
Regulation and Modulation of Human DNA Polymerase δ Activity and Function

Abstract
1. Introduction
1.1. Brief Historical Background
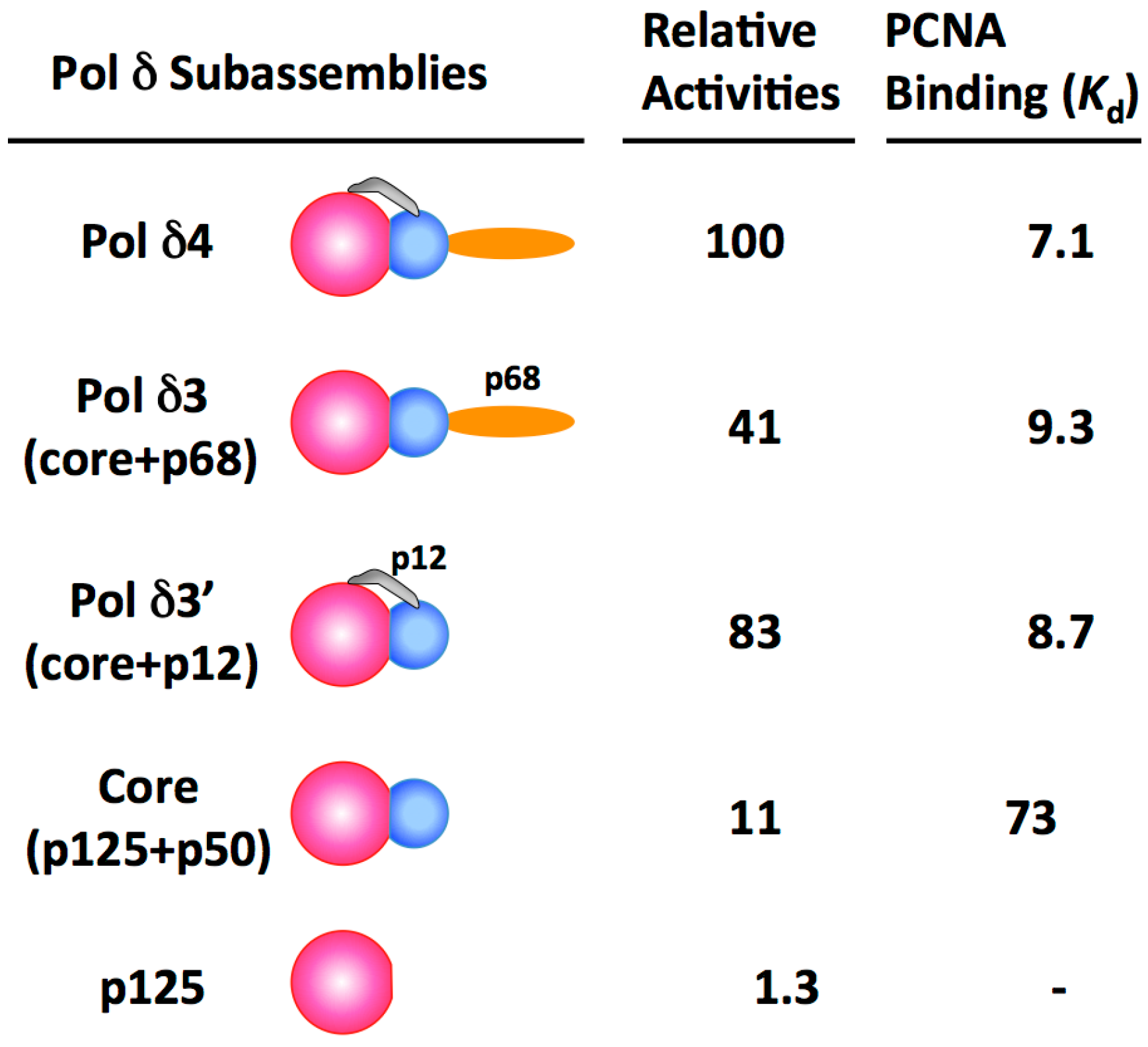
1.2. Properties of Human Pol δ4 and Its Subassemblies and the Roles of the p68 and p12 Subunits
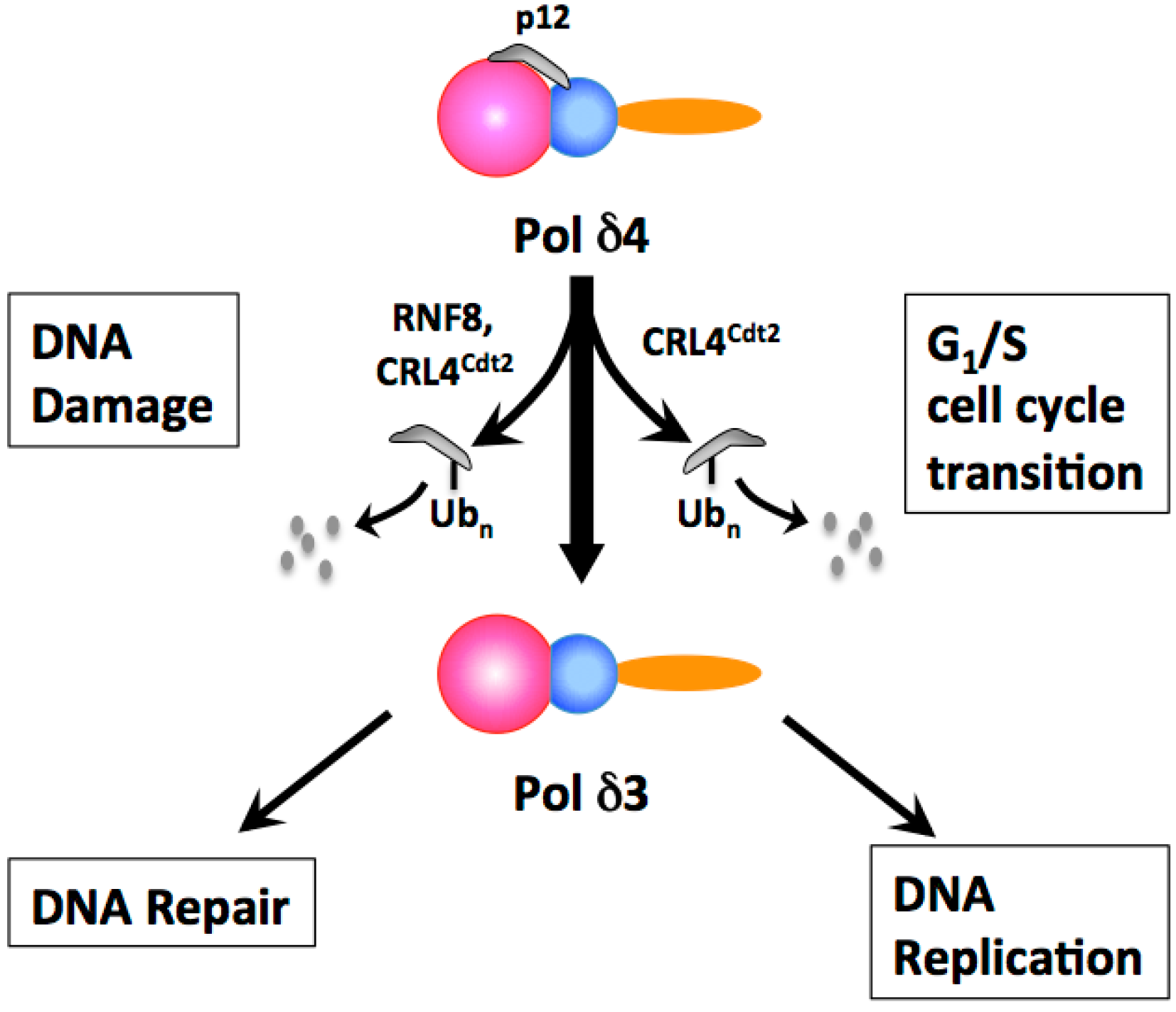
2. Alteration in Subunit Composition by the Degradation of the p12 Subunit Is the Key Mechanism for the Regulation of Human Pol δ
2.1. The Degradation of the p12 Subunit of Pol δ in Response to DNA Damage
- p12 is rapidly lost in a variety of cell types, in a UV flux- and time-dependent manner, followed by a slower recovery over 24 h.
- Treatment with alkylating agents such as methyl methanesulfonate (MMS) or agents inducing replication stress (hydroxyurea and aphidicolin) also caused p12 degradation.
- The loss of p12 is due to an accelerated rate of proteasomal degradation initiated by its polyubiquitination.
- Degradation of p12 is dependent on ATR signaling, but not on ATM, as shown by the use of ATR or ATM depleted cells.
- The p12 subunit of Pol δ is selectively targeted, and similar changes are not observed for the other three subunits.
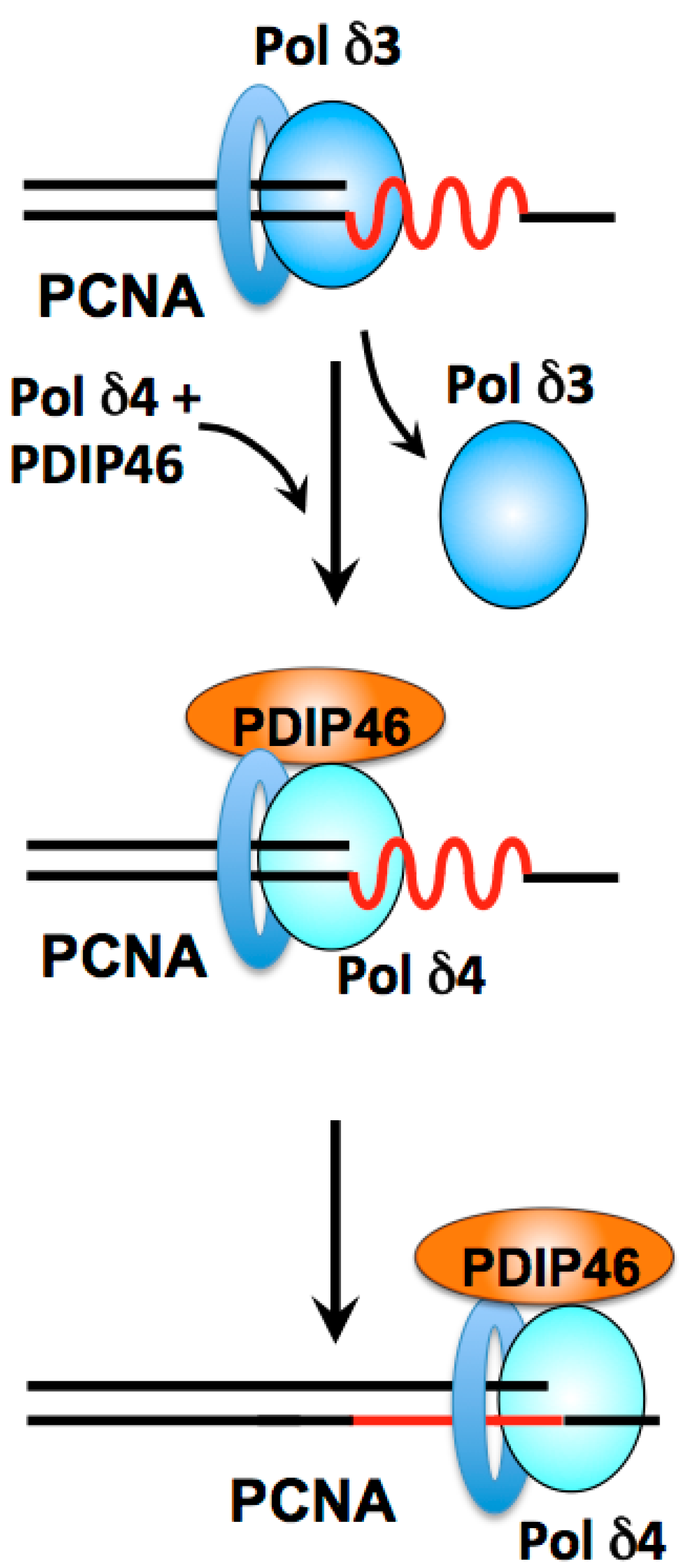
- Loss of the p12 subunit leads to the in vivo conversion of Pol δ4 to the heterotrimer, Pol δ3.
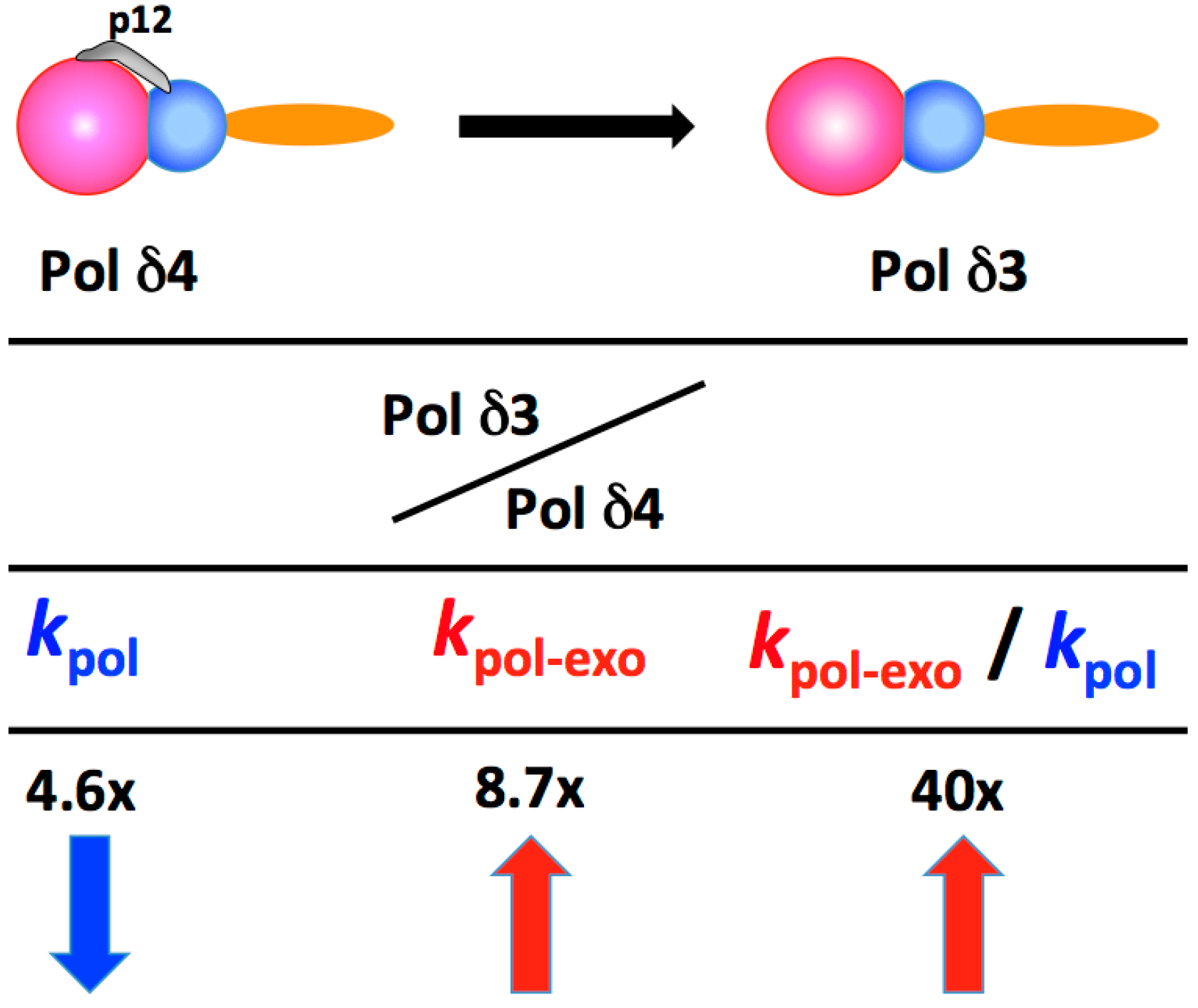
2.2. Pol δ3 Exhibits Altered Behaviors from Pol δ4 in Lesion Bypass and in Extension of Mismatched Primers that Represent a Gain of Function
2.3. Spatiotemporal Analysis of the Recruitment of Pol δ to Sites of UV Damage Indicates Pol δ3 Is in the Right Place at the Right Time
2.4. Conversion of Pol δ4 to Pol δ3 May Facilitate the Switch between Pol δ and Pol η
2.5. Does the Plasticity of Pol δ Subunit Composition Extend to Other Subunits besides p12?
2.6. RNF8 Is Involved in DNA Damage-Induced p12 Degradation
2.7. Degradation of p12 by CRL4Cdt2
2.8. Mechanism and Characteristics of Okazaki Fragment Processing by Pol δ4 and Pol δ3
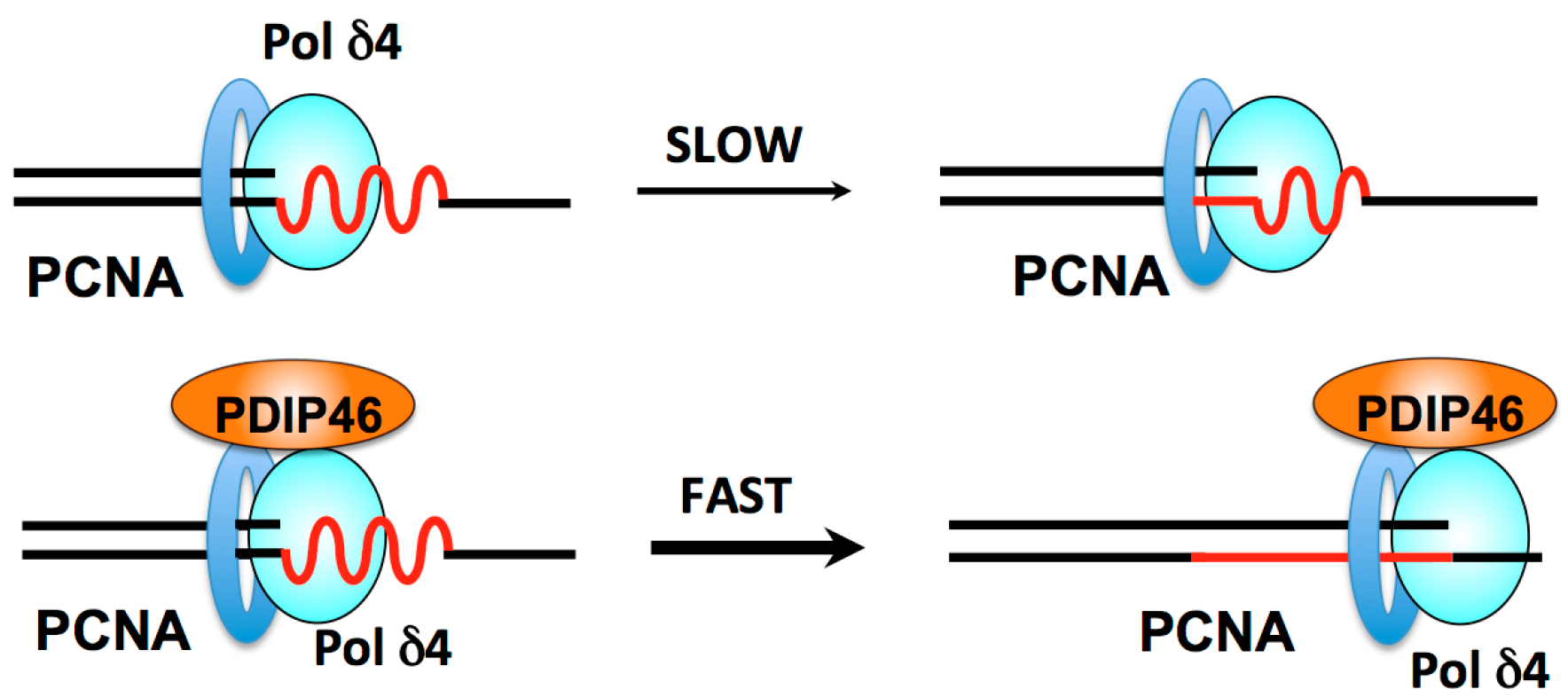
3. Role of the Pol δ Binding Protein PDIP46/Poldip3 in DNA Replication and Repair
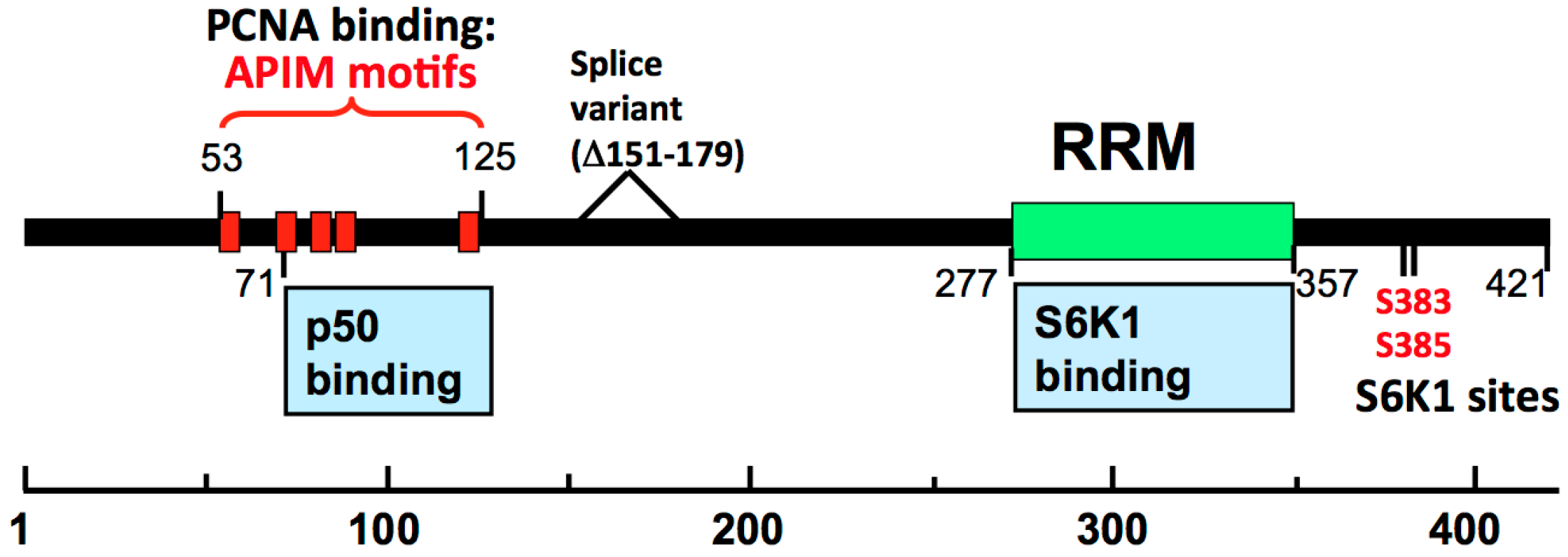
3.1. Mapping of the Interaction Sites between PDIP46 and Pol δ /PCNA Reveals that These Are Located in a Region Separate from Those Involved in S6K1 Binding
3.2. Evidence that PDIP46 Is Associated with Pol δ In Vivo
3.3. PDIP46 Is a Potent Activator of Pol δ
3.4. Future Horizons: Accommodating Two Forms of Pol δ and PDIP46 at the Replication Fork
3.4.1. Roles of Pol δ3, Pol δ4 and PDIP46 in Lagging Strand Synthesis
3.4.2. Roles of Pol δ4 and PDIP46 in Leading Strand Synthesis
4. PDIP38/Poldip2: A Multi-Faceted Protein
4.1. PDIP38 Is a Mitochondrial Protein with Multiple Subcellular Localizations
4.2. Interaction of PDIP38/Poldip2 with Pol η and Other TLS Polymerases: Involvement of PDIP38 in the DNA Damage Tolerance Pathway
4.3. PDIP38 Responds to Genotoxic and Transcriptional Stress by Translocation to the Spliceosomes/Nuclear Speckles and Is Involved in Regulation of the Alternative Splicing of Mdm2
4.4. PDIP38 Binds to p22phox and Regulates the Activity of the Nox4/p22phox NADPH Oxidase
4.5. Summary
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kornberg, A. Ten commandments: Lessons from the enzymology of DNA replication. J. Bacteriol. 2000, 182, 3613–3618. [Google Scholar] [CrossRef] [PubMed]
- Barnes, R.; Eckert, K. Maintenance of Genome Integrity: How Mammalian Cells Orchestrate Genome Duplication by Coordinating Replicative and Specialized DNA Polymerases. Genes 2017, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, E.; Golemis, E.A.; Arora, S. POLD1: Central mediator of DNA replication and repair, and implication in cancer and other pathologies. Gene 2016, 590, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Weissbach, A. Eukaryotic DNA polymerases. Annu. Rev. Biochem. 1977, 46, 25–47. [Google Scholar] [CrossRef] [PubMed]
- Brutlag, D.; Kornberg, A. Enzymatic synthesis of deoxyribonucleic acid. 36. A proofreading function for the 3′ leads to 5′ exonuclease activity in deoxyribonucleic acid polymerases. J. Biol. Chem. 1972, 247, 241–248. [Google Scholar] [PubMed]
- Muzyczka, N.; Poland, R.L.; Bessman, M.J. Studies on the biochemical basis of spontaneous mutation. I. A comparison of the deoxyribonucleic acid polymerases of mutator, antimutator, and wild type strains of bacteriophage T4. J. Biol. Chem. 1972, 247, 7116–7122. [Google Scholar] [PubMed]
- Reha-Krantz, L.J. DNA polymerase proofreading: Multiple roles maintain genome stability. Biochim. Biophys. Acta 2010, 1804, 1049–1063. [Google Scholar] [CrossRef] [PubMed]
- Byrnes, J.J.; Downey, K.M.; Black, V.; Esserman, L.; So, A.G. Selective Inhibition of the 3′ to 5′ Exonuclease Activity Associated with Mammalian DNA Polymerase δ. In Miami Winter Symposium: Cancer Enzymology; Schultz, J., Ahmad, F., Eds.; Academic Press: Cambridge, MA, USA, 1976; Volume 12, pp. 245–264. [Google Scholar]
- Byrnes, J.J.; Downey, K.M.; Black, V.L.; So, A.G. A new mammalian DNA polymerase with 3′ to 5′ exonuclease activity: DNA polymerase δ. Biochemistry 1976, 15, 2817–2823. [Google Scholar] [CrossRef] [PubMed]
- Byrnes, J.J.; Downey, K.M.; Que, B.G.; Lee, M.Y.; Black, V.L.; So, A.G. Selective inhibition of the 3′ to 5′ exonuclease activity associated with DNA polymerases: A mechanism of mutagenesis. Biochemistry 1977, 16, 3740–3746. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; Byrnes, J.J.; Downey, K.M.; So, A.G. Mechanism of inhibition of deoxyribonucleic acid synthesis by 1-beta-D-arabinofuranosyladenosine triphosphate and its potentiation by 6-mercaptopurine ribonucleoside 5′-monophosphate. Biochemistry 1980, 19, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; Tan, C.K.; So, A.G.; Downey, K.M. Purification of deoxyribonucleic acid polymerase δ from calf thymus: Partial characterization of physical properties. Biochemistry 1980, 19, 2096–2101. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; Tan, C.K.; Downey, K.M.; So, A.G. Structural and functional properties of calf thymus DNA polymerase δ. Prog. Nucleic Acid Res. Mol. Biol. 1981, 26, 83–96. [Google Scholar] [PubMed]
- Lee, M.Y.; Tan, C.K.; Downey, K.M.; So, A.G. Further studies on calf thymus DNA polymerase δ purified to homogeneity by a new procedure. Biochemistry 1984, 23, 1906–1913. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; Toomey, N.L.; Wright, G.E. Differential inhibition of human placental DNA polymerases δ and α by BuPdGTP and BuAdATP. Nucleic Acids Res. 1985, 13, 8623–8630. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; Toomey, N.L. Human placental DNA polymerase δ: Identification of a 170-kilodalton polypeptide by activity staining and immunoblotting. Biochemistry 1987, 26, 1076–1085. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y. Isolation of multiple forms of DNA polymerase δ: Evidence of proteolytic modification during isolation. Biochemistry 1988, 27, 5188–5193. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; Alejandro, R.; Toomey, N.L. Immunochemical studies of DNA polymerase δ: Relationships with DNA polymerase α. Arch. Biochem. Biophys. 1989, 272, 1–9. [Google Scholar] [CrossRef]
- Lee, M.Y.; Jiang, Y.Q.; Zhang, S.J.; Toomey, N.L. Characterization of human DNA polymerase δ and its immunochemical relationships with DNA polymerase α and epsilon. J. Biol. Chem. 1991, 266, 2423–2429. [Google Scholar] [PubMed]
- Wong, S.W.; Syvaoja, J.; Tan, C.K.; Downey, K.M.; So, A.G.; Linn, S.; Wang, T.S. DNA polymerases α and δ are immunologically and structurally distinct. J. Biol. Chem. 1989, 264, 5924–5928. [Google Scholar] [PubMed]
- Zhang, J.; Chung, D.W.; Tan, C.K.; Downey, K.M.; Davie, E.W.; So, A.G. Primary structure of the catalytic subunit of calf thymus DNA polymerase δ: Sequence similarities with other DNA polymerases. Biochemistry 1991, 30, 11742–11750. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.; Jiang, Y.; Zhang, S.J.; Zhang, P.; Zeng, R.X.; Lee, M.Y. Structural and functional relationships of human DNA polymerases. Chromosoma 1992, 102, S121–S127. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.L.; Chang, L.S.; Zhang, P.; Hao, H.; Zhu, L.; Toomey, N.L.; Lee, M.Y. Molecular cloning of the cDNA for the catalytic subunit of human DNA polymerase δ. Nucleic Acids Res. 1992, 20, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Crute, J.J.; Wahl, A.F.; Bambara, R.A. Purification and characterization of two new high molecular weight forms of DNA polymerase δ. Biochemistry 1986, 25, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Syvaoja, J.; Linn, S. Characterization of a large form of DNA polymerase δ from HeLa cells that is insensitive to proliferating cell nuclear antigen. J. Biol. Chem. 1989, 264, 2489–2497. [Google Scholar] [PubMed]
- Burgers, P.M.; Bambara, R.A.; Campbell, J.L.; Chang, L.M.; Downey, K.M.; Hubscher, U.; Lee, M.Y.; Linn, S.M.; So, A.G.; Spadari, S. Revised nomenclature for eukaryotic DNA polymerases. Eur. J. Biochem. 1990, 191, 617–618. [Google Scholar] [CrossRef] [PubMed]
- Pospiech, H.; Syvaoja, J.E. DNA polymerase epsilon—More than a polymerase. Sci. World J. 2003, 3, 87–104. [Google Scholar] [CrossRef] [PubMed]
- Prelich, G.; Tan, C.K.; Kostura, M.; Mathews, M.B.; So, A.G.; Downey, K.M.; Stillman, B. Functional identity of proliferating cell nuclear antigen and a DNA polymerase-δ auxiliary protein. Nature 1987, 326, 517–520. [Google Scholar] [CrossRef] [PubMed]
- Choe, K.N.; Moldovan, G.L. Forging Ahead through Darkness: PCNA, Still the Principal Conductor at the Replication Fork. Mol. Cell 2017, 65, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.; Liu, L.; Leon, A.; Mazloum, N.; Lee, M.Y. Evidence that DNA polymerase δ isolated by immunoaffinity chromatography exhibits high-molecular weight characteristics and is associated with the KIAA0039 protein and RPA. Biochemistry 2000, 39, 7245–7254. [Google Scholar] [CrossRef] [PubMed]
- Hughes, P.; Tratner, I.; Ducoux, M.; Piard, K.; Baldacci, G. Isolation and identification of the third subunit of mammalian DNA polymerase δ by PCNA-affinity chromatography of mouse FM3A cell extracts. Nucleic Acids Res. 1999, 27, 2108–2114. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Mo, J.; Rodriguez-Belmonte, E.M.; Lee, M.Y. Identification of a fourth subunit of mammalian DNA polymerase δ. J. Biol. Chem. 2000, 275, 18739–18744. [Google Scholar] [CrossRef] [PubMed]
- Garg, P.; Burgers, P.M. DNA polymerases that propagate the eukaryotic DNA replication fork. Crit. Rev. Biochem. Mol. Biol. 2005, 40, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Gerik, K.J.; Li, X.; Pautz, A.; Burgers, P.M. Characterization of the two small subunits of Saccharomyces cerevisiae DNA polymerase δ. J. Biol. Chem. 1998, 273, 19747–19755. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, N.; Watt, A.; Fantes, P.A.; MacNeill, S.A. Cdm1, the smallest subunit of DNA polymerase d in the fission yeast Schizosaccharomyces pombe, is non-essential for growth and division. Curr. Genet. 1998, 34, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Zuo, S.; Gibbs, E.; Kelman, Z.; Wang, T.S.; O’Donnell, M.; MacNeill, S.A.; Hurwitz, J. DNA polymerase δ isolated from Schizosaccharomyces pombe contains five subunits. Proc. Natl. Acad. Sci. USA 1997, 94, 11244–11249. [Google Scholar] [CrossRef]
- Zuo, S.; Bermudez, V.; Zhang, G.; Kelman, Z.; Hurwitz, J. Structure and activity associated with multiple forms of Schizosaccharomyces pombe DNA polymerase δ. J. Biol. Chem. 2000, 275, 5153–5162. [Google Scholar] [CrossRef] [PubMed]
- MacNeill, S.A.; Moreno, S.; Reynolds, N.; Nurse, P.; Fantes, P.A. The fission yeast Cdc1 protein, a homologue of the small subunit of DNA polymerase δ, binds to Pol3 and Cdc27. EMBO J. 1996, 15, 4613–4628. [Google Scholar] [PubMed]
- Bermudez, V.P.; MacNeill, S.A.; Tappin, I.; Hurwitz, J. The influence of the Cdc27 subunit on the properties of the Schizosaccharomyces pombe DNA polymerase δ. J. Biol. Chem. 2002, 277, 36853–36862. [Google Scholar] [CrossRef] [PubMed]
- Johansson, E.; Majka, J.; Burgers, P.M. Structure of DNA polymerase δ from Saccharomyces cerevisiae. J. Biol. Chem. 2001, 276, 43824–43828. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Mo, J.Y.; Perez, A.; Leon, A.; Liu, L.; Mazloum, N.; Xu, H.; Lee, M.Y. Direct interaction of proliferating cell nuclear antigen with the p125 catalytic subunit of mammalian DNA polymerase δ. J. Biol. Chem. 1999, 274, 26647–26653. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Sun, Y.; Hsu, H.; Zhang, L.; Zhang, Y.; Lee, M.Y. The interdomain connector loop of human PCNA is involved in a direct interaction with human polymerase δ. J. Biol. Chem. 1998, 273, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Roos, G.; Jiang, Y.; Landberg, G.; Nielsen, N.H.; Zhang, P.; Lee, M.Y. Determination of the epitope of an inhibitory antibody to proliferating cell nuclear antigen. Exp. Cell Res. 1996, 226, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.J.; Zeng, X.R.; Zhang, P.; Toomey, N.L.; Chuang, R.Y.; Chang, L.S.; Lee, M.Y. A conserved region in the amino terminus of DNA polymerase δ is involved in proliferating cell nuclear antigen binding. J. Biol. Chem. 1995, 270, 7988–7992. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xie, B.; Zhou, Y.; Rahmeh, A.; Trusa, S.; Zhang, S.; Gao, Y.; Lee, E.Y.; Lee, M.Y. Functional roles of p12, the fourth subunit of human DNA polymerase δ. J. Biol. Chem. 2006, 281, 14748–14755. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Tan, C.K.; Zhou, J.Q.; You, M.; Carastro, L.M.; Downey, K.M.; So, A.G. Direct interaction of proliferating cell nuclear antigen with the small subunit of DNA polymerase δ. J. Biol. Chem. 2002, 277, 24340–24345. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, Q.; Chen, H.; Li, X.; Mai, W.; Chen, K.; Zhang, S.; Lee, E.Y.; Lee, M.Y.; Zhou, Y. p50, the Small Subunit of DNA Polymerase Δ, Is Required for Mediation of the Interaction of Polymerase Δ Subassemblies with PCNA. PLoS ONE 2011, 6, e27092. [Google Scholar] [CrossRef] [PubMed]
- Rahmeh, A.A.; Zhou, Y.; Xie, B.; Li, H.; Lee, E.Y.; Lee, M.Y. Phosphorylation of the p68 Subunit of Pol Δ Acts as a Molecular Switch to Regulate Its Interaction with PCNA. Biochemistry 2012, 51, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Podust, V.N.; Chang, L.S.; Ott, R.; Dianov, G.L.; Fanning, E. Reconstitution of human DNA polymerase δ using recombinant baculoviruses: The p12 subunit potentiates DNA polymerizing activity of the four-subunit enzyme. J. Biol. Chem. 2002, 277, 3894–3901. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Mazloum, N.; Liu, L.; Rahmeh, A.; Li, H.; Lee, M.Y. Reconstitution and characterization of the human DNA polymerase δ four-subunit holoenzyme. Biochemistry 2002, 41, 13133–13142. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Meng, X.; Zhang, S.; Lee, E.Y.; Lee, M.Y. Characterization of human DNA polymerase δ and its subassemblies reconstituted by expression in the multibac system. PLoS ONE 2012, 7, e39156. [Google Scholar] [CrossRef] [PubMed]
- Masuda, Y.; Suzuki, M.; Piao, J.; Gu, Y.; Tsurimoto, T.; Kamiya, K. Dynamics of human replication factors in the elongation phase of DNA replication. Nucleic Acids Res. 2007, 35, 6904–6916. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhang, S.J.; Wu, S.M.; Lee, M.Y. Immunoaffinity purification of DNA polymerase δ. Arch. Biochem. Biophys. 1995, 320, 297–304. [Google Scholar] [CrossRef]
- Podust, V.N.; Georgaki, A.; Strack, B.; Hubscher, U. Calf thymus RF-C as an essential component for DNA polymerase δ and epsilon holoenzymes function. Nucleic Acids Res. 1992, 20, 4159–4165. [Google Scholar] [CrossRef] [PubMed]
- Burgers, P.M.; Gerik, K.J. Structure and processivity of two forms of Saccharomyces cerevisiae DNA polymerase δ. J. Biol. Chem. 1998, 273, 19756–19762. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, S.; Zheng, R.; Yue, F.; Lin, S.H.; Rahmeh, A.A.; Lee, E.Y.; Zhang, Z.; Lee, M.Y. PDIP46 (DNA polymerase δ interacting protein 46) is an activating factor for human DNA polymerase δ. Oncotarget 2016, 7, 6294–6313. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhou, Y.; Xie, B.; Zhang, S.; Rahmeh, A.; Huang, H.S.; Lee, M.Y.; Lee, E.Y. Protein phosphatase-1 is targeted to DNA polymerase δ via an interaction with the p68 subunit. Biochemistry 2008, 47, 11367–11376. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; Zhang, S.; Lin, S.H.; Chea, J.; Wang, X.; LeRoy, C.; Wong, A.; Zhang, Z.; Lee, E.Y. Regulation of human DNA polymerase Δ in the cellular responses to DNA damage. Environ. Mol. Mutagen. 2012, 53, 683–698. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; Zhang, S.; Lin, S.H.; Wang, X.; Darzynkiewicz, Z.; Zhang, Z.; Lee, E.Y. The tail that wags the dog: p12, the smallest subunit of DNA polymerase δ, is degraded by ubiquitin ligases in response to DNA damage and during cell cycle progression. Cell Cycle 2014, 13, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhou, Y.; Trusa, S.; Meng, X.; Lee, E.Y.; Lee, M.Y. A novel DNA damage response: Rapid degradation of the p12 subunit of DNA polymerase δ. J. Biol. Chem. 2007, 282, 15330–15340. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhao, H.; Darzynkiewicz, Z.; Zhou, P.; Zhang, Z.; Lee, E.Y.; Lee, M.Y. A novel function of CRL4Cdt2: Regulation of the subunit structure of DNA polymerase δ in response to DNA damage and during the S phase. J. Biol. Chem. 2013, 288, 29950–29961. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, W.K. The human intra-S checkpoint response to UVC-induced DNA damage. Carcinogenesis 2010, 31, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Cimprich, K.A.; Cortez, D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Flynn, R.L.; Zou, L. ATR: A master conductor of cellular responses to DNA replication stress. Trends Biochem. Sci. 2011, 36, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Conti, C.; Seiler, J.A.; Pommier, Y. The mammalian DNA replication elongation checkpoint: Implication of Chk1 and relationship with origin firing as determined by single DNA molecule and single cell analyses. Cell Cycle 2007, 6, 2760–2767. [Google Scholar] [CrossRef] [PubMed]
- Warren, J.J.; Forsberg, L.J.; Beese, L.S. The structural basis for the mutagenicity of O(6)-methyl-guanine lesions. Proc. Natl. Acad. Sci. USA 2006, 103, 19701–19706. [Google Scholar] [CrossRef] [PubMed]
- Hsu, G.W.; Ober, M.; Carell, T.; Beese, L.S. Error-prone replication of oxidatively damaged DNA by a high-fidelity DNA polymerase. Nature 2004, 431, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Zhou, Y.; Zhang, S.; Lee, E.Y.; Frick, D.N.; Lee, M.Y. DNA damage alters DNA polymerase δ to a form that exhibits increased discrimination against modified template bases and mismatched primers. Nucleic Acids Res. 2009, 37, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.A. Conformational coupling in DNA polymerase fidelity. Annu. Rev. Biochem. 1993, 62, 685–713. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, T.A.; Bebenek, K. DNA replication fidelity. Annu. Rev. Biochem. 2000, 69, 497–529. [Google Scholar] [CrossRef] [PubMed]
- Steitz, T.A. DNA polymerases: Structural diversity and common mechanisms. J. Biol. Chem. 1999, 274, 17395–17398. [Google Scholar] [CrossRef] [PubMed]
- Shamoo, Y.; Steitz, T.A. Building a replisome from interacting pieces: Sliding clamp complexed to a peptide from DNA polymerase and a polymerase editing complex. Cell 1999, 99, 155–166. [Google Scholar] [CrossRef]
- Donlin, M.J.; Patel, S.S.; Johnson, K.A. Kinetic partitioning between the exonuclease and polymerase sites in DNA error correction. Biochemistry 1991, 30, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Khare, V.; Eckert, K.A. The proofreading 3′-5′ exonuclease activity of DNA polymerases: A kinetic barrier to translesion DNA synthesis. Mutat. Res. 2002, 510, 45–54. [Google Scholar] [CrossRef]
- Meng, X.; Zhou, Y.; Lee, E.Y.; Lee, M.Y.; Frick, D.N. The p12 subunit of human polymerase δ modulates the rate and fidelity of DNA synthesis. Biochemistry 2010, 49, 3545–3554. [Google Scholar] [CrossRef] [PubMed]
- Ogi, T.; Limsirichaikul, S.; Overmeer, R.M.; Volker, M.; Takenaka, K.; Cloney, R.; Nakazawa, Y.; Niimi, A.; Miki, Y.; Jaspers, N.G.; et al. Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Mol. Cell 2010, 37, 714–727. [Google Scholar] [CrossRef] [PubMed]
- Terai, K.; Shibata, E.; Abbas, T.; Dutta, A. Degradation of p12 Subunit by CRL4Cdt2 E3 Ligase Inhibits Fork Progression after DNA Damage. J. Biol. Chem. 2013, 288, 30509–30514. [Google Scholar] [CrossRef] [PubMed]
- Bekker-Jensen, S.; Mailand, N. Assembly and function of DNA double-strand break repair foci in mammalian cells. DNA Repair 2010, 9, 1219–1228. [Google Scholar] [CrossRef] [PubMed]
- Mailand, N.; Bekker-Jensen, S.; Faustrup, H.; Melander, F.; Bartek, J.; Lukas, C.; Lukas, J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 2007, 131, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Huen, M.S.; Grant, R.; Manke, I.; Minn, K.; Yu, X.; Yaffe, M.B.; Chen, J. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 2007, 131, 901–914. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.W.; Elledge, S.J. The DNA damage response: Ten years after. Mol. Cell 2007, 28, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Jetten, A.M. RAP80 and RNF8, key players in the recruitment of repair proteins to DNA damage sites. Cancer Lett. 2008, 271, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Sirbu, B.M.; Cortez, D. DNA damage response: Three levels of DNA repair regulation. Cold Spring Harb. Perspect. Biol. 2013, 5, a012724. [Google Scholar] [CrossRef] [PubMed]
- Chea, J.; Zhang, S.; Zhao, H.; Zhang, Z.; Lee, E.Y.; Darzynkiewicz, Z.; Lee, M.Y. Spatiotemporal recruitment of human DNA polymerase δ to sites of UV damage. Cell Cycle 2012, 11, 2885–2895. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.L.; Xu, F.; Xiao, W. Eukaryotic DNA damage tolerance and translesion synthesis through covalent modifications of PCNA. Cell Res. 2008, 18, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Bozza, W.; Zhuang, Z. Ubiquitination of PCNA and its essential role in eukaryotic translesion synthesis. Cell Biochem. Biophys. 2011, 60, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Ghosal, G.; Chen, J. DNA damage tolerance: A double-edged sword guarding the genome. Transl. Cancer Res. 2013, 2, 107–129. [Google Scholar] [PubMed]
- Sale, J.E.; Lehmann, A.R.; Woodgate, R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol. 2012, 13, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Vaisman, A.; Woodgate, R. Translesion DNA polymerases in eukaryotes: What makes them tick? Crit. Rev. Biochem. Mol. Biol. 2017, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Hoege, C.; Pfander, B.; Moldovan, G.L.; Pyrowolakis, G.; Jentsch, S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 2002, 419, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Kannouche, P.L.; Wing, J.; Lehmann, A.R. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: A possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell 2004, 14, 491–500. [Google Scholar] [CrossRef]
- Kannouche, P.L.; Lehmann, A.R. Ubiquitination of PCNA and the polymerase switch in human cells. Cell Cycle 2004, 3, 1011–1013. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, S.; Lin, S.H.; Wang, X.; Wu, L.; Lee, E.Y.; Lee, M.Y. Structure of monoubiquitinated PCNA: Implications for DNA polymerase switching and Okazaki fragment maturation. Cell Cycle 2012, 11, 2128–2136. [Google Scholar] [CrossRef] [PubMed]
- Murga, M.; Lecona, E.; Kamileri, I.; Diaz, M.; Lugli, N.; Sotiriou, S.K.; Anton, M.E.; Mendez, J.; Halazonetis, T.D.; Fernandez-Capetillo, O. POLD3 Is Haploinsufficient for DNA Replication in Mice. Mol. Cell 2016, 63, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Yoshikiyo, K.; Guilbaud, G.; Tsurimoto, T.; Murai, J.; Tsuda, M.; Phillips, L.G.; Narita, T.; Nishihara, K.; Kobayashi, K.; et al. The POLD3 subunit of DNA polymerase δ can promote translesion synthesis independently of DNA polymerase ζ. Nucleic Acids Res. 2015, 43, 1671–1683. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.E.; Prakash, L.; Prakash, S. Pol31 and Pol32 subunits of yeast DNA polymerase δ are also essential subunits of DNA polymerase ζ. Proc. Natl. Acad. Sci. USA 2012, 109, 12455–12460. [Google Scholar] [CrossRef] [PubMed]
- Baranovskiy, A.G.; Lada, A.G.; Siebler, H.; Zhang, Y.; Pavlov, Y.I.; Tahirov, T.H. DNA polymerases δ and ζ switching by sharing the accessory subunits of DNA polymerase δ. J. Biol. Chem. 2012, 287, 17281–17287. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhou, Y.; Sarkeshik, A.; Yates, J.R.; Thomson, T.; Zhang, Z.; Lee, E.Y.; Lee, M.Y. Identification of RNF8 as a Ubiquitin Ligase Involved in Targeting the p12 Subunit of DNA Polymerase δ for Degradation in Response to DNA Damage. J. Biol. Chem. 2013, 288, 2941–2950. [Google Scholar] [CrossRef] [PubMed]
- Brzovic, P.S.; Klevit, R.E. Ubiquitin transfer from the E2 perspective: Why is UbcH5 so promiscuous? Cell Cycle 2006, 5, 2867–2873. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Elledge, S.J. Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proc. Natl. Acad. Sci. USA 2007, 104, 20759–20763. [Google Scholar] [CrossRef] [PubMed]
- Marteijn, J.A.; Bekker-Jensen, S.; Mailand, N.; Lans, H.; Schwertman, P.; Gourdin, A.M.; Dantuma, N.P.; Lukas, J.; Vermeulen, W. Nucleotide excision repair-induced H2A ubiquitination is dependent on MDC1 and RNF8 and reveals a universal DNA damage response. J. Cell Biol. 2009, 186, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Chea, J.; Meng, X.; Zhou, Y.; Lee, E.Y.; Lee, M.Y. PCNA is ubiquitinated by RNF8. Cell Cycle 2008, 7, 3399–3404. [Google Scholar] [CrossRef] [PubMed]
- Abbas, T.; Dutta, A. CRL4Cdt2: Master coordinator of cell cycle progression and genome stability. Cell Cycle 2011, 10, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Hannah, J.; Zhou, P. Distinct and overlapping functions of the cullin E3 ligase scaffolding proteins CUL4A and CUL4B. Gene 2015, 573, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Havens, C.G.; Shobnam, N.; Guarino, E.; Centore, R.C.; Zou, L.; Kearsey, S.E.; Walter, J.C. Direct Role for proliferating cell nuclear antigen (PCNA) in substrate recognition by the E3 Ubiquitin ligase CRL4-Cdt2. J. Biol. Chem. 2012, 287, 11410–11421. [Google Scholar] [CrossRef] [PubMed]
- Havens, C.G.; Walter, J.C. Mechanism of CRL4(Cdt2), a PCNA-dependent E3 ubiquitin ligase. Genes Dev. 2011, 25, 1568–1582. [Google Scholar] [CrossRef] [PubMed]
- Arias, E.E.; Walter, J.C. Replication-dependent destruction of Cdt1 limits DNA replication to a single round per cell cycle in Xenopus egg extracts. Genes Dev. 2005, 19, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Nishitani, H.; Sugimoto, N.; Roukos, V.; Nakanishi, Y.; Saijo, M.; Obuse, C.; Tsurimoto, T.; Nakayama, K.I.; Nakayama, K.; Fujita, M.; et al. Two E3 ubiquitin ligases, SCF-Skp2 and DDB1-Cul4, target human Cdt1 for proteolysis. EMBO J. 2006, 25, 1126–1136. [Google Scholar] [CrossRef] [PubMed]
- Higa, L.A.; Banks, D.; Wu, M.; Kobayashi, R.; Sun, H.; Zhang, H. L2DTL/CDT2 interacts with the CUL4/DDB1 complex and PCNA and regulates CDT1 proteolysis in response to DNA damage. Cell Cycle 2006, 5, 1675–1680. [Google Scholar] [CrossRef] [PubMed]
- Soria, G.; Gottifredi, V. PCNA-coupled p21 degradation after DNA damage: The exception that confirms the rule? DNA Repair 2010, 9, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Bendjennat, M.; Boulaire, J.; Jascur, T.; Brickner, H.; Barbier, V.; Sarasin, A.; Fotedar, A.; Fotedar, R. UV irradiation triggers ubiquitin-dependent degradation of p21(WAF1) to promote DNA repair. Cell 2003, 114, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Higa, L.A.; Mihaylov, I.S.; Banks, D.P.; Zheng, J.; Zhang, H. Radiation-mediated proteolysis of CDT1 by CUL4-ROC1 and CSN complexes constitutes a new checkpoint. Nat. Cell Biol. 2003, 5, 1008–1015. [Google Scholar] [CrossRef] [PubMed]
- Abbas, T.; Sivaprasad, U.; Terai, K.; Amador, V.; Pagano, M.; Dutta, A. PCNA-dependent regulation of p21 ubiquitylation and degradation via the CRL4Cdt2 ubiquitin ligase complex. Genes Dev. 2008, 22, 2496–2506. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; McCall, C.M.; Ohta, T.; Xiong, Y. Targeted ubiquitination of CDT1 by the DDB1-CUL4A-ROC1 ligase in response to DNA damage. Nat. Cell Biol. 2004, 6, 1003–1009. [Google Scholar] [CrossRef] [PubMed]
- Hannah, J.; Zhou, P. Regulation of DNA damage response pathways by the cullin-RING ubiquitin ligases. DNA Repair 2009, 8, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.J.; Milhollen, M.A.; Smith, P.G.; Narayanan, U.; Dutta, A. NEDD8-targeting drug MLN4924 elicits DNA rereplication by stabilizing Cdt1 in S phase, triggering checkpoint activation, apoptosis, and senescence in cancer cells. Cancer Res. 2010, 70, 10310–10320. [Google Scholar] [CrossRef] [PubMed]
- Seiler, J.A.; Conti, C.; Syed, A.; Aladjem, M.I.; Pommier, Y. The intra-S-phase checkpoint affects both DNA replication initiation and elongation: Single-cell and -DNA fiber analyses. Mol. Cell Biol. 2007, 27, 5806–5818. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Zhang, S.; Xu, D.; Lee, M.Y.; Zhang, Z.; Lee, E.Y.; Darzynkiewicz, Z. Expression of the p12 subunit of human DNA polymerase δ (Pol δ), CDK inhibitor p21(WAF1), Cdt1, cyclin A, PCNA and Ki-67 in relation to DNA replication in individual cells. Cell Cycle 2014, 13, 3529–3540. [Google Scholar] [CrossRef] [PubMed]
- Darzynkiewicz, Z.; Zhao, H.; Zhang, S.; Lee, M.Y.; Lee, E.Y.; Zhang, Z. Initiation and termination of DNA replication during S phase in relation to cyclins D1, E and A, p21WAF1, Cdt1 and the p12 subunit of DNA polymerase δ revealed in individual cells by cytometry. Oncotarget 2015, 6, 11735–11750. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, L.; Bambara, R.A. Okazaki fragment metabolism. Cold Spring Harb. Perspect. Biol. 2013, 5, a10173. [Google Scholar] [CrossRef] [PubMed]
- Garg, P.; Stith, C.M.; Sabouri, N.; Johansson, E.; Burgers, P.M. Idling by DNA polymerase δ maintains a ligatable nick during lagging-strand DNA replication. Genes Dev. 2004, 18, 2764–2773. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, L.; Bambara, R.A. FLAP Endonuclease 1. Ann. Rev. Biochem. 2013, 82, 119–138. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.H.; Ayyagari, R.; Resnick, M.A.; Gordenin, D.A.; Burgers, P.M. Okazaki fragment maturation in yeast. II. Cooperation between the polymerase and 3′-5′-exonuclease activities of Pol δ in the creation of a ligatable nick. J. Biol. Chem. 2003, 278, 1626–1633. [Google Scholar] [CrossRef] [PubMed]
- Burgers, P.M. It’s all about flaps: DNA2 and checkpoint activation. Cell Cycle 2011, 10, 2417–2418. [Google Scholar] [CrossRef] [PubMed]
- Howes, T.R.; Tomkinson, A.E. DNA ligase I, the replicative DNA ligase. Subcell. Biochem. 2012, 62, 327–341. [Google Scholar] [PubMed]
- Kelly, R.B.; Cozzarelli, N.R.; Deutscher, M.P.; Lehman, I.R.; Kornberg, A. Enzymatic synthesis of deoxyribonucleic acid. XXXII. Replication of duplex deoxyribonucleic acid by polymerase at a single strand break. J. Biol. Chem. 1970, 245, 39–45. [Google Scholar] [PubMed]
- Lin, S.H.; Wang, X.; Zhang, S.; Zhang, Z.; Lee, E.Y.; Lee, M.Y. Dynamics of Enzymatic Interactions during Short Flap Human Okazaki Fragment Processing by Two Forms of Human DNA Polymerase δ. DNA Repair 2013, 12, 922–935. [Google Scholar] [CrossRef] [PubMed]
- Beattie, T.R.; Bell, S.D. Coordination of multiple enzyme activities by a single PCNA in archaeal Okazaki fragment maturation. EMBO J. 2012, 31, 1556–1567. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.S.; Wong, I.; Johnson, K.A. Pre-steady-state kinetic analysis of processive DNA replication including complete characterization of an exonuclease-deficient mutant. Biochemistry 1991, 30, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Langston, L.D.; O’Donnell, M. DNA polymerase δ is highly processive with proliferating cell nuclear antigen and undergoes collision release upon completing DNA. J. Biol. Chem. 2008, 283, 29522–29531. [Google Scholar] [CrossRef] [PubMed]
- Burgers, P.M.; Kunkel, T.A. Eukaryotic DNA Replication Fork. Annu. Rev. Biochem. 2017, 86, 417–438. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.M.; Akashi, T.; Masuda, Y.; Kamiya, K.; Takahashi, T.; Suzuki, M. Roles of POLD4, smallest subunit of DNA polymerase δ, in nuclear structures and genomic stability of human cells. Biochem. Biophys. Res. Commun. 2010, 391, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Suzuki, M.; Zeng, Y.; Zhang, H.; Yang, D.; Lin, H. Downregulation of POLD4 in Calu6 cells results in G1-S blockage through suppression of the Akt-Skp2-p27 pathway. Bioorg. Med. Chem. Lett. 2014, 24, 1780–1783. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Rodriguez-Belmonte, E.M.; Mazloum, N.; Xie, B.; Lee, M.Y. Identification of a novel protein, PDIP38, that interacts with the p50 subunit of DNA polymerase δ and proliferating cell nuclear antigen. J. Biol. Chem. 2003, 278, 10041–10047. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Tan, C.K.; Downey, K.M.; So, A.G. A tumor necrosis factor α- and interleukin 6-inducible protein that interacts with the small subunit of DNA polymerase δ and proliferating cell nuclear antigen. Proc. Natl. Acad. Sci. USA 2001, 98, 11979–11984. [Google Scholar] [CrossRef] [PubMed]
- Richardson, C.J.; Broenstrup, M.; Fingar, D.C.; Julich, K.; Ballif, B.A.; Gygi, S.; Blenis, J. SKAR is a specific target of S6 kinase 1 in cell growth control. Curr. Biol. 2004, 14, 1540–1549. [Google Scholar] [CrossRef] [PubMed]
- Magnuson, B.; Ekim, B.; Fingar, D.C. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem. J. 2012, 441, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Richardson, C.J.; Schalm, S.S.; Blenis, J. PI3-kinase and TOR: PIKTORing cell growth. Semin. Cell Dev. Biol. 2004, 15, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Banko, M.I.; Krzyzanowski, M.K.; Turcza, P.; Maniecka, Z.; Kulis, M.; Kozlowski, P. Identification of amino acid residues of ERH required for its recruitment to nuclear speckles and replication foci in HeLa cells. PLoS ONE 2013, 8, e74885. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.M.; Yoon, S.O.; Richardson, C.J.; Julich, K.; Blenis, J. SKAR links pre-mRNA splicing to mTOR/S6K1-mediated enhanced translation efficiency of spliced mRNAs. Cell 2008, 133, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Fingar, D.C.; Salama, S.; Tsou, C.; Harlow, E.; Blenis, J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 2002, 16, 1472–1487. [Google Scholar] [CrossRef] [PubMed]
- Smyk, A.; Szuminska, M.; Uniewicz, K.A.; Graves, L.M.; Kozlowski, P. Human enhancer of rudimentary is a molecular partner of PDIP46/SKAR, a protein interacting with DNA polymerase δ and S6K1 and regulating cell growth. FEBS J. 2006, 273, 4728–4741. [Google Scholar] [CrossRef] [PubMed]
- Weng, M.T.; Luo, J. The enigmatic ERH protein: Its role in cell cycle, RNA splicing and cancer. Protein Cell 2013, 4, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Weng, M.T.; Tung, T.H.; Lee, J.H.; Wei, S.C.; Lin, H.L.; Huang, Y.J.; Wong, J.M.; Luo, J.; Sheu, J.C. Enhancer of rudimentary homolog regulates DNA damage response in hepatocellular carcinoma. Sci. Rep. 2015, 5, 9357. [Google Scholar] [CrossRef] [PubMed]
- Kavanaugh, G.; Zhao, R.; Guo, Y.; Mohni, K.N.; Glick, G.; Lacy, M.E.; Hutson, M.S.; Ascano, M.; Cortez, D. Enhancer of Rudimentary Homolog affects the replication stress response through regulation of RNA processing. Mol. Cell Biol. 2015, 35, 2979–2990. [Google Scholar] [CrossRef] [PubMed]
- Gilljam, K.M.; Feyzi, E.; Aas, P.A.; Sousa, M.M.; Muller, R.; Vagbo, C.B.; Catterall, T.C.; Liabakk, N.B.; Slupphaug, G.; Drablos, F.; et al. Identification of a novel, widespread, and functionally important PCNA-binding motif. J. Cell Biol. 2009, 186, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Gilljam, K.M.; Muller, R.; Liabakk, N.B.; Otterlei, M. Nucleotide excision repair is associated with the replisome and its efficiency depends on a direct interaction between XPA and PCNA. PLoS ONE 2012, 7, e49199. [Google Scholar] [CrossRef] [PubMed]
- Bacquin, A.; Pouvelle, C.; Siaud, N.; Perderiset, M.; Salome-Desnoulez, S.; Tellier-Lebegue, C.; Lopez, B.; Charbonnier, J.B.; Kannouche, P.L. The helicase FBH1 is tightly regulated by PCNA via CRL4(Cdt2)-mediated proteolysis in human cells. Nucleic Acids Res. 2013, 41, 6501–6513. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Samson, L.D.; Hubscher, U.; van Loon, B. The interaction between ALKBH2 DNA repair enzyme and PCNA is direct, mediated by the hydrophobic pocket of PCNA and perturbed in naturally-occurring ALKBH2 variants. DNA Repair 2015, 35, 13–18. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, M.; Li, H. The Eukaryotic Replisome Goes Under the Microscope. Curr. Biol. 2016, 26, R247–R256. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.H.; Farina, A.; Bermudez, V.P.; Tappin, I.; Du, F.; Galal, W.C.; Hurwitz, J. Interaction between human Ctf4 and the Cdc45/Mcm2-7/GINS (CMG) replicative helicase. Proc. Natl. Acad. Sci. USA 2013, 110, 19760–19765. [Google Scholar] [CrossRef] [PubMed]
- Villa, F.; Simon, A.C.; Ortiz Bazan, M.A.; Kilkenny, M.L.; Wirthensohn, D.; Wightman, M.; Matak-Vinkovic, D.; Pellegrini, L.; Labib, K. Ctf4 Is a Hub in the Eukaryotic Replisome that Links Multiple CIP-Box Proteins to the CMG Helicase. Mol. Cell 2016, 63, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Li, H.; Wang, Q.; Xie, S.; Rahmeh, A.; Dai, W.; Lee, M.Y. Further characterization of human DNA polymerase δ interacting protein 38. J. Biol. Chem. 2005, 280, 22375–22384. [Google Scholar] [CrossRef] [PubMed]
- Pursell, Z.F.; Isoz, I.; Lundstrom, E.B.; Johansson, E.; Kunkel, T.A. Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science 2007, 317, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Nick McElhinny, S.A.; Gordenin, D.A.; Stith, C.M.; Burgers, P.M.; Kunkel, T.A. Division of labor at the eukaryotic replication fork. Mol. Cell 2008, 30, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Larrea, A.A.; Lujan, S.A.; Nick McElhinny, S.A.; Mieczkowski, P.A.; Resnick, M.A.; Gordenin, D.A.; Kunkel, T.A. Genome-wide model for the normal eukaryotic DNA replication fork. Proc. Natl. Acad. Sci. USA 2010, 107, 17674–17679. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.E.; Klassen, R.; Prakash, L.; Prakash, S. A Major Role of DNA Polymerase δ in Replication of Both the Leading and Lagging DNA Strands. Mol. Cell 2015, 59, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Eckert, K.A.; Hile, S.E. Every microsatellite is different: Intrinsic DNA features dictate mutagenesis of common microsatellites present in the human genome. Mol. Carcinog. 2009, 48, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Le, H.P.; Masuda, Y.; Tsurimoto, T.; Maki, S.; Katayama, T.; Furukohri, A.; Maki, H. Short CCG repeat in huntingtin gene is an obstacle for replicative DNA polymerases, potentially hampering progression of replication fork. Genes Cells 2015, 20, 817–833. [Google Scholar] [CrossRef] [PubMed]
- Hile, S.E.; Wang, X.; Lee, M.Y.; Eckert, K.A. Beyond translesion synthesis: Polymerase к fidelity as a potential determinant of microsatellite stability. Nucleic Acids Res. 2012, 40, 1636–1647. [Google Scholar] [CrossRef] [PubMed]
- Baptiste, B.A.; Eckert, K.A. DNA polymerase к microsatellite synthesis: Two distinct mechanisms of slippage-mediated errors. Environ. Mol. Mutagen. 2012, 53, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Rey, L.; Sidorova, J.M.; Puget, N.; Boudsocq, F.; Biard, D.S.; Monnat, R.J.J.; Cazaux, C.; Hoffmann, J.S. Human DNA polymerase eta is required for common fragile site stability during unperturbed DNA replication. Mol. Cell Biol. 2009, 29, 3344–3354. [Google Scholar] [CrossRef] [PubMed]
- Bergoglio, V.; Boyer, A.S.; Walsh, E.; Naim, V.; Legube, G.; Lee, M.Y.; Rey, L.; Rosselli, F.; Cazaux, C.; Eckert, K.A.; et al. DNA synthesis by Pol eta promotes fragile site stability by preventing under-replicated DNA in mitosis. J. Cell Biol. 2013, 201, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Exposito, L.; Bournique, E.; Bergoglio, V.; Bose, A.; Barroso-Gonzalez, J.; Zhang, S.; Roncaioli, J.L.; Lee, M.; Wallace, C.T.; Watkins, S.C.; et al. Proteomic Profiling Reveals a Specific Role for Translesion DNA Polymerase eta in the Alternative Lengthening of Telomeres. Cell Rep. 2016, 17, 1858–1871. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, V.P.; Farina, A.; Raghavan, V.; Tappin, I.; Hurwitz, J. Studies on Human DNA Polymerase epsilon and GINS Complex and Their Role in DNA Replication. J. Biol. Chem. 2011, 286, 28963–28977. [Google Scholar] [CrossRef] [PubMed]
- Ganai, R.A.; Zhang, X.P.; Heyer, W.D.; Johansson, E. Strand displacement synthesis by yeast DNA polymerase epsilon. Nucleic Acids Res. 2016, 44, 8229–8240. [Google Scholar] [CrossRef] [PubMed]
- Yeeles, J.T.; Janska, A.; Early, A.; Diffley, J.F. How the Eukaryotic Replisome Achieves Rapid and Efficient DNA Replication. Mol. Cell 2017, 65, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.C.; Janska, A.; Goswami, P.; Renault, L.; Abid Ali, F.; Kotecha, A.; Diffley, J.F.X.; Costa, A. CMG-Pol epsilon dynamics suggests a mechanism for the establishment of leading-strand synthesis in the eukaryotic replisome. Proc. Natl. Acad. Sci. USA 2017, 114, 4141–4146. [Google Scholar] [CrossRef] [PubMed]
- Kurat, C.F.; Yeeles, J.T.; Patel, H.; Early, A.; Diffley, J.F. Chromatin Controls DNA Replication Origin Selection, Lagging-Strand Synthesis, and Replication Fork Rates. Mol. Cell 2017, 65, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Yeeles, J.T.; Deegan, T.D.; Janska, A.; Early, A.; Diffley, J.F. Regulated eukaryotic DNA replication origin firing with purified proteins. Nature 2015, 519, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Schauer, G.D.; O’Donnell, M.E. Quality control mechanisms exclude incorrect polymerases from the eukaryotic replication fork. Proc. Natl. Acad. Sci. USA 2017, 114, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Yurieva, O.; O’Donnell, M. Reconstitution of a eukaryotic replisome reveals the mechanism of asymmetric distribution of DNA polymerases. Nucleus 2016, 7, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Hedglin, M.; Pandey, B.; Benkovic, S.J. Stability of the human polymerase δ holoenzyme and its implications in lagging strand DNA synthesis. Proc. Natl. Acad. Sci. USA 2016, 113, E1777–E1786. [Google Scholar] [CrossRef] [PubMed]
- Beattie, T.R.; Kapadia, N.; Nicolas, E.; Uphoff, S.; Wollman, A.J.; Leake, M.C.; Reyes-Lamothe, R. Frequent exchange of the DNA polymerase during bacterial chromosome replication. Elife 2017, 6, e21763. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Kelly, T.J. Simian virus 40 DNA replication in vitro. Proc. Natl. Acad. Sci. USA 1984, 81, 6973–6977. [Google Scholar] [CrossRef] [PubMed]
- Waga, S.; Stillman, B. Anatomy of a DNA replication fork revealed by reconstitution of SV40 DNA replication in vitro. Nature 1994, 369, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Rytkonen, A.K.; Vaara, M.; Nethanel, T.; Kaufmann, G.; Sormunen, R.; Laara, E.; Nasheuer, H.P.; Rahmeh, A.; Lee, M.Y.; Syvaoja, J.E.; et al. Distinctive activities of DNA polymerases during human DNA replication. FEBS J. 2006, 273, 2984–3001. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, E.C.; Lehmann, A.R.; Fuchs, R.P. Trading places: How do DNA polymerases switch during translesion DNA synthesis? Mol. Cell 2005, 18, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, A.R. Translesion synthesis in mammalian cells. Exp. Cell Res. 2006, 312, 2673–2676. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Guilliam, T.A.; Doherty, A.J. PrimPol-Prime Time to Reprime. Genes 2017, 8, 20. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.H.; Pandian, V.; Ramraj, S.; Natarajan, M.; Aravindan, S.; Herman, T.S.; Aravindan, N. Acquired genetic alterations in tumor cells dictate the development of high-risk neuroblastoma and clinical outcomes. BMC Cancer 2015, 15, 514. [Google Scholar] [CrossRef] [PubMed]
- Human Protein Atlas. Available online: http://www.proteinatlas.org/ENSG00000100227-POLDIP3/cancer (accessed on 9 April 2017).
- COSMIC (Catalogue of Somatic Mutations in Cancer). Available online: http://cancer.sanger.ac.uk/cosmic (accessed on 9 April 2017).
- Gakh, O.; Cavadini, P.; Isaya, G. Mitochondrial processing peptidases. Biochim. Biophys. Acta 2002, 1592, 63–77. [Google Scholar] [CrossRef]
- Cheng, X.; Kanki, T.; Fukuoh, A.; Ohgaki, K.; Takeya, R.; Aoki, Y.; Hamasaki, N.; Kang, D. PDIP38 associates with proteins constituting the mitochondrial DNA nucleoid. J. Biochem. 2005, 138, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Arakaki, N.; Nishihama, T.; Kohda, A.; Owaki, H.; Kuramoto, Y.; Abe, R.; Kita, T.; Suenaga, M.; Himeda, T.; Kuwajima, M.; et al. Regulation of mitochondrial morphology and cell survival by Mitogenin I and mitochondrial single-stranded DNA binding protein. Biochim. Biophys. Acta 2006, 1760, 1364–1372. [Google Scholar] [CrossRef] [PubMed]
- Klaile, E.; Muller, M.M.; Kannicht, C.; Otto, W.; Singer, B.B.; Reutter, W.; Obrink, B.; Lucka, L. The cell adhesion receptor carcinoembryonic antigen-related cell adhesion molecule 1 regulates nucleocytoplasmic trafficking of DNA polymerase δ-interacting protein 38. J. Biol. Chem. 2007, 282, 26629–26640. [Google Scholar] [CrossRef] [PubMed]
- Klaile, E.; Kukalev, A.; Obrink, B.; Muller, M.M. PDIP38 is a novel mitotic spindle-associated protein that affects spindle organization and chromosome segregation. Cell Cycle 2008, 7, 3180–3186. [Google Scholar] [CrossRef] [PubMed]
- Tissier, A.; Janel-Bintz, R.; Coulon, S.; Klaile, E.; Kannouche, P.; Fuchs, R.P.; Cordonnier, A.M. Crosstalk between replicative and translesional DNA polymerases: PDIP38 interacts directly with Poleta. DNA Repair 2010, 9, 922–928. [Google Scholar] [CrossRef] [PubMed]
- Maga, G.; Crespan, E.; Markkanen, E.; Imhof, R.; Furrer, A.; Villani, G.; Hubscher, U.; van Loon, B. DNA polymerase δ-interacting protein 2 is a processivity factor for DNA polymerase lambda during 8-oxo-7,8-dihydroguanine bypass. Proc. Natl. Acad. Sci. USA 2013, 110, 18850–18855. [Google Scholar] [CrossRef] [PubMed]
- Bebenek, K.; Pedersen, L.C.; Kunkel, T.A. Structure-Function Studies of DNA Polymerase lambda. Biochemistry 2014, 53, 2781–2792. [Google Scholar] [CrossRef] [PubMed]
- Mentegari, E.; Kissova, M.; Bavagnoli, L.; Maga, G.; Crespan, E. DNA Polymerases lambda and beta: The Double-Edged Swords of DNA Repair. Genes 2016, 7, 57. [Google Scholar] [CrossRef] [PubMed]
- Waters, L.S.; Minesinger, B.K.; Wiltrout, M.E.; D’Souza, S.; Woodruff, R.V.; Walker, G.C. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol. Mol. Biol. Rev. 2009, 73, 134–154. [Google Scholar] [CrossRef] [PubMed]
- Braithwaite, E.K.; Kedar, P.S.; Stumpo, D.J.; Bertocci, B.; Freedman, J.H.; Samson, L.D.; Wilson, S.H. DNA polymerases beta and lambda mediate overlapping and independent roles in base excision repair in mouse embryonic fibroblasts. PLoS ONE 2010, 5, e12229. [Google Scholar] [CrossRef] [PubMed]
- Braithwaite, E.K.; Kedar, P.S.; Lan, L.; Polosina, Y.Y.; Asagoshi, K.; Poltoratsky, V.P.; Horton, J.K.; Miller, H.; Teebor, G.W.; Yasui, A.; et al. DNA polymerase lambda protects mouse fibroblasts against oxidative DNA damage and is recruited to sites of DNA damage/repair. J. Biol. Chem. 2005, 280, 31641–31647. [Google Scholar] [CrossRef] [PubMed]
- Hubscher, U.; Maga, G. DNA replication and repair bypass machines. Curr. Opin. Chem. Biol. 2011, 15, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Guilliam, T.A.; Bailey, L.J.; Brissett, N.C.; Doherty, A.J. PolDIP2 interacts with human PrimPol and enhances its DNA polymerase activities. Nucleic Acids Res. 2016, 44, 3317–3329. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.; Zhang, S.; Mordue, D.; Wu, J.M.; Zhang, Z.; Darzynkiewicz, Z.; Lee, E.Y.; Lee, M.Y. PDIP38 is translocated to the spliceosomes/nuclear speckles in response to UV-induced DNA damage and is required for UV-induced alternative splicing of MDM2. Cell Cycle 2013, 12, 3184–3193. [Google Scholar] [CrossRef] [PubMed]
- Spector, D.L.; Lamond, A.I. Nuclear speckles. Cold Spring Harb. Perspect. Biol. 2011, 3, a000646. [Google Scholar] [CrossRef] [PubMed]
- Campalans, A.; Amouroux, R.; Bravard, A.; Epe, B.; Radicella, J.P. UVA irradiation induces relocalisation of the DNA repair protein hOGG1 to nuclear speckles. J. Cell Sci. 2007, 120, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, M.; Sanchez, G.; Barbier, J.; Corcos, L.; Auboeuf, D. The emerging role of pre-messenger RNA splicing in stress responses: Sending alternative messages and silent messengers. RNA Biol. 2011, 8, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Giono, L.E.; Nieto Moreno, N.; Cambindo Botto, A.E.; Dujardin, G.; Munoz, M.J.; Kornblihtt, A.R. The RNA Response to DNA Damage. J. Mol. Biol. 2016, 428, 2636–2651. [Google Scholar] [CrossRef] [PubMed]
- Bartel, F.; Taubert, H.; Harris, L.C. Alternative and aberrant splicing of MDM2 mRNA in human cancer. Cancer Cell 2002, 2, 9–15. [Google Scholar] [CrossRef]
- Chandler, D.S.; Singh, R.K.; Caldwell, L.C.; Bitler, J.L.; Lozano, G. Genotoxic stress induces coordinately regulated alternative splicing of the p53 modulators MDM2 and MDM4. Cancer Res. 2006, 66, 9502–9508. [Google Scholar] [CrossRef] [PubMed]
- Jeyaraj, S.; O’Brien, D.M.; Chandler, D.S. MDM2 and MDM4 splicing: An integral part of the cancer spliceome. Front. Biosci. 2009, 14, 2647–2656. [Google Scholar] [CrossRef]
- Okoro, D.R.; Rosso, M.; Bargonetti, J. Splicing up mdm2 for cancer proteome diversity. Genes Cancer 2012, 3, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.N.; Hancock, A.R.; Vogel, H.; Donehower, L.A.; Bradley, A. Overexpression of Mdm2 in mice reveals a p53-independent role for Mdm2 in tumorigenesis. Proc. Natl. Acad. Sci. USA 1998, 95, 15608–15612. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, R.; Tada, M.; Nozaki, M.; Zhang, C.L.; Sawamura, Y.; Abe, H. Short alternative splice transcripts of the mdm2 oncogene correlate to malignancy in human astrocytic neoplasms. Cancer Res. 1998, 58, 609–613. [Google Scholar] [PubMed]
- Naro, C.; Bielli, P.; Pagliarini, V.; Sette, C. The interplay between DNA damage response and RNA processing: the unexpected role of splicing factors as gatekeepers of genome stability. Front. Genet. 2015, 6, 142. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, A.; Biamonti, G. Pre-mRNA processing factors meet the DNA damage response. Front. Genet. 2013, 4, 102. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D.; Neish, A.S. Nox enzymes and new thinking on reactive oxygen: A double-edged sword revisited. Annu. Rev. Pathol. 2014, 9, 119–145. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D.; Kawahara, T.; Diebold, B. Regulation of Nox and Duox enzymatic activity and expression. Free Radic. Biol. Med. 2007, 43, 319–331. [Google Scholar] [CrossRef]
- Brown, D.I.; Griendling, K.K. Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circ. Res. 2015, 116, 531–549. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Lambeth, J.D. Nox enzymes, ROS, and chronic disease: An example of antagonistic pleiotropy. Free Radic. Biol. Med. 2007, 43, 332–347. [Google Scholar] [CrossRef]
- Nisimoto, Y.; Diebold, B.A.; Cosentino-Gomes, D.; Lambeth, J.D. Nox4: A hydrogen peroxide-generating oxygen sensor. Biochemistry 2014, 53, 5111–5120. [Google Scholar] [CrossRef] [PubMed]
- Lassegue, B.; Griendling, K.K. NADPH Oxidases: Functions and Pathologies in the Vasculature. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Lyle, A.N.; Deshpande, N.N.; Taniyama, Y.; Seidel-Rogol, B.; Pounkova, L.; Du, P.; Papaharalambus, C.; Lassegue, B.; Griendling, K.K. Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ. Res. 2009, 105, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Datla, S.R.; McGrail, D.J.; Vukelic, S.; Huff, L.P.; Lyle, A.N.; Pounkova, L.; Lee, M.; Seidel-Rogol, B.; Khalil, M.K.; Hilenski, L.L.; et al. Poldip2 controls vascular smooth muscle cell migration by regulating focal adhesion turnover and force polarization. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H945–H957. [Google Scholar] [CrossRef] [PubMed]
- Sutliff, R.L.; Hilenski, L.L.; Amanso, A.M.; Parastatidis, I.; Dikalova, A.E.; Hansen, L.; Datla, S.R.; Long, J.S.; El-Ali, A.M.; Joseph, G.; et al. Polymerase Delta Interacting Protein 2 Sustains Vascular Structure and Function. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2154–2161. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.I.; Lassegue, B.; Lee, M.; Zafari, R.; Long, J.S.; Saavedra, H.I.; Griendling, K.K. Poldip2 knockout results in perinatal lethality, reduced cellular growth and increased autophagy of mouse embryonic fibroblasts. PLoS ONE 2014, 9, e96657. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Human | p125 | p50 | p68 | p12 |
|---|---|---|---|---|
| Schizosaccharomyces pombe | Pol3 | Cdc1 | Cdc27 | Cdm1 |
| Saccharomyces cerevisiae | Pol3 | Pol31 | Pol32 | - |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, M.Y.W.T.; Wang, X.; Zhang, S.; Zhang, Z.; Lee, E.Y.C. Regulation and Modulation of Human DNA Polymerase δ Activity and Function. Genes 2017, 8, 190. https://doi.org/10.3390/genes8070190
Lee MYWT, Wang X, Zhang S, Zhang Z, Lee EYC. Regulation and Modulation of Human DNA Polymerase δ Activity and Function. Genes. 2017; 8(7):190. https://doi.org/10.3390/genes8070190
Chicago/Turabian StyleLee, Marietta Y. W. T., Xiaoxiao Wang, Sufang Zhang, Zhongtao Zhang, and Ernest Y. C. Lee. 2017. "Regulation and Modulation of Human DNA Polymerase δ Activity and Function" Genes 8, no. 7: 190. https://doi.org/10.3390/genes8070190
APA StyleLee, M. Y. W. T., Wang, X., Zhang, S., Zhang, Z., & Lee, E. Y. C. (2017). Regulation and Modulation of Human DNA Polymerase δ Activity and Function. Genes, 8(7), 190. https://doi.org/10.3390/genes8070190