High-Throughput Study of the Effects of Celastrol on Activated Fibroblast-Like Synoviocytes from Patients with Rheumatoid Arthritis

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Isolation and Primary Culture of RA–FLSs
2.2. Immunofluorescence Staining
2.3. Cell Proliferation Assay
2.4. In Vitro Invasion Assay
2.5. Flow Cytometry Assay
2.6. DNA Microarray
2.7. RNA Extraction and Real-Time PCR Analysis
2.8. ELISA Assay
2.9. Western Blot Analysis
2.10. Statistical Analysis
3. Results
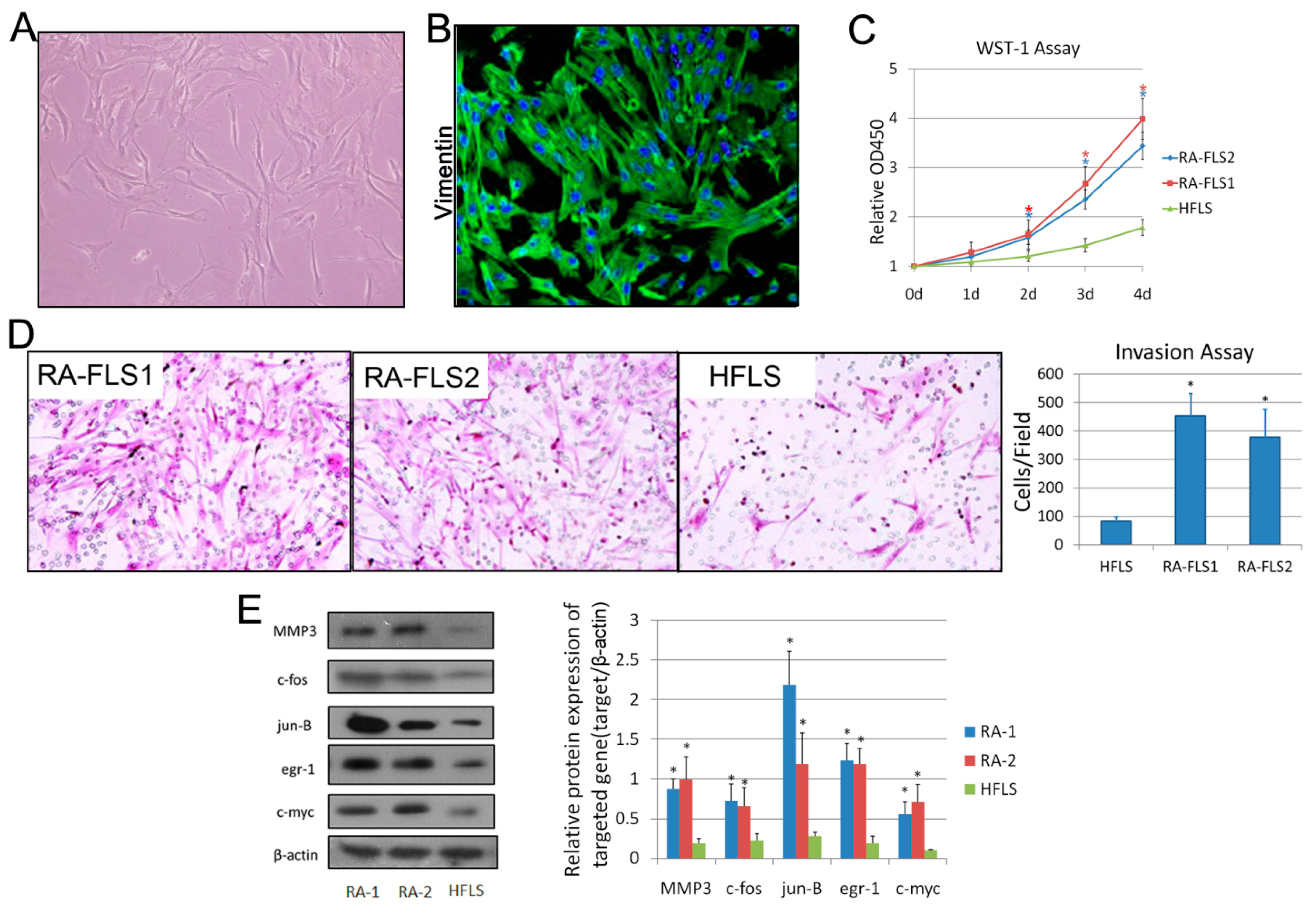
3.1. Isolation and Identification of FLSs from Patients with RA
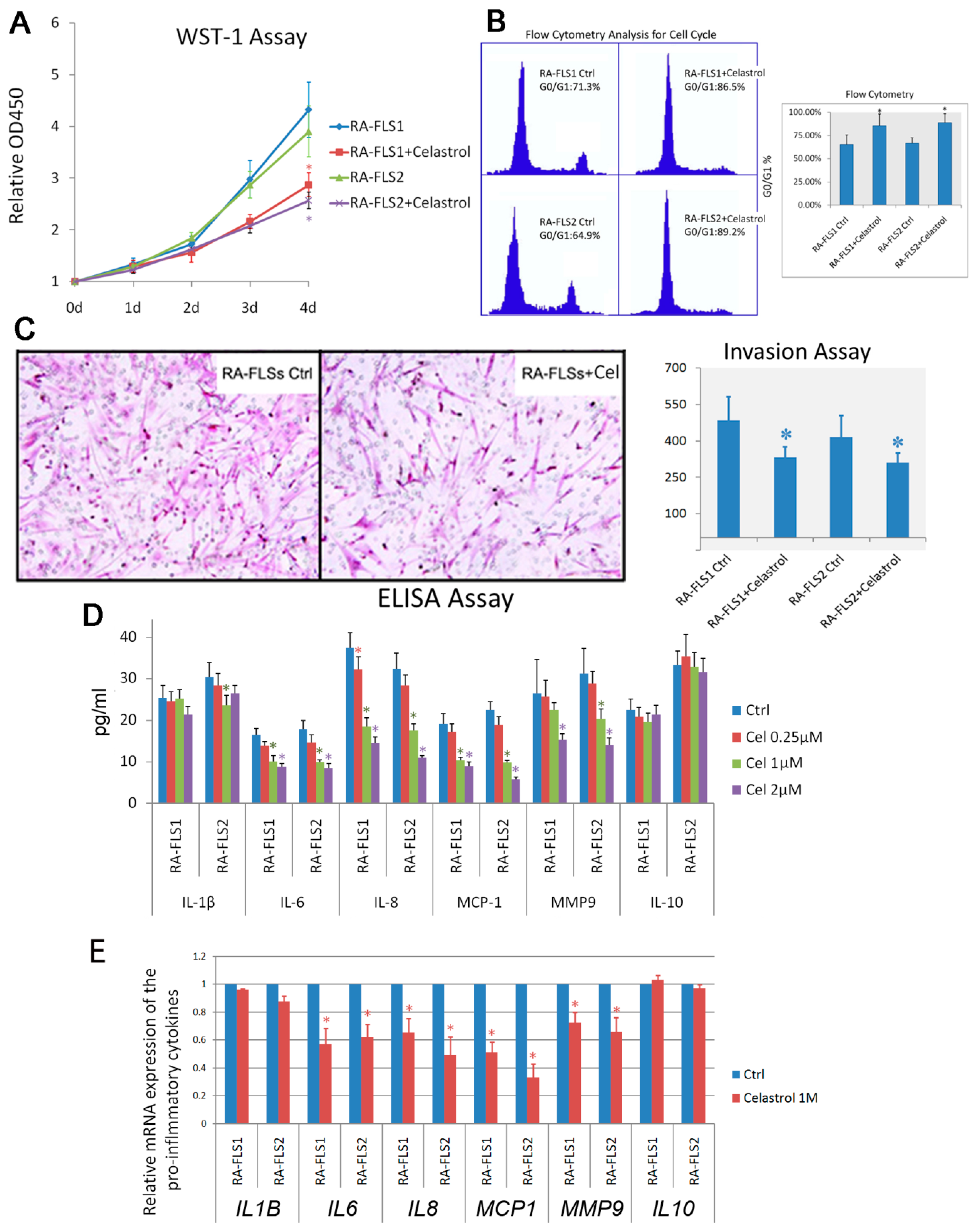
3.2. Treatment of Celastrol Attenuated the Activation Status of RA–FLSs
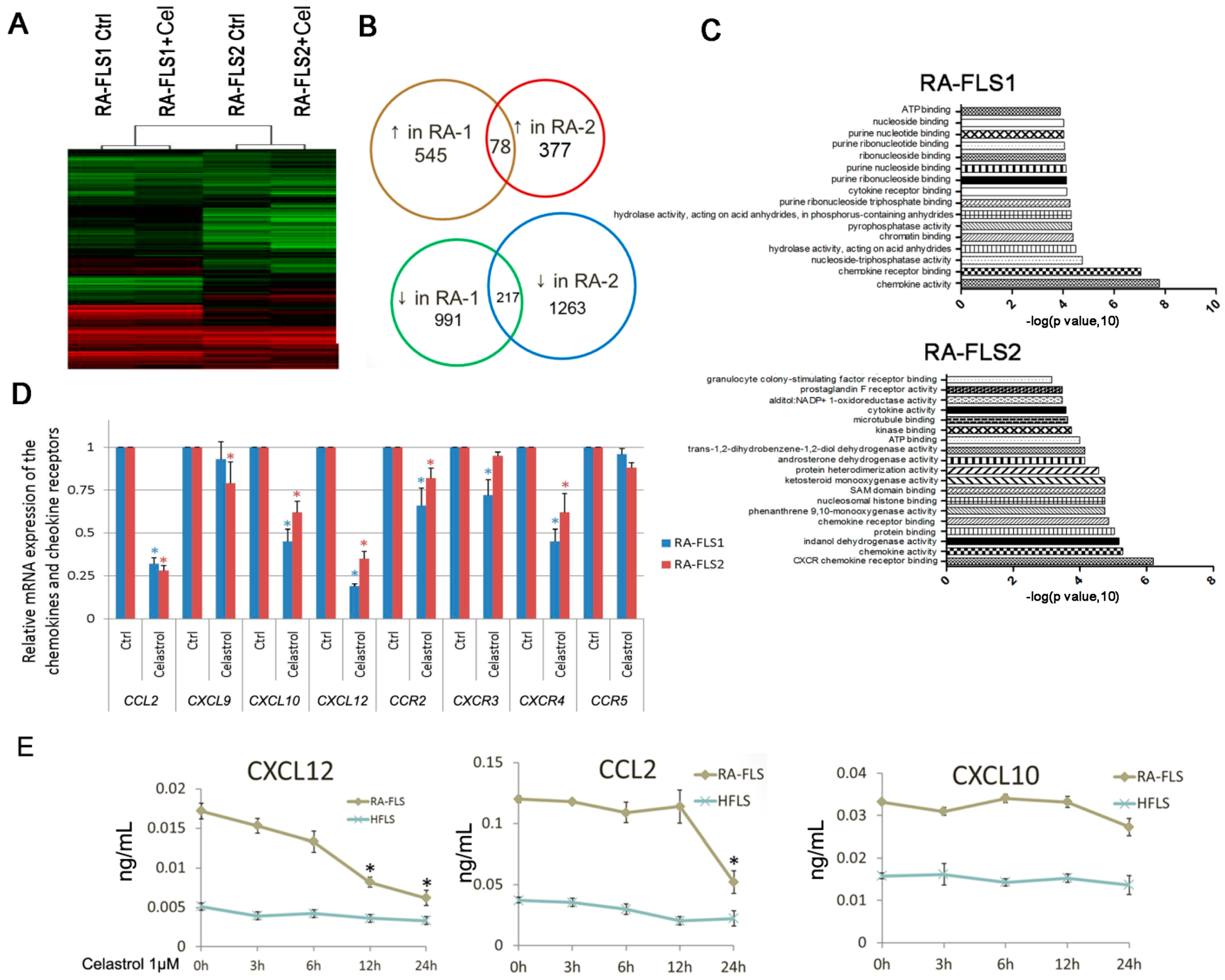
3.3. Expression of Several Chemokine and Chemokine Receptors Was Influenced by Celastrol Treatment in RA–FLSs
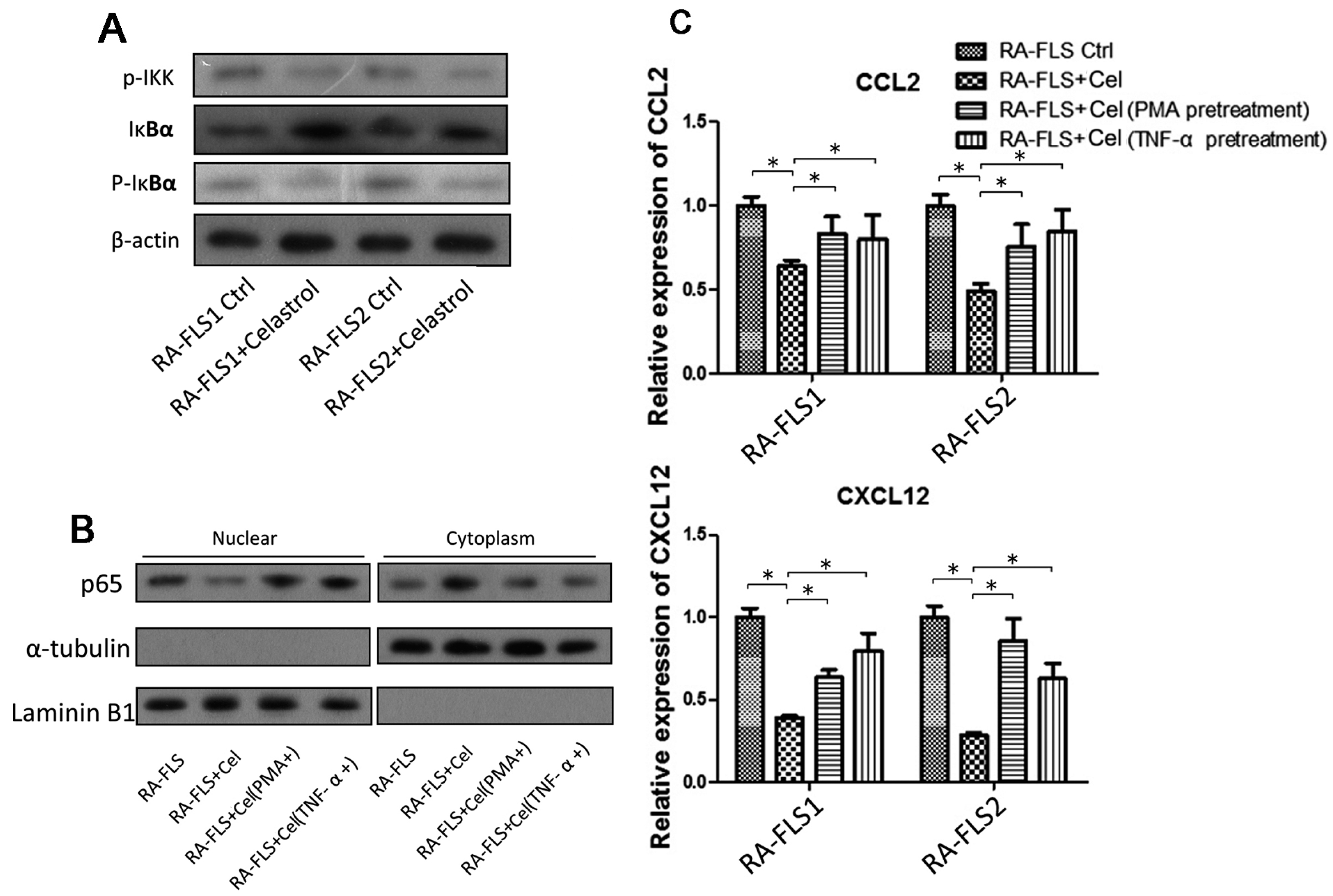
3.4. NF-κB Signaling Pathway Was Partially Involved in the Celastrol-Mediated Regulation of CCL2 and CXCL12
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wordsworth, P. Rheumatoid arthritis. Curr. Opin. Immunol. 1992, 4, 766–769. [Google Scholar] [CrossRef]
- Trentham, D.E. The immunopathogenesis of rheumatoid arthritis. J. Rheumatol. 1989, 12, 7–10. [Google Scholar]
- Izquierdo, E.; Cañete, J.D.; Celis, R.; Del Rey, M.J.; Usategui, A.; Marsal, S.; Sanmarti, R.; Criado, G.; Pablos, J.L. Synovial fibroblast hyperplasia in rheumatoid arthritis: Clinicopathologic correlations and partial reversal by anti-tumor necrosis factor therapy. Arthritis Rheumatol. 2011, 63, 2575–2583. [Google Scholar] [CrossRef] [PubMed]
- Henderson, B.; Revell, P.A.; Edwards, J.C. Synovial lining cell hyperplasia in rheumatoid arthritis: Dogma and fact. Ann. Rheumatol. Dis. 1988, 47, 348–349. [Google Scholar] [CrossRef]
- Huber, L.C.; Distler, O.; Tarner, I.; Gay, R.E.; Gay, S.; Pap, T. Synovial fibroblasts: Key players in rheumatoid arthritis. Rheumatology 2006, 45, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Mor, A.; Abramson, S.B.; Pillinger, M.H. The fibroblast-like synovial cell in rheumatoid arthritis: A key player in inflammation and joint destruction. Clin. Immunol. 2005, 115, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Firestein, G.S. Invasive fibroblast-like synoviocytes in rheumatoid arthritis. Passive responders or transformed aggressors? Arthritis Rheumatol. 1996, 39, 1781–1790. [Google Scholar] [CrossRef]
- Klein, K.; Gay, S. Epigenetic modifications in rheumatoid arthritis, a review. Curr. Opin. Pharmacol. 2012, 13, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Klein, K.; Ospelt, C.; Gay, S. Epigenetic contributions in the development of rheumatoid arthritis. Arthritis Res. Ther. 2012, 14, 227. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, Y.; Kozawa, E.; Urakawa, H.; Arai, E.; Futamura, N.; Zhuo, L.; Kimata, K.; Ishiguro, N.; Nishida, Y. Suppression of hyaluronan synthesis alleviates inflammatory responses in murine arthritis and in human rheumatoid synovial fibroblasts. Arthritis Rheumatol. 2013, 65, 1160–1170. [Google Scholar] [CrossRef] [PubMed]
- Huh, J.E.; Seo, B.K.; Park, Y.C.; Kim, J.I.; Lee, J.D.; Choi, D.Y.; Baek, Y.H.; Park, D.S. WIN-34B, a new herbal medicine, inhibits the inflammatory response by inactivating IκB-α phosphorylation and mitogen activated protein kinase pathways in fibroblast-like synoviocytes. J. Ethnopharmacol. 2012, 143, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Feng, X.; Tan, W.; Gu, W.; Guo, D.; Zhang, M.; Wang, F. IL-29 enhances Toll-like receptor-mediated IL-6 and IL-8 production by the synovial fibroblasts from rheumatoid arthritis patients. Arthritis Res. Ther. 2013, 15, R170. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Chen, J.W.; Gao, J.S.; Li, L.; Xie, X. Resveratrol inhibits TNF-α-induced IL-1β, MMP-3 production in human rheumatoid arthritis fibroblast-like synoviocytes via modulation of PI3kinase/Akt pathway. Rheumatol. Int. 2013, 33, 1829–1835. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.R.; Xu, L.F.; Ma, L.; Wang, D.H.; Gao, J.W. Studies on pharmacological actions of total glycosides in Tripterygium wilfordii. Zhongguo Yi Xue Ke Xue Yuan Xue Bao 1983, 5, 1–8. [Google Scholar] [PubMed]
- Ni, L.; Zhang, X.M.; Zhou, X.; Ma, J.; Li, C.J.; Li, L.; Zhang, T.T.; Zhang, D.M. Megastigmane Glycosides from the Leaves of Tripterygium wilfordii. Nat. Prod. Commun. 2015, 10, 2023–2026. [Google Scholar] [PubMed]
- Tao, X.; Sun, Y.; Zhang, N. Treatment of rheumatoid arthritis with low doses of multi-glycosides of Tripterygium wilfordii. Zhong Xi Yi Jie He Za Zhi 1990, 10, 261–262. [Google Scholar]
- Xu, Z.; Wu, G.; Wei, X.; Chen, X.; Wang, Y.; Chen, L. Celastrol induced DNA damage, cell cycle arrest, and apoptosis in human rheumatoid fibroblast-like synovial cells. Am. J. Chin. Med. 2013, 41, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Li, G.Q.; Zhang, Y.; Liu, D.; Qian, Y.Y.; Zhang, H.; Guo, S.Y.; Sunagawa, M.; Hisamitsu, T.; Liu, Y.Q. Celastrol inhibits interleukin-17A-stimulated rheumatoid fibroblast-like synoviocyte migration and invasion through suppression of NF-κB-mediated matrix metalloproteinase-9 expression. Int. Immunopharmacol. 2012, 14, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Bhambhani, M. American Rheumatism Association (ARA) criteria for the classification of rheumatoid arthritis (RA). J. Assoc. Physicians India 1996, 44, 89. [Google Scholar] [PubMed]
- Ishida, N.; Hara, T.; Kamura, T.; Yoshida, M.; Nakayama, K.; Nakayama, K.I. Phosphorylation of p27Kip1 on serine 10 is required for its binding to CRM1 and nuclear export. J. Biol. Chem. 2002, 277, 14355–14358. [Google Scholar] [CrossRef] [PubMed]
- Berg, K.D.; Tamas, R.M.; Riemann, A.; Niels-Christiansen, L.L.; Hansen, G.H.; Michael Danielsen, E. Caveolae in fibroblast-like synoviocytes: Static structures associated with vimentin-based intermediate filaments. Histochem. Cell Biol. 2009, 131, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Trabandt, A.; Aicher, W.K.; Gay, R.E.; Sukhatme, V.P.; Nilson-Hamilton, M.; Hamilton, R.T.; McGhee, J.R.; Fassbender, H.G.; Gay, S. Expression of the collagenolytic and Ras-induced cysteine proteinase cathepsin L and proliferation-associated oncogenes in synovial cells of MRL/I mice and patients with rheumatoid arthritis. Matrix 1990, 10, 349–361. [Google Scholar] [CrossRef]
- Ritchlin, C.T.; Winchester, R.J. Potential mechanisms for coordinate gene activation in the rheumatoid synoviocyte: Implications and hypotheses. Springer Semin. Immunopathol. 1989, 11, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Case, J.P.; Lafyatis, R.; Remmers, E.F.; Kumkumian, G.K.; Wilder, R.L. Transin/stromelysin expression in rheumatoid synovium. A transformation-associated metalloproteinase secreted by phenotypically invasive synoviocytes. Am. J. Pathol. 1989, 135, 1055–1064. [Google Scholar] [PubMed]
- Trabandt, A.; Aicher, W.K.; Gay, R.E.; Sukhatme, V.P.; Fassbender, H.G.; Gay, S. Spontaneous expression of immediately-early response genes c-fos and egr-1 in collagenase-producing rheumatoid synovial fibroblasts. Rheumatol. Int. 1992, 12, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Scheinman, R. NF-κB and rheumatoid arthritis: Will understanding genetic risk lead to a therapeutic reward? Forum Immunopathol. Dis. Ther. 2013, 4, 93–110. [Google Scholar] [CrossRef] [PubMed]
- Van Loo, G.; Beyaert, R. Negative regulation of NF-κB and its involvement in rheumatoid arthritis. Arthritis Res. Ther. 2011, 13, 221. [Google Scholar] [CrossRef] [PubMed]
- Roman-Blas, J.A.; Jimenez, S.A. Targeting NF-κB: A promising molecular therapy in inflammatory arthritis. Int. Rev. Immunol. 2008, 27, 351–374. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, R.E.; Foxwell, B.M. Signalling, inflammation and arthritis: NF-κB and its relevance to arthritis and inflammation. Rheumatology 2008, 47, 584–590. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse |
|---|---|---|
| IL-1β | GCTTATTACAGTGGCAATGAGGAT | TAGTGGTGGTCGGAGATTCG |
| IL-6 | GCCACTCACCTCTTCAGAAC | GCAAGTCTCCTCATTGAATCCA |
| IL-8 | AGGACAAGAGCCAGGAAGAA | GGGTGGAAAGGTTTGGAGTATG |
| CCL2 | CTGTGCCTGCTGCTCATAG | CTTGCTGCTGGTGATTCTTCT |
| MMP-3 | CAGCAAGGCATAGAGACAACAT | CGCACAGCAACAGTAGGATT |
| IL-10 | GCCAAGCCTTGTCTGAGATG | GCATTCTTCACCTGCTCCAC |
| CXCL9 | CCCTGTTTCTTCCACAGTGC | GCACCTGCTCTGAGACAATG |
| CXCL10 | AAGGATGGACCACACAGAGG | AGTAGCAGCTGATTTGGTGAC |
| CXCL12 | TGGGCACATTGATCTGGGAT | CAGGTACAGGGCATGGATGA |
| CCR2 | CAGTTGCTGAGAAGCCTGAC | AGAACGAGATGTGGACAGCA |
| CXCR3 | AGGTGCCCTCTTCAACATCA | GCTGGGTGGCATGAACTATG |
| CXCR4 | GAGGCCCTAGCTTTCTTCCA | GAATGTCCACCTCGCTTTCC |
| CCR5 | CTTCTGGGCTCCCTACAACA | GTCACCTGCATAGCTTGGTC |
| ACTB | GACCTGACTGACTACCTCATGAAGAT | GTCACACTTCATGATGGAGTTGAAGG |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, Z.; He, D.; Yu, B.; Liu, F.; Zuo, J.; Li, Y.; Lin, Q.; Zhou, X.; Wang, Q. High-Throughput Study of the Effects of Celastrol on Activated Fibroblast-Like Synoviocytes from Patients with Rheumatoid Arthritis. Genes 2017, 8, 221. https://doi.org/10.3390/genes8090221
Fang Z, He D, Yu B, Liu F, Zuo J, Li Y, Lin Q, Zhou X, Wang Q. High-Throughput Study of the Effects of Celastrol on Activated Fibroblast-Like Synoviocytes from Patients with Rheumatoid Arthritis. Genes. 2017; 8(9):221. https://doi.org/10.3390/genes8090221
Chicago/Turabian StyleFang, Zhengyu, Dongyi He, Bo Yu, Feng Liu, Jianping Zuo, Yuxia Li, Qi Lin, Xiaodong Zhou, and Qingwen Wang. 2017. "High-Throughput Study of the Effects of Celastrol on Activated Fibroblast-Like Synoviocytes from Patients with Rheumatoid Arthritis" Genes 8, no. 9: 221. https://doi.org/10.3390/genes8090221
APA StyleFang, Z., He, D., Yu, B., Liu, F., Zuo, J., Li, Y., Lin, Q., Zhou, X., & Wang, Q. (2017). High-Throughput Study of the Effects of Celastrol on Activated Fibroblast-Like Synoviocytes from Patients with Rheumatoid Arthritis. Genes, 8(9), 221. https://doi.org/10.3390/genes8090221