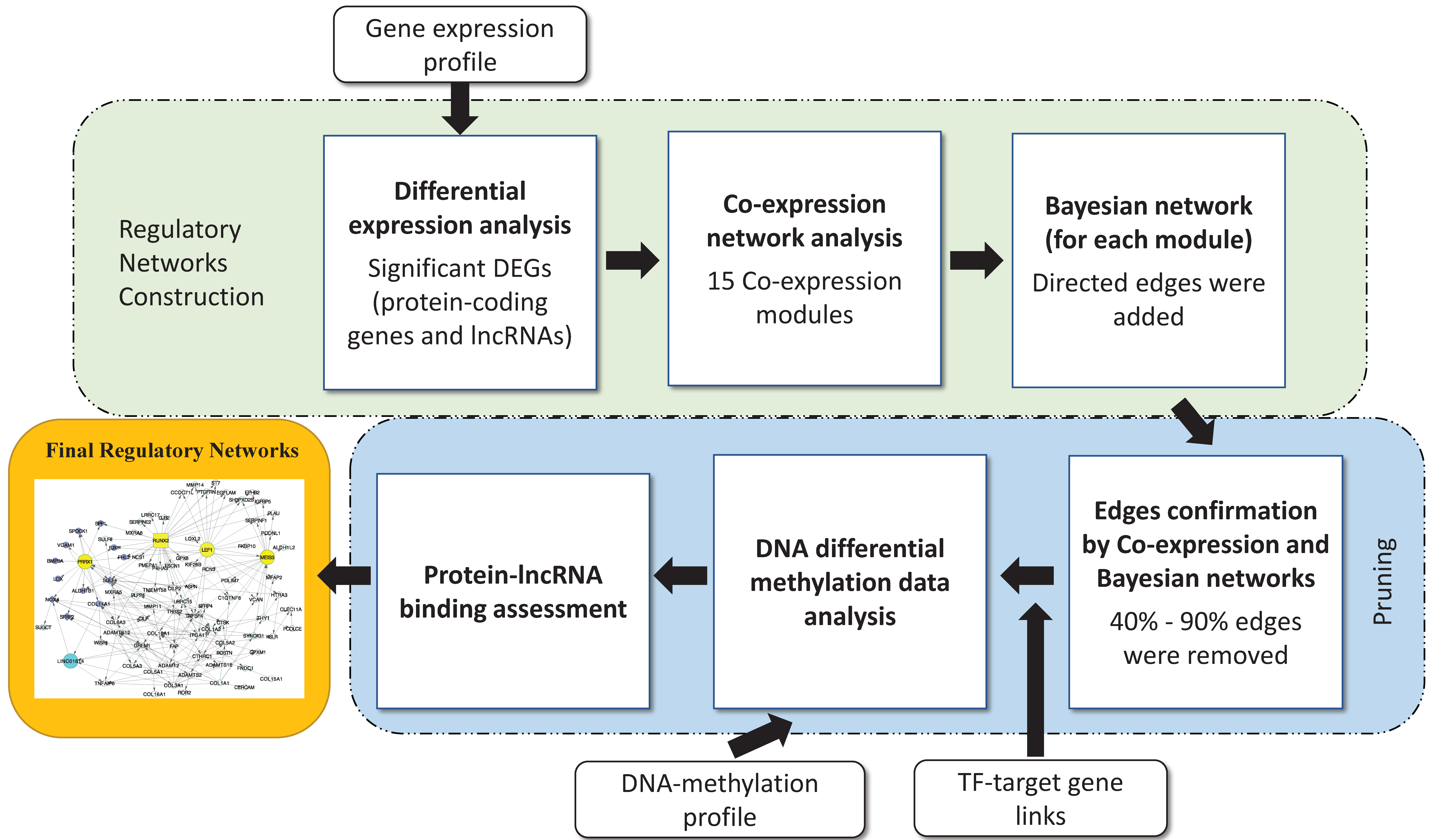
2.3. Regulatory Network Building
2.4. Driver Somatic Mutation Identification
2.5. Gene Enrichment Analysis
3. Results
3.1. Differential Analysis of Multidimensional Genomic Profiles of Lung Adenocarcinoma
3.2. Disease Regulatory Network Identification
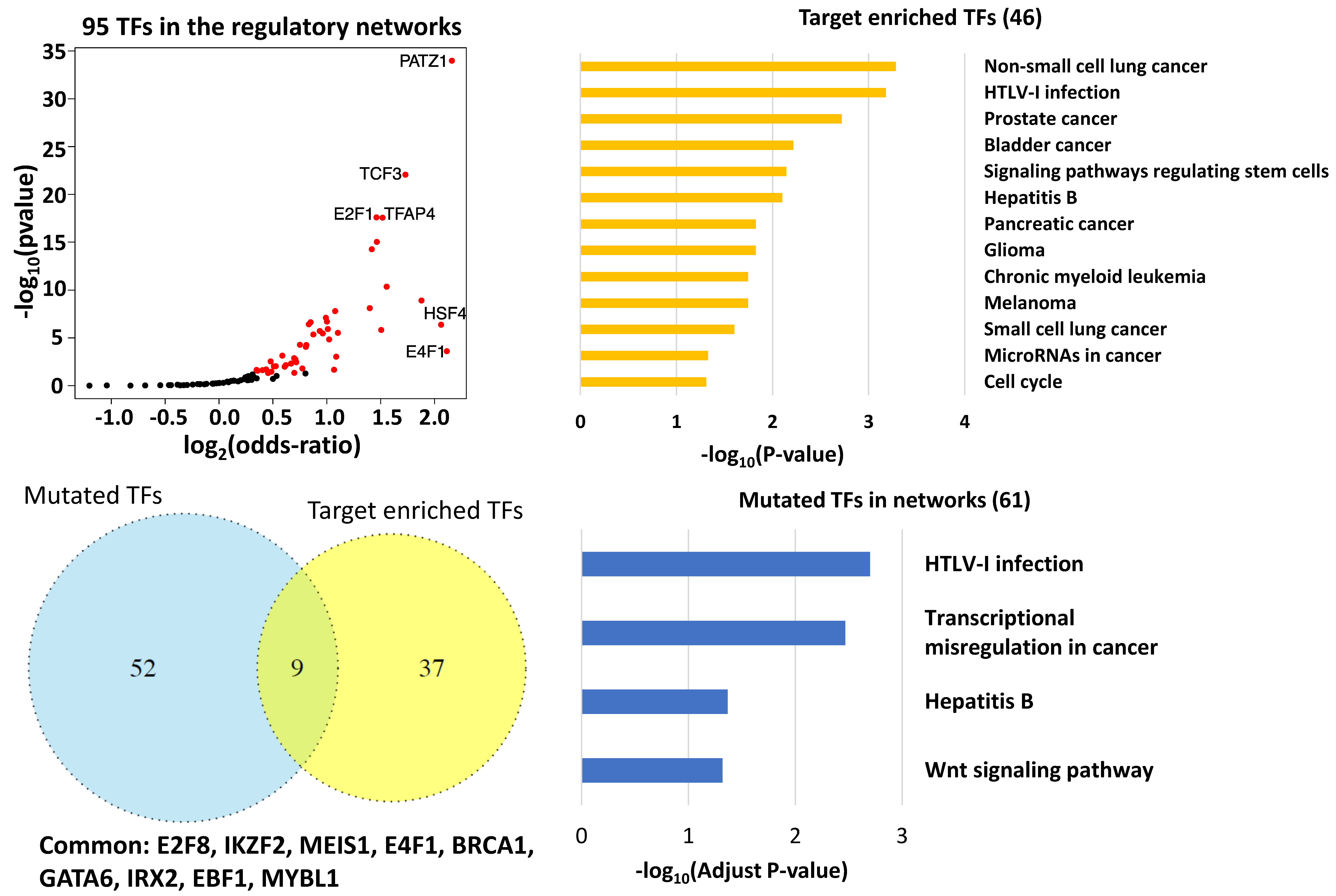
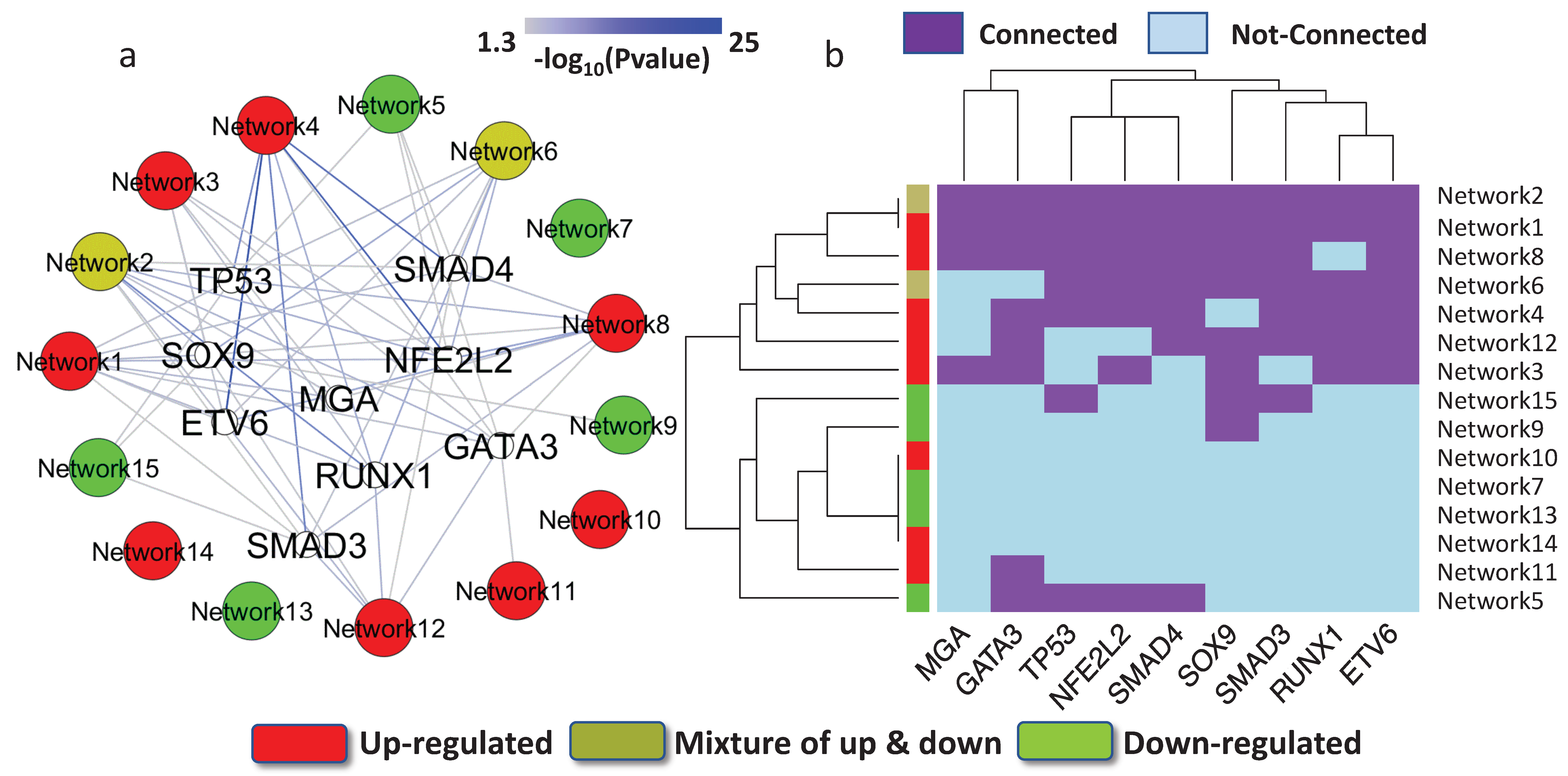
3.3. Key Regulatory Elements in the Networks
3.4. Key Regulatory Elements Outside the Networks
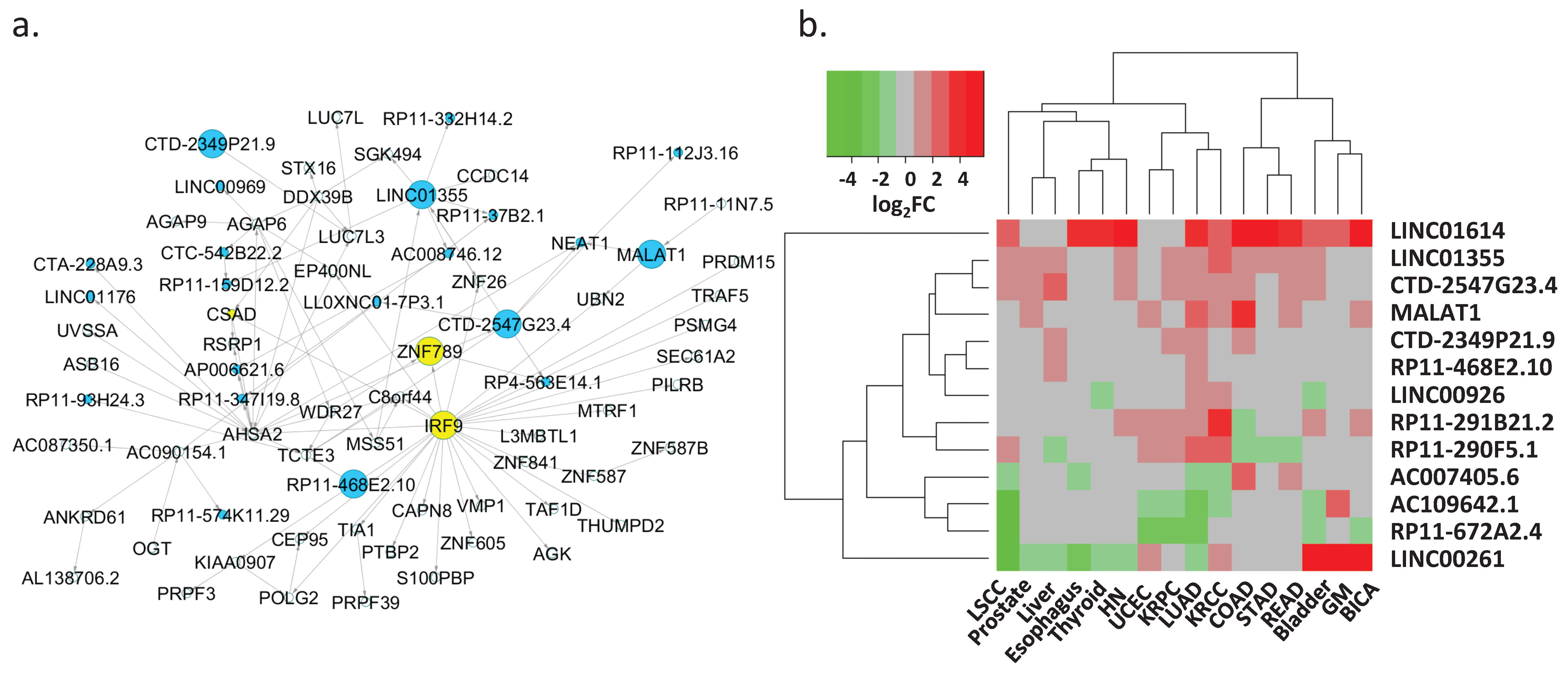
3.5. Regulation of Key lncRNAs
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- American Cancer Society. Key Statistics on Lung Cancer. Available online: www.cancer.org/cancer/non-small-cell-lung-cancer/about/key-statistics.html (accessed on 15 August 2017).
- Brown, T. Silica exposure, smoking, silicosis and lung cancer-complex interactions. Occup. Med. 2009, 59, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Govindan, R.; Ding, L.; Griffith, M.; Subramanian, J.; Dees, N.D.; Kanchi, K.L.; Maher, C.A.; Fulton, R.; Fulton, L.; Wallis, J.; et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell 2012, 150, 1121–1134. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Schiller, J.H.; Gazdar, A.F. Lung cancer in never smokers—A different disease. Nat. Rev. Cancer 2007, 7, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Jiang, Z.; Liu, J.; Haverty, P.M.; Guan, Y.; Stinson, J.; Yue, P.; Zhang, Y.; Pant, K.P.; Bhatt, D.; et al. The mutation spectrum revealed by paired genome sequences from a lung cancer patient. Nature 2010, 465, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 2007, 7, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-Y.; Jan, Y.-H.; Juan, Y.-H.; Yang, C.-J.; Huang, M.-S.; Yu, C.-J.; Yang, P.-C.; Hsiao, M.; Hsu, T.-L.; Wong, C.-H. Fucosyltransferase 8 as a functional regulator of nonsmall cell lung cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Carbone, D.P.; Gandara, D.R.; Antonia, S.J.; Zielinski, C.; Paz-Ares, L. Non–small-cell lung cancer: Role of the immune system and potential for immunotherapy. J. Thorac. Oncol. 2015, 10, 974–984. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Li, S.; Zhang, F.; Xi, Y.; Wang, L.; Bi, Y.; Li, D. Long non-coding RNA NEAT1 promotes non-small cell lung cancer progression through regulation of MIR-377-3p-e2f3 pathway. Oncotarget 2016, 7, 51784. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-C.; Jen, J.; Lai, W.-W.; Tang, Y.-A.; Wang, Y.-C.; Lu, Y.-H. Oct4 transcriptionally regulates the expression of long non-coding RNAs NEAT1 and MALAT1 to promote lung cancer progression. Mol. Cancer 2017, 16, 104. [Google Scholar]
- Iyer, M.K.; Niknafs, Y.S.; Malik, R.; Singhal, U.; Sahu, A.; Hosono, Y.; Barrette, T.R.; Prensner, J.R.; Evans, J.R.; Zhao, S.; et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015, 47, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Xie, J.; Shen, C.; Cheng, D.; Shi, Y.; Wu, Z.; Deng, X.; Chen, H.; Shen, B.; Peng, C.; et al. Upregulation of long noncoding RNA ZEB1-AS1 promotes tumor metastasis and predicts poor prognosis in hepatocellular carcinoma. Oncogene 2016, 35, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Guo, W.; Xu, X.-J.; Su, F.; Wang, Y.; Zhang, Y.; Wang, Q.; Zhu, L. Melanoma long non-coding RNA signature predicts prognostic survival and directs clinical risk-specific treatments. J. Dermatol. Sci. 2017, 85, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.I.; Young, R.A. Transcriptional regulation and its misregulation in disease. Cell 2013, 152, 1237–1251. [Google Scholar] [CrossRef] [PubMed]
- Sławek, S.; Szmyt, K.; Fularz, M.; Dziudzia, J.; Boruczkowski, M.; Sikora, J.; Kaczmarek, M. Pluripotency transcription factors in lung cancer—a review. Tumor Biol. 2016, 37, 4241–4249. [Google Scholar] [CrossRef] [PubMed]
- Qu, H.; Li, R.; Liu, Z.; Zhang, J.; Luo, R. Prognostic value of cancer stem cell marker CD133 expression in non-small cell lung cancer: A systematic review. Int. J. Clin. Exp. Pathol. 2013, 6, 2644. [Google Scholar] [PubMed]
- Semenova, E.A.; Kwon, M.-C.; Monkhorst, K.; Song, J.-Y.; Bhaskaran, R.; Krijgsman, O.; Kuilman, T.; Peters, D.; Buikhuisen, W.A.; Smit, E.F.; et al. Transcription factor NFIB is a driver of small cell lung cancer progression in mice and marks metastatic disease in patients. Cell Rep. 2016, 16, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Tagne, J.-B.; Mohtar, O.R.; Campbell, J.D.; Lakshminarayanan, M.; Huang, J.; Hinds, A.C.; Lu, J.; Ramirez, M.I. Transcription factor and microRNA interactions in lung cells: An inhibitory link between NK2 HOMEOBOX 1, MIR-200C and the developmental and oncogenic factors NFIB and MYB. Respir. Res. 2015, 16, 22. [Google Scholar] [CrossRef] [PubMed]
- Mitra, R.; Edmonds, M.D.; Sun, J.; Zhao, M.; Yu, H.; Eischen, C.M.; Zhao, Z. Reproducible combinatorial regulatory networks elucidate novel oncogenic microRNAs in non-small cell lung cancer. RNA 2014, 20, 1356–1368. [Google Scholar] [CrossRef] [PubMed]
- Cogill, S.B.; Wang, L. Co-expression network analysis of human lncRNAs and cancer genes. Cancer Inform. 2014, 13, 49. [Google Scholar] [CrossRef] [PubMed]
- Hamed, M.; Spaniol, C.; Zapp, A.; Helms, V. Integrative network-based approach identifies key genetic elements in breast invasive carcinoma. BMC Genomics 2015, 16 (Suppl. 5), S2. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Genomic Data Commons Data Portal. Available online: https://portal.gdc.cancer.gov/ (accessed on 6 June 2017).
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.; Du, P.; Bilke, S.; Triche, T.; Bootwalla, M. Methylumi: Handle Illumina methylation data. R package version 2.4. 0. 2012. Available online: www.bioconductor.org/packages/release/bioc/html/methylumi.html (accessed on 10 July 2017).
- Du, P.; Zhang, X.; Huang, C.-C.; Jafari, N.; Kibbe, W.A.; Hou, L.; Lin, S.M. Comparison of beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinform. 2010, 11, 587. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. Edger: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Tibshirani, R. Finding consistent patterns: A nonparametric approach for identifying differential expression in RNA-seq data. Stat. Methods Med. Res. 2013, 22, 519–536. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Knuth, D.E. Postscript about NP-hard problems. ACM SIGACT News 1974, 6, 15–16. [Google Scholar] [CrossRef]
- Gámez, J.A.; Mateo, J.L.; Puerta, J.M. Learning bayesian networks by hill climbing: Efficient methods based on progressive restriction of the neighborhood. Data Min. Knowl. Discov. 2011, 22, 106–148. [Google Scholar] [CrossRef]
- Scutari, M. Learning bayesian networks with the bnlearn R package. J. Stat. Softw. 2010, 35, 1–22. [Google Scholar]
- Mathelier, A.; Fornes, O.; Arenillas, D.J.; Chen, C.-Y.; Denay, G.; Lee, J.; Shi, W.; Shyr, C.; Tan, G.; Worsley-Hunt, R.; et al. JASPAR 2016: A major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2016, 44, D110–D115. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-M.; Liu, T.; Liu, C.-J.; Song, S.; Zhang, X.; Liu, W.; Jia, H.; Xue, Y.; Guo, A.-Y. AnimalTFDB 2.0: A resource for expression, prediction and functional study of animal transcription factors. Nucleic Acids Res. 2014, 43, D76–D81. [Google Scholar] [CrossRef] [PubMed]
- Marbach, D.; Lamparter, D.; Quon, G.; Kellis, M.; Kutalik, Z.; Bergmann, S. Tissue-specific regulatory circuits reveal variable modular perturbations across complex diseases. Nat. Methods 2016, 13, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Xuan, Z.; Zhao, F.; Zhang, M.Q. TRED: A transcriptional regulatory element database, new entries and other development. Nucleic Acids Res. 2007, 35, D137–D140. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Ren, S.; Lu, M.; Zhang, Y.; Zhu, D.; Zhang, X.; Li, T. Computational prediction of associations between long non-coding RNAs and proteins. BMC Genom. 2013, 14, 651. [Google Scholar] [CrossRef] [PubMed]
- Douville, C.; Carter, H.; Kim, R.; Niknafs, N.; Diekhans, M.; Stenson, P.D.; Cooper, D.N.; Ryan, M.; Karchin, R. CRAVAT: Cancer-related analysis of variants toolkit. Bioinformatics 2013, 29, 647–648. [Google Scholar] [CrossRef] [PubMed]
- Carter, H.; Chen, S.; Isik, L.; Tyekucheva, S.; Velculescu, V.E.; Kinzler, K.W.; Vogelstein, B.; Karchin, R. Cancer-specific high-throughput annotation of somatic mutations: Computational prediction of driver missense mutations. Cancer Res. 2009, 69, 6660–6667. [Google Scholar] [CrossRef] [PubMed]
- Douville, C.; Masica, D.L.; Stenson, P.D.; Cooper, D.N.; Gygax, D.M.; Kim, R.; Ryan, M.; Karchin, R. Assessing the pathogenicity of insertion and deletion variants with the variant effect scoring tool (VEST-Indel). Hum. Mutat. 2016, 37, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Selamat, S.A.; Galler, J.S.; Joshi, A.D.; Fyfe, M.N.; Campan, M.; Siegmund, K.D.; Kerr, K.M.; Laird-Offringa, I.A. DNA methylation changes in atypical adenomatous hyperplasia, adenocarcinoma in situ, and lung adenocarcinoma. PLoS ONE 2011, 6, e21443. [Google Scholar] [CrossRef] [PubMed]
- Zöchbauer-Müller, S.; Minna, J.D.; Gazdar, A.F. Aberrant DNA methylation in lung cancer: Biological and clinical implications. Oncologist 2002, 7, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Eroglu, B.; Cho, W.; Yamaguchi, Y.; Moskophidis, D.; Mivechi, N.F. Inactivation of heat shock factor HSF4 induces cellular senescence and suppresses tumorigenesis in vivo. Mol. Cancer Res. 2012, 10, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Xiong, D.; Sun, X.; Wang, J.; Hao, M.; Ding, T.; Xiao, G.; Wang, X.; Mao, Y.; Fu, Y.; et al. Signification of hypermethylated in cancer 1 (HIC1) as tumor suppressor gene in tumor progression. Cancer Microenviron. 2012, 5, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Rodier, G.; Kirsh, O.; Baraibar, M.; Houles, T.; Lacroix, M.; Delpech, H.; Hatchi, E.; Arnould, S.; Severac, D.; Dubois, E.; et al. The transcription factor E4F1 coordinates CHK1-dependent checkpoint and mitochondrial functions. Cell Rep. 2015, 11, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Hung, J.-J.; Hsueh, C.-T.; Chen, K.-H.; Hsu, W.-H.; Wu, Y.-C. Clinical significance of E2F1 protein expression in non-small cell lung cancer. Exp. Hematol. Oncol. 2012, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- El-Aarag, S.A.; Mahmoud, A.; Hashem, M.H.; Elkader, H.A.; Hemeida, A.E.; El Hefnawi, M. In silico identification of potential key regulatory factors in smoking-induced lung cancer. BMC Med. Genom. 2017, 10, 40. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.W. Measles virus for cancer therapy. In Measles; Griffin, D.E., Oldstone, M.B.A., Eds.; Springer: Heidelberg, Germany, 2009; pp. 213–241. [Google Scholar]
- Taylor, J.M.; Nicot, C. HTLV-1 and apoptosis: Role in cellular transformation and recent advances in therapeutic approaches. Apoptosis 2008, 13, 733. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-A.; Platt, J.; Lee, J.W.; López-Giráldez, F.; Herbst, R.S.; Koo, J.S. E2F8 as a novel therapeutic target for lung cancer. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Huang, K.; Guo, H.; Cui, G. MEIS1 regulates proliferation of non-small-cell lung cancer cells. J. Thorac. Dis. 2014, 6, 850. [Google Scholar] [PubMed]
- Reguart, N.; Cardona, A.F.; Carrasco, E.; Gomez, P.; Taron, M.; Rosell, R. BRCA1: A new genomic marker for non–small-cell lung cancer. Clin. Lung Cancer 2008, 9, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Cheung, W.K.; Zhao, M.; Liu, Z.; Stevens, L.E.; Cao, P.D.; Fang, J.E.; Westbrook, T.F.; Nguyen, D.X. Control of alveolar differentiation by the lineage transcription factors GATA6 and HOPX inhibits lung adenocarcinoma metastasis. Cancer Cell 2013, 23, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Arai, E.; Kohno, T.; Takahashi, Y.; Miyata, S.; Tsuta, K.; Watanabe, S.I.; Soejima, K.; Betsuyaku, T.; Kanai, Y. Epigenetic clustering of lung adenocarcinomas based on DNA methylation profiles in adjacent lung tissue: Its correlation with smoking history and chronic obstructive pulmonary disease. Int. J. Cancer 2014, 135, 319–334. [Google Scholar] [CrossRef] [PubMed]
- Liao, D. Emerging roles of the EBF family of transcription factors in tumor suppression. Mol. Cancer Res. 2009, 7, 1893–1901. [Google Scholar] [CrossRef] [PubMed]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 2016, 45, D777–D783. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fan, H.; Fang, S.; Wang, L.; Chen, L.; Jin, Y.; Jiang, W.; Lin, Z.; Shi, Y.; Zhan, C.; et al. Mutations and expression of the NFE2L2/KEAP1/CUL3 pathway in Chinese patients with lung squamous cell carcinoma. J. Thorac. Dis. 2016, 8, 1639. [Google Scholar] [CrossRef] [PubMed]
- Rotblat, B.; Melino, G.; Knight, R.A. NRF2 and p53: Januses in cancer? Oncotarget 2012, 3, 1272. [Google Scholar] [CrossRef] [PubMed]
- White, N.M.; Cabanski, C.R.; Silva-Fisher, J.M.; Dang, H.X.; Govindan, R.; Maher, C.A. Transcriptome sequencing reveals altered long intergenic non-coding RNAs in lung cancer. Genome Biol. 2014, 15, 429. [Google Scholar] [CrossRef] [PubMed]
- Li, C.M.-C.; Gocheva, V.; Oudin, M.J.; Bhutkar, A.; Wang, S.Y.; Date, S.R.; Ng, S.R.; Whittaker, C.A.; Bronson, R.T.; Snyder, E.L.; et al. Foxa2 and Cdx2 cooperate with Nkx2-1 to inhibit lung adenocarcinoma metastasis. Gene. Dev. 2015, 29, 1850–1862. [Google Scholar] [CrossRef] [PubMed]
- Metelli, A.; Wu, B.X.; Fugle, C.W.; Rachidi, S.; Sun, S.; Zhang, Y.; Wu, J.; Tomlinson, S.; Howe, P.H.; Yang, Y.; et al. Surface expression of TGF-β docking receptor GARP promotes oncogenesis and immune tolerance in breast cancer. Cancer Res. 2016, 76, 7106–7117. [Google Scholar] [CrossRef] [PubMed]
- Johnsson, P.; Lipovich, L.; Grandér, D.; Morris, K.V. Evolutionary conservation of long non-coding RNAs; sequence, structure, function. Biochim. Biophys. Acta 2014, 1840, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.J.; Zhang, Q.C.; Georgiev, P.; Ilik, I.A.; Akhtar, A.; Chang, H.Y. Rapid evolutionary turnover underlies conserved lncRNA–genome interactions. Gene. Dev. 2016, 30, 191–207. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, H.; Fang, S.; Kang, Y.; Hao, Y.; Li, Z.; Bu, D.; Sun, N.; Zhang, M.Q.; Chen, R. NONCODE 2016: An informative and valuable data source of long non-coding RNAs. Nucleic Acids Res. 2016, 44, D203–D208. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.M.; Li, D.; El-Deeb, A.S.; Wolff, R.A.; Bondy, M.L.; Davila, M.; Abbruzzese, J.L. Association between hepatitis B virus and pancreatic cancer. J. Clin. Oncol. 2008, 26, 4557–4562. [Google Scholar] [CrossRef] [PubMed]
- Perz, J.F.; Armstrong, G.L.; Farrington, L.A.; Hutin, Y.J.; Bell, B.P. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J. Hepatol. 2006, 45, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.V.; King, L.Y.; Chung, R.T. Hepatitis C virus–associated cancer. Annu. Rev. Pathol. 2015, 10, 345–370. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Key lncRNA | Target Protein-Coding Gene(s) 1 | Target lncRNA(s) 1 | Differentially Expressed in Cancer Types 2 |
|---|---|---|---|
| LINC00261 | FOXA2 (0.74; 0.7) | NA | BICA, Bladder, Esophagus, GM, HN, KRCC, Liver, LSCC, LUAD, Prostate, Thyroid, UCEC |
| MALAT1 | UBN2 (0.49; 0.83) | NEAT1 (0.77; 0.84) | BICA, COAD, KRCC, LUAD, Prostate, READ, UCEC |
| LINC01614 | TNFAIP6 (0.64; 0.64), NOX4 (0.75; 0.71) | NA | BICA, Bladder, COAD, Esophagus, GM, HN, KRCC, LSCC, LUAD, READ, STAD, Thyroid |
| AC007405.6 | ERICH2 (0.83; 0.81) | NA | COAD, Esophagus, KRCC, LSCC, LUAD, READ |
| AC109642.1 | FMO2 (0.82; 0.81), ANGPT1 (0.79; 0.77) | NA | Bladder, GM, KRCC, KRPC, LSCC, LUAD, UCEC |
| RP11-672A2.4 | LRRC32 (0.93; 0.92) | NA | BICA, Bladder, KRPC, LSCC, LUAD, UCEC |
| LINC01355 | WDR2 (0.72; 0.83), SGK494 (0.67; 0.70), CCDC14 (0.71; 0.84), ZNF26 (0.66; 0.85), PCGF3 (0.62; 0.72), AC087350.1 (0.54; 0.72) | RP11-159D12.2 (0.68; 0.78), AC008746.12 (0.77; 0.85), RP11-332H14.2 (0.64; 0.65) | Bladder, COAD, HN, KRCC, KRPC, Liver, LSCC, LUAD, Prostate, READ, STAD |
| CTD-2547G23.4 | TCTE3 (0.78; 0.81), HCG27 (0.71; 0.72) | NEAT1 (0.47; 0.70), LL0XNC01-7P3.1 (0.81; 0.82), LINC01355 (0.77; 0.85), RP4-563E14.1 (0.70; 0.82), RP11-112J3.16 (0.73; 0.72) | Bladder, COAD, HN, KRCC, KRPC, Liver, LSCC, LUAD, Prostate, READ |
| CTD-2349P21.9 | LUC7L3 (0.66; 0.65) | NA | COAD, KRPC, Liver, LUAD |
| RP11-468E2.10 | TCTE3 (0.65; 0.70) | NA | Liver, LUAD |
| LINC00926 | TRAF3IP3 (0.67; 0.70), TNFRSF13C (0.77; 0.80), FDCSP (0.44; 0.38) | NA | Bladder, KRCC, LUAD, Thyroid |
| RP11-290F5.1 | FCRL5 (0.84; 0.84), PIM2 (0.84; 0.77), DERL3 (0.76; 0.66) | NA | COAD, KRCC, KRPC, Liver, LSCC, LUAD, READ, STAD, UCEC |
| RP11-291B21.2 | ZNF683 (0.80; 0.75) | AC002331.1 (0.74; 0.71) | BICA, Bladder, COAD, HN, KRCC, KRPC, LUAD, UCEC |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, D.; Yang, W.; Zhang, J.; Yang, J.Y.; Guan, R.; Yang, M.Q. Transcription Factor and lncRNA Regulatory Networks Identify Key Elements in Lung Adenocarcinoma. Genes 2018, 9, 12. https://doi.org/10.3390/genes9010012
Li D, Yang W, Zhang J, Yang JY, Guan R, Yang MQ. Transcription Factor and lncRNA Regulatory Networks Identify Key Elements in Lung Adenocarcinoma. Genes. 2018; 9(1):12. https://doi.org/10.3390/genes9010012
Chicago/Turabian StyleLi, Dan, William Yang, Jialing Zhang, Jack Y. Yang, Renchu Guan, and Mary Qu Yang. 2018. "Transcription Factor and lncRNA Regulatory Networks Identify Key Elements in Lung Adenocarcinoma" Genes 9, no. 1: 12. https://doi.org/10.3390/genes9010012
APA StyleLi, D., Yang, W., Zhang, J., Yang, J. Y., Guan, R., & Yang, M. Q. (2018). Transcription Factor and lncRNA Regulatory Networks Identify Key Elements in Lung Adenocarcinoma. Genes, 9(1), 12. https://doi.org/10.3390/genes9010012