1. Introduction
The interphotoreceptor matrix proteoglycans-1 and -2, also known as sialoprotein associated with cones and rods (SPACR) and SPACRCAN (a proteoglyCan related to SPACR), respectively, are large glycosylated protein components of the insoluble interphotoreceptor matrix (IPM) [
1]. The IPM is a specialized type of extracellular matrix that surrounds the photoreceptor inner and outer segments and the apical processes of the retinal pigment epithelium (RPE). It serves several important functions, including communication between photoreceptors, RPE and Müller cells, by regulating trafficking of signaling molecules, nutrients and metabolites and the maintenance of retinal adhesion and photoreceptor alignment [
1,
2] IMPG1 and IMPG2 have hyaluronan (HA) binding motifs of the receptor for HA-mediated motility (RHAMM) type [
1,
3]. It is believed that secreted IMPG1 and IMPG2 bind HA and that this interaction stabilizes or modulates the IPM scaffold [
1].
First evidence for an involvement of the interphotoreceptor matrix proteoglycans in the etiology of retinal degenerative disease was given when the heterozygous c.1736T > C/p.(Leu579Pro) mutation in the
IMPG1 gene was shown to cause autosomal dominant benign concentric annular macular dystrophy (BCAMD) in a Dutch family [
4]. This phenotype is characterized by an initial parafoveal hypopigmentation and good visual acuity which then progresses to a retinitis pigmentosa-like manifestation. Subsequently, autosomal recessive mutations in the
IMPG2 gene were identified to be causally associated with early-onset retinitis pigmentosa (RP) [
5]. The RP phenotype is frequently accompanied by early macular abnormalities, ranging from minor pigment alterations to profound chorioretinal atrophy [
6].
Recently, mutations in
IMPG1 and
IMPG2 have been reported to also play a causative role in autosomal dominant and recessive vitelliform macular dystrophy (VMD) [
7,
8]. Vitelliform lesions have been defined by Gass in 1974 as bilateral, round or oval, yellow, symmetrical, singular, subretinal lesions, typically one-third to one-half disc diameter in size [
9]. Best disease is caused by dominant (mostly missense) mutations in the
BEST1 gene [
10,
11]. Characteristic features of this disease entity include a diminished Arden ratio measured by electrooculography (EOG) as well as the typical egg yolk-like vitelliform macular lesion, which can already occur during childhood [
12]. In contrast, adult-onset VMD (AVMD) becomes symptomatic not before the fourth or fifth decade of life with a relatively mild and slowly progressive loss of central visual acuity [
12,
13]. Another main clinical aspect in AVMD, and an important difference to Best disease, is a normal or only slightly subnormal light induced rise in ocular potential [
12,
14]. AVMD is genetically more heterogeneous than Best disease and has in some instances been linked to dominant mutations in
BEST1 but also in the
PRPH2 gene [
15].
In the present study, we aimed to further investigate the role of
IMPG1 and
IMPG2 sequence variations in the development of vitelliform macular disease in our large patient cohort. We have screened the
IMPG1 and
IMPG2 genes in a total of 106 unrelated patients diagnosed with Best disease or AVMD but found negative after
BEST1 or
PRPH2 mutation testing. We identified two patients with novel causative mutations in the
IMPG1 gene. In addition, three novel stop mutations and four novel missense mutations as well as the c.3230G > T/p.(Cys1077Phe) mutation reported before [
8] were found in the
IMPG2 gene to be heterozygously present in eight patients. To assess the pathogenicity of these mutations we performed bioinformatic evaluation, in vitro splice assays, database searches and family segregation as well as comparative clinical characterization of the patients.
2. Subjects and Methods
2.1. Patient Recruitment
The study included a total of 106 patients with suspected VMD or AVMD who underwent routine genetic testing at the Institute of Human Genetics Regensburg and which were tested negative for mutations in the coding exons/flanking intronic sequences of BEST1 and PRPH2. A positive family history of VMD was reported for 15 cases, 46 individuals had a negative family history and no further information on the familial status of retinal degeneration was available for 45 patients. Seven unaffected family members were available for carrier testing. In accordance with the German Genetic Diagnostics Act written informed consent for genetic analysis was obtained from all subjects. This study adhered to the tenets of the Declaration of Helsinki. The study was also approved by the Ethics Committee of the University Regensburg, Germany (ID 17-514-104, date of approval 24.03.2017).
2.2. Clinical and Functional Ophthalmological Evaluation
Clinical, imaging, and electrophysiological findings were available and reviewed for ten index patients carrying mutations in the IMPG1 or IMPG2 gene as well as for seven family members. Age at initial diagnosis corresponds to the patient´s first visit to the respective University eye clinic in Regensburg, Bonn, Freiburg, Köln, Tübingen and Berlin, where the diagnosis of vitelliform macular dystrophy was established. Best-corrected visual acuity (VA) was assessed with standard charts under standard clinic conditions and results were transferred into logarithm of the Minimum Angle of Resolution (logMAR).
Electrooculograms (EOG) were recorded in six of ten index patients according to the standards of the International Society of Clinical Electrophysiology of Vision (ISCEV) [
16]. A light peak-to-dark trough ratio of <1.5 was defined as abnormally low, of >2.0 as normal, of between 1.5 and 2.0 as borderline, in line with the ISCEV recommendations [
16]. Moreover, in six of ten patients, multifocal electroretinograms (mfERG) were conducted following ISCEV standards [
17].
Multimodal retinal imaging included color fundus photography, obtained with the Zeiss FF450+ or Visucam (Carl Zeiss Meditec AG, Jena, Germany) and spectral domain-optical coherence tomography (SD-OCT) as well as fundus autofluorescence (FAF) imaging, performed with a combined Heidelberg Retina Angiograph and SD-OCT Spectralis device (Heidelberg Engineering, Heidelberg, Germany).
2.3. Molecular Analysis
For DNA extractions, ethlylenediaminetetraacetic acid (EDTA) peripheral blood samples were obtained from all patients and family members. Next-generation sequencing (NGS) was performed using the ION TorrentTM semiconductor technology (Thermo Fisher Scientific, Dreieich, Germany) upon multiplex-PCR amplification of the gene fragments by exon flanking oligonucleotide primer pairs (sequences available upon request) using the QIAGEN Multiplex PCR Master Mix (Qiagen, Hilden, Germany). In brief, the multiplexed PCR reactions of each patient were pooled after quantification in the MultiNA Shimadzu system (Shimadzu Biotech, Duisburg, Germany) and processed using the Ion Xpress™ Plus Fragment Library kit (Thermo Fisher Scientific, Dreieich, Germany) according to the manufacturers recommendations. The barcoded DNA libraries were purified with AMPure beads (Beckman Coulter, Krefeld, Germany) and the resulting library concentration was determined using an Agilent 2100 bioanalyzer (Agilent, Waldbronn, Germany). The emulsion PCR was carried out on the Ion OneTouch™ 2 System and the samples were loaded on a 318 Ion Torrent™ System Chip and sequenced with an ION Personal Genome Machine (Thermo Fisher Scientific, Dreieich, Germany). NGS data were analyzed with the CLC Genomics Workbench (CLC bio, Aarhus, Denmark). The mean coverage for the amplicons ranged from 56× to 5420×, with all bases sequenced at least 34×. Potentially disease-relevant variants were confirmed by Sanger chain-terminating dideoxynucleotide sequencing.
2.4. Bioinformatics Analysis
Variants were classified as recommended by the American College of Medical Genetics and Genomics (ACMG) standards and guidelines and the Association for Molecular Pathology (AMP) Clinical Practice Guidelines. The recommendations were based on population data, computational data and previous publications. Variant frequencies were taken from the Browser of the Exome Aggregation Consortium (ExAC), variants with a minor allele frequency (MAF) > 0.005 were excluded from further analysis. Rare missense mutations were tested for disease relevance by prediction algorithms of the programs MutationTaster, SIFT and PolyPhen-2. When at least two of the three programs predicted a pathogenic effect, the variants were classified as “likely pathogenic”. Stop mutations were classified as pathogenic. Intronic and synonymous variants were further analyzed for a potential effect on correct splicing by using the interface provided by the software package Alamut (Interactive Biosoftware, Rouen, France, Alamut Visual 2.7.1) which combines five algorithms (SpliceSiteFnder, MaxEntScan, NNSPLICE, GeneSplicer and Human Splicing Finder). All tools provide prediction scores for the normal and the mutant allele.
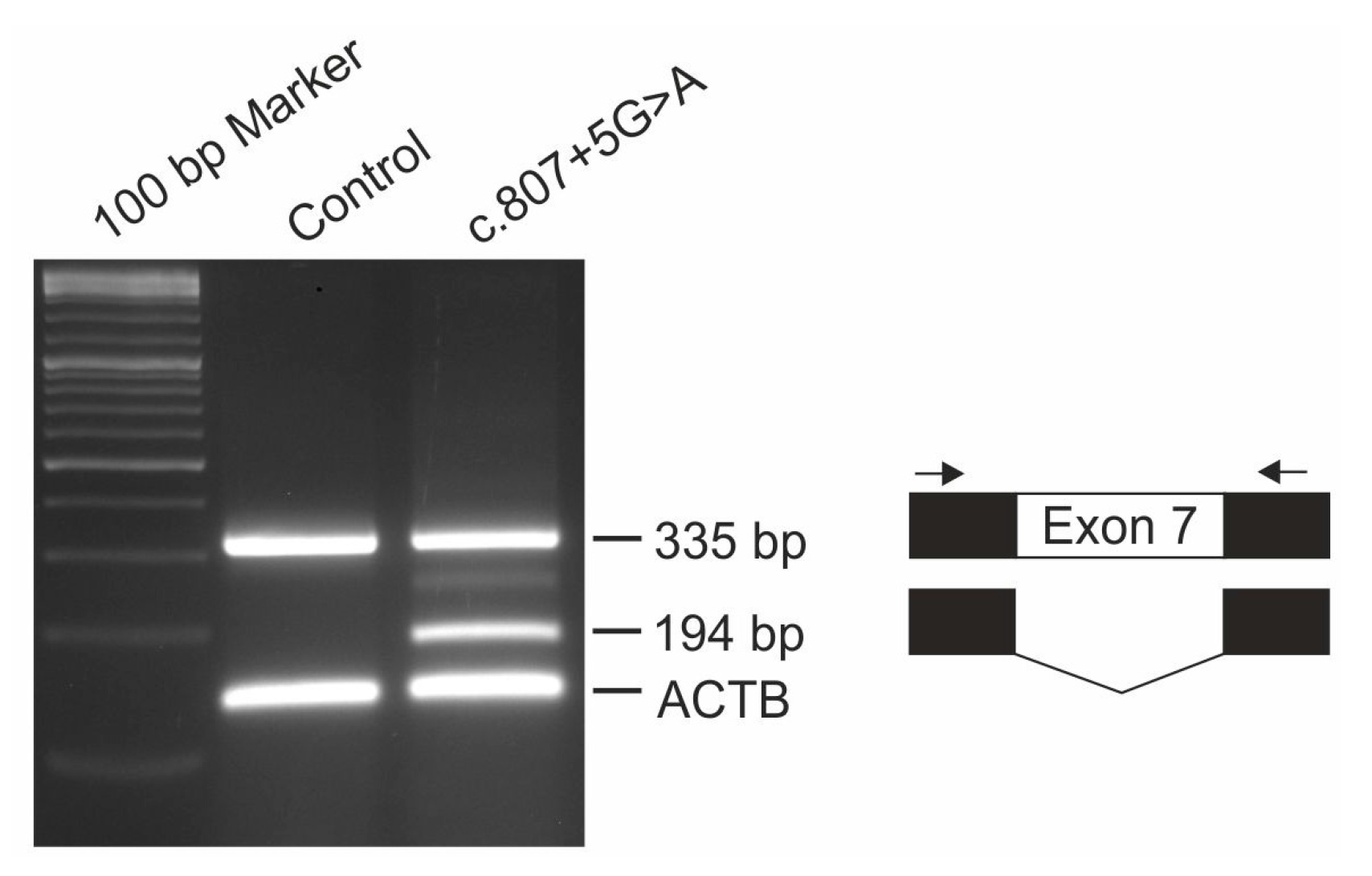
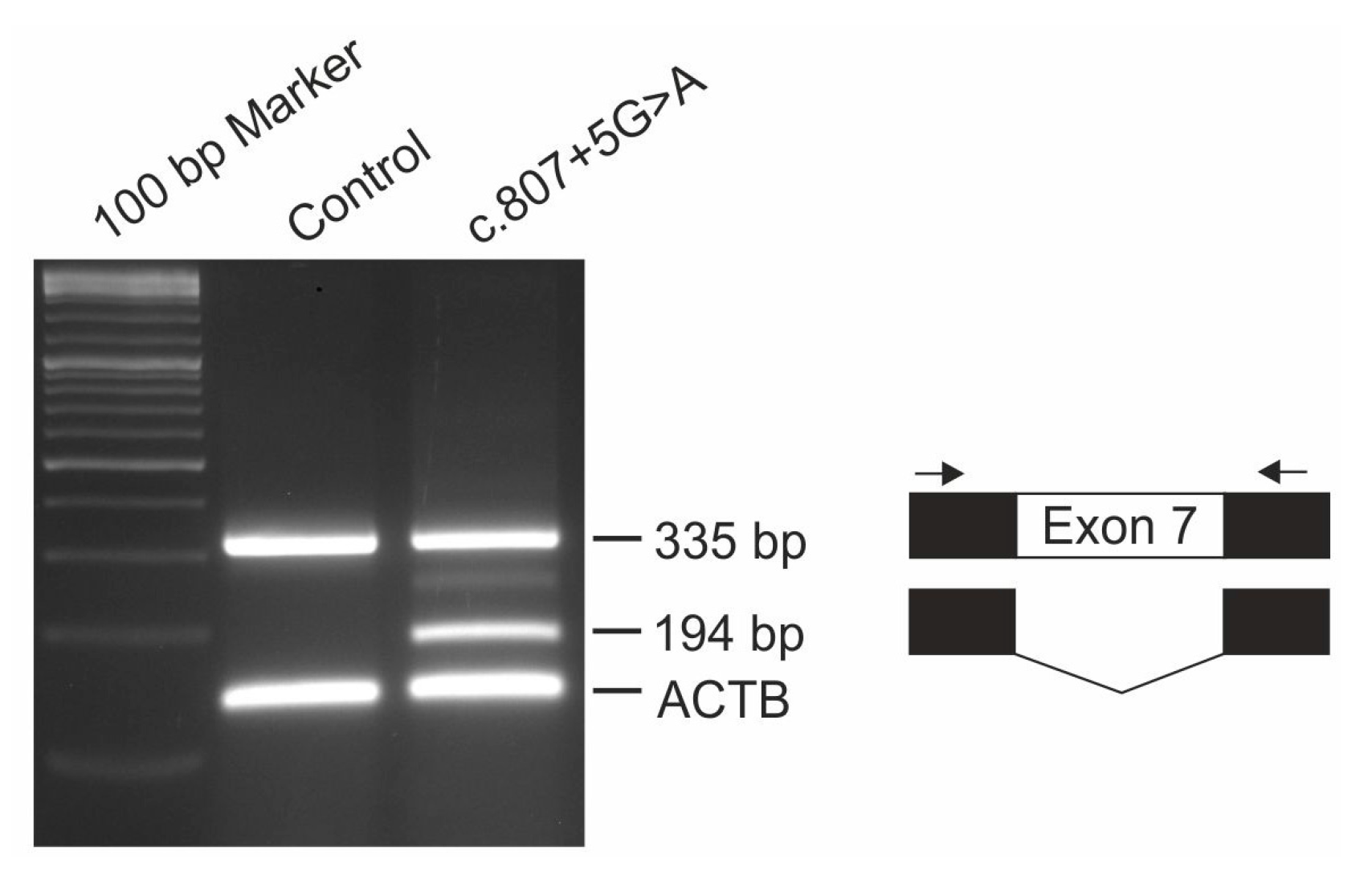
2.5. Minigene Assay
Functional consequences of IMPG1 splicing variant c.807 + 5G > A was analyzed with the pSPL3b-based exon trapping system. Briefly, exon 7 of the IMPG1 gene was PCR amplified from genomic DNA of patient #2 and control using a 7:1 ratio of Taq:Pfu polymerase and oligonucleotide primer pairs flanked with restriction sites for NotI and BamHI (5’-GCG GCC GCG CCT TCA TAA TCC ACT TCT TGA-3’; 5’-GGA TCC GGG CCC ATT GTA ATT TTG GT-3’) and cloned into the pSPL3b vector (kindly provided by Dr. M. Gessler, University Würzburg, Germany). HEK293 cells were transfected in duplicate with 3 µg of construct DNA using TransIT®-LT1 Transfection Reagent (Mirus, Madison, WI, USA), harvested after 48 hours and total RNA was extracted using RNeasy Micro Kit (Qiagen, Hilden, Germany). RT-PCR was performed with the RevertAid™ H First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, MA, USA) and a pSPL3-specific SD6 (5’-TCT GAG TCA CCT GGA CAA CC-3’) and SA4 (5’-CAC CTG AGG AGT GAA TTG GTC G-3’) primer pair in combination with oligonucleotide primers for the housekeeping gene ACTB (5’-GAC ATC CGC AAA GAC CTG TA-3’; 5’-CAG GAG AGC AAT GAT CTT GA-3’). PCR products were separated by electrophoresis on a 2% agarose gel, excised, cloned and Sanger sequenced.
4. Discussion
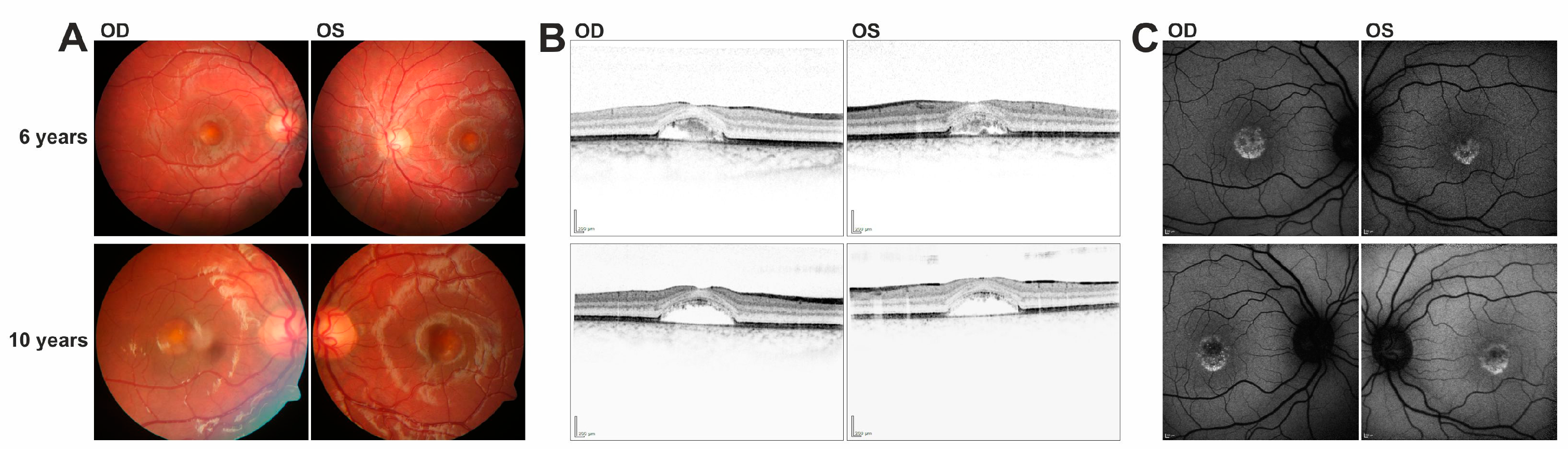
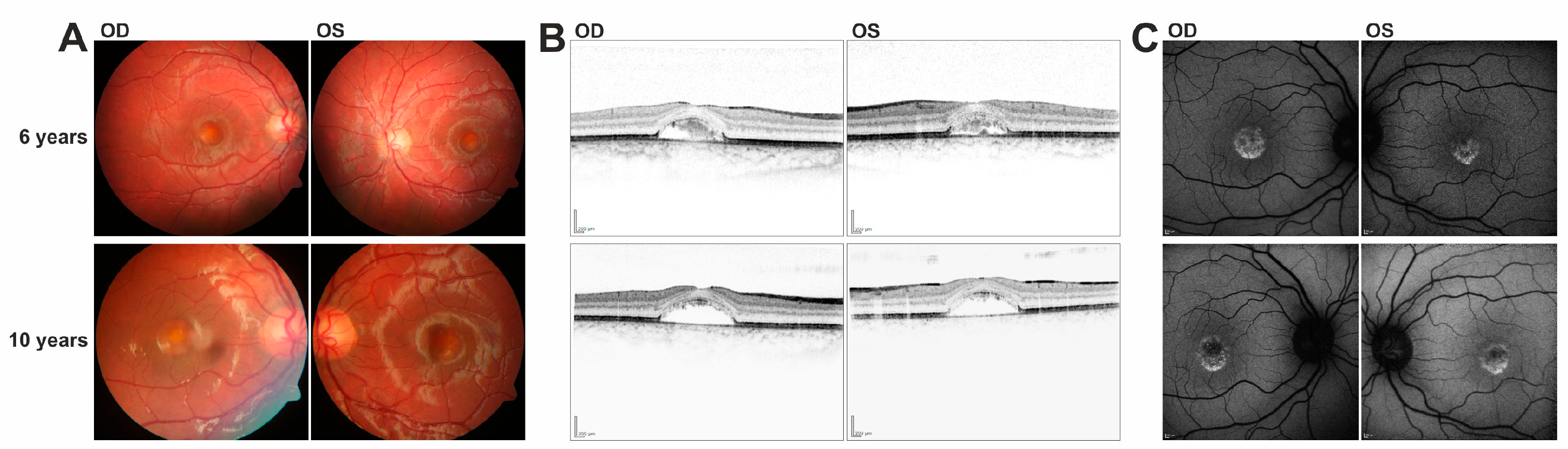
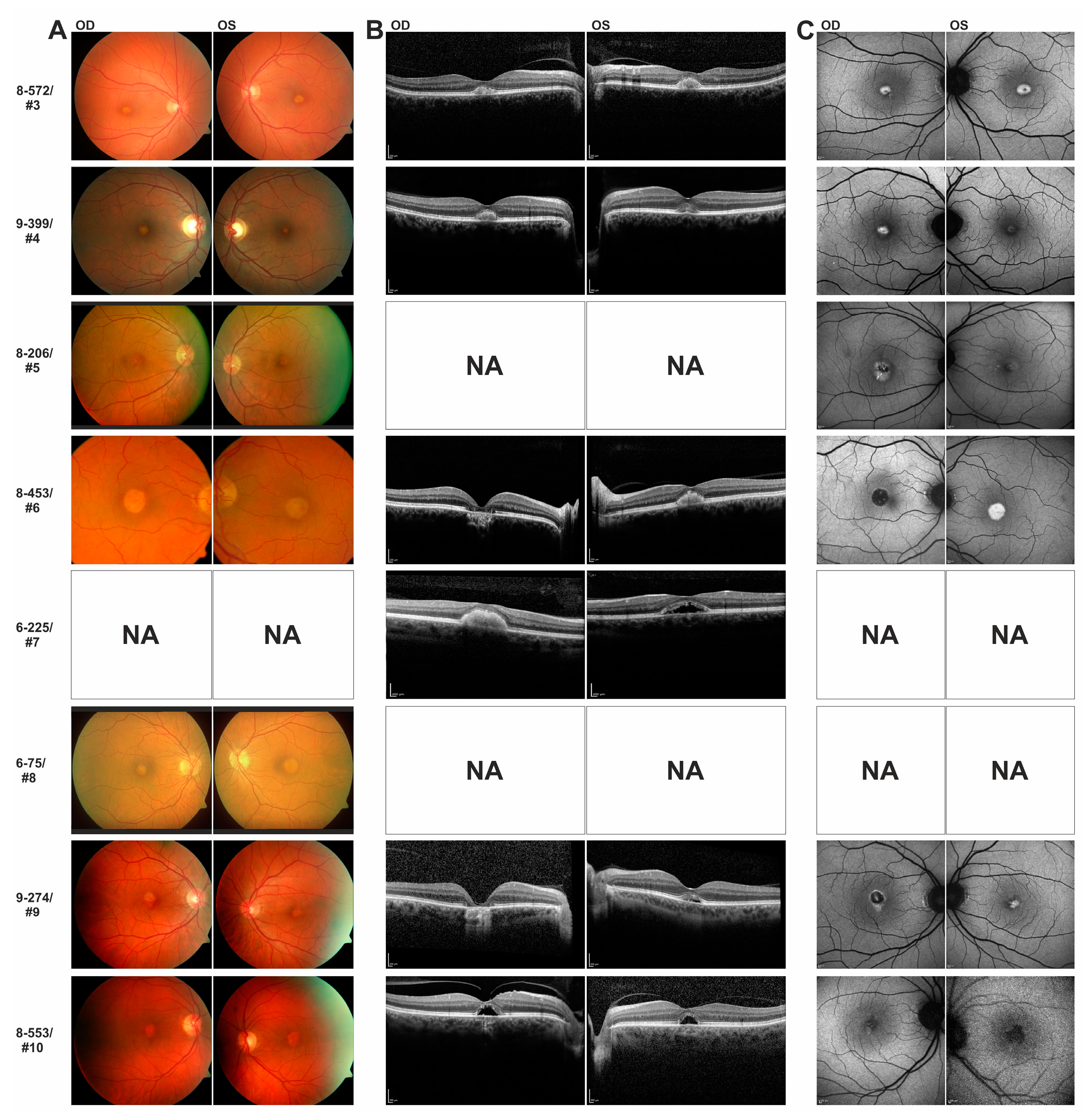
In this study we have identified a series of novel mutations in the IMPG1/IMPG2 genes and provide supportive evidence for their causativity in the development of vitelliform macular dystrophy. Patients with IMPG1/IMPG2 mutations described here reveal strikingly similar phenotypic characteristics, including retinal lesions which can be divided into presumably consecutive stages: (i) singular vitelliform lesions in the central macula with detachment of the neurosensory retina in SD-OCT, with hyperreflective material located above the seemingly preserved Bruch’s membrane/RPE; (ii) resorption of the hyperreflective material leaving behind a dome-shaped, optically empty cavity; alternatively, the foveal cavity formed by retinal detachment may become successively filled with material; (iii) collapse of the cavity and central retinal atrophy with loss of RPE. This stage of disease lead to the most pronounced loss of visual acuity.
Previous reports of clinical features in
IMPG-associated VMD [
7,
8] are largely consistent with our findings including mild to moderate vision impairment, normal or borderline EOG and regular or slightly abnormal ERG findings as well as the appearance of the vitelliform macular lesions in color fundus images and FAF with subretinal material located strictly above the preserved RPE as depicted by SD-OCT. All our patients had bilateral singular, unifocal deposits without associated drusen-like lesions at the posterior pole. It has been proposed that satellite drusen-like lesions in the foveal zone are characteristic for
IMPG1-linked VMD [
8]. Since the ages of onset in the patients with satellite drusen ranged between 27 and 54 and the patients at examination were thus significantly older than our two patients with
IMPG1-mutations (they are now in their teens), it remains to be seen whether they will also develop similar drusen-like abnormalities in adulthood.
An earlier study reported on a c.713T > G mutation in exon 7 of the
IMPG1 gene in three families with autosomal dominant VMD [
7]. Here, we have detected a similar mutation affecting c.713, a thymine to guanine exchange, in a simplex case of VMD. Both mutations lead to the substitution of the moderately conserved, hydrophobic amino acid leucine at position 238 to either a positively charged arginine [p.(Leu238Arg)] or to a cyclic proline [p.(Leu238Pro)]. Modelling of the SEAI domain of IMPG1 indicated that an arginine at position 238 destabilizes the protein [
7] and mutations which result in an introduction of a proline are known to significantly affect protein stability [
18]. Thus, the current finding of a second missense mutation affecting amino acid p.(Leu238) in a patient with VMD supports the causative role of defective IMPG1 molecules in disease via an as yet unknown dominant-negative effect or haploinsufficiency.
The second novel mutation in the
IMPG1 gene described in this study, a c.807 + 5G > A mutation in intron 7 causing aberrant splicing of the
IMPG1 mRNA, was found in a young Turkish boy in a homozygous state. Another splice mutation in intron 7, c.807 + 1G > T affecting the canonical GT donor splice sequence, has previously been described in a consanguineous Italian family [
7]. Affected individuals of this family showed multifocal vitelliform deposits in addition to the prominent central lesion. Since the fundus images of these patients were taken at the age of 29 and 33, it will be interesting to observe whether our 13 year old patient will develop similar lesions when disease progresses with age. Interestingly, heterozygous carriers of the c.807 + 1G > T mutation show a slightly decreased EOG Arden ratio and tiny extramacular deposits in fundus examination suggesting that heterozygous carriers of the c.807 + 1G > T variant display a mild subclinical phenotype due to haploinsufficiency. Both heterozygous 35 and 40 year old parents of patient #2 are clinically asymptomatic with full visual acuity and regular findings in multimodal retinal imaging (data not shown). Our finding that a significant fraction of pre-mRNAs harboring the c.807 + 5G > A mutation are correctly spliced suggests that the level of functional IMPG1 is sufficient to prevent even subclinical abnormalities in heterozygous carriers.
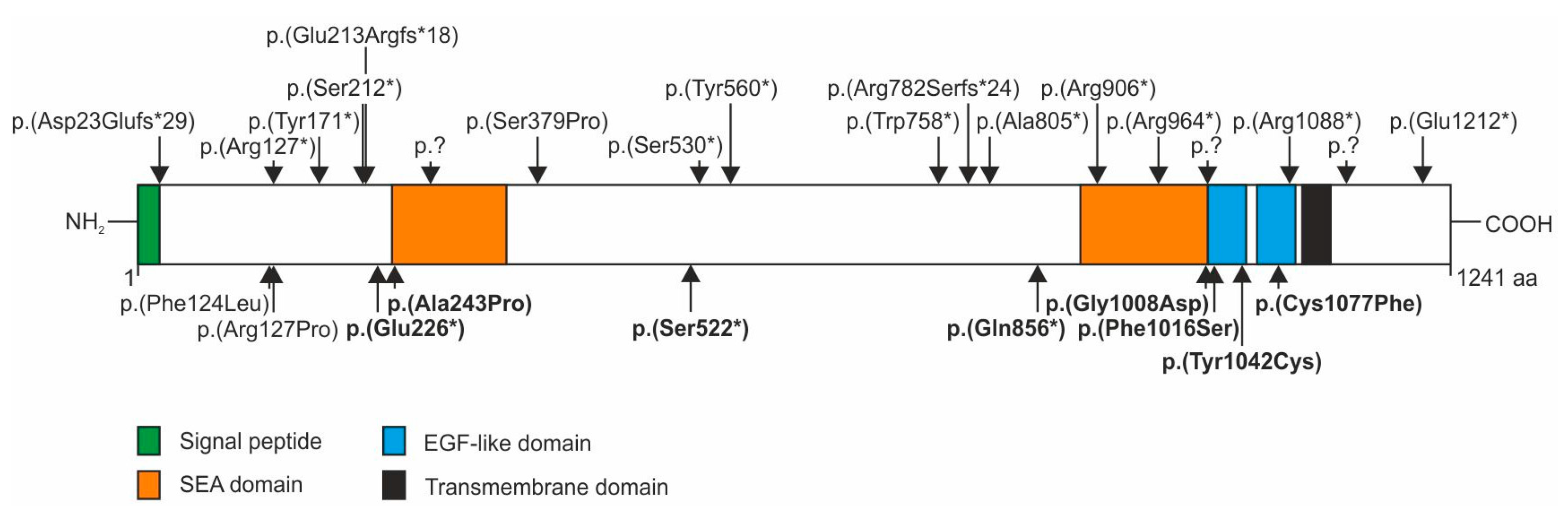
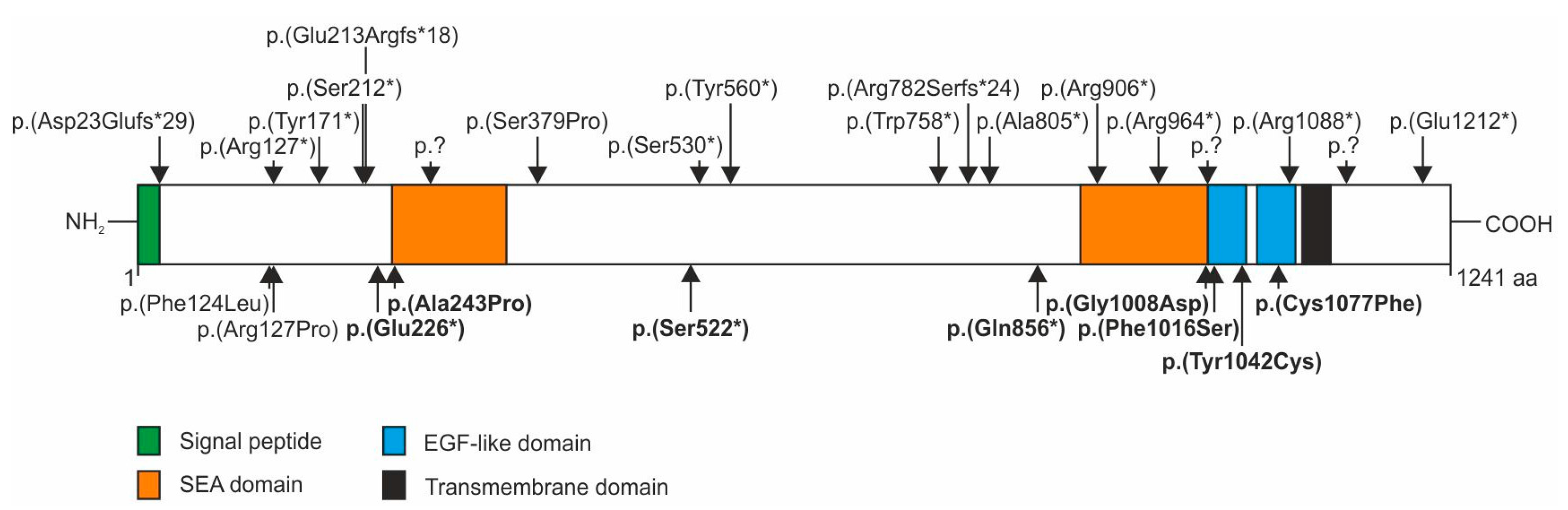
Homozygous or compound heterozygous nonsense, frameshift or splice site mutations in the
IMPG2 gene causing loss of IMPG2 function have repeatedly been described in patients with autosomal recessive retinitis pigmentosa (RP) (
Figure 3, upper panel). These mutations are scattered throughout the gene but are preferentially found in the N-terminal part (23-276 aa), two central regions (530-560 aa and 758-805 aa) and the C-terminal part (964-1212 aa) of IMPG2. In addition, a homozygous p.(Phe124Leu) mutation was reported in a patient with mild maculopathy [
5], a p.(Arg137Pro) mutation in a patient with Goldmann-Favre syndrome [
19] and a heterozygous p.(Cys1077Phe) mutation in a patient with VMD [
8]. These sequence variants as well as the stop and missense mutations found in our VMD patients are located in the N-terminal, the first central and C-terminal “hotspots”. The VMD-associated stop mutations p.(Glu226*), p.(Ser522*) and p.(Gln856*) in exon 7 and 13 of the
IMPG2 gene are likely targets for nonsense mediated decay (NMD) or lead to truncated proteins with impaired function and are therefore considered null mutations. So far, the evidence for a possible VMD causing effect of IMPG2 haploinsufficiency is based on the absence of the respective stop mutations in a large control population and the typical IMPG-linked phenotype of our three index patients. A recent report about patients with
IMPG2-associated RP revealed that three quarters of the patients (n = 13) had profound macular abnormalities including bull’s eye maculopathy and macular atrophy [
6]. These macular lesions are described as an early generalized loss of the outer retinal layers prior to RPE degeneration and are thus very different from the foveal subretinal deposits seen in our VMD patients. Nevertheless, it would be important to study the heterozygous parents of these and other
IMPG2-associated RP patients for subtle ophthalmological abnormalities such as minor foveal deposits.
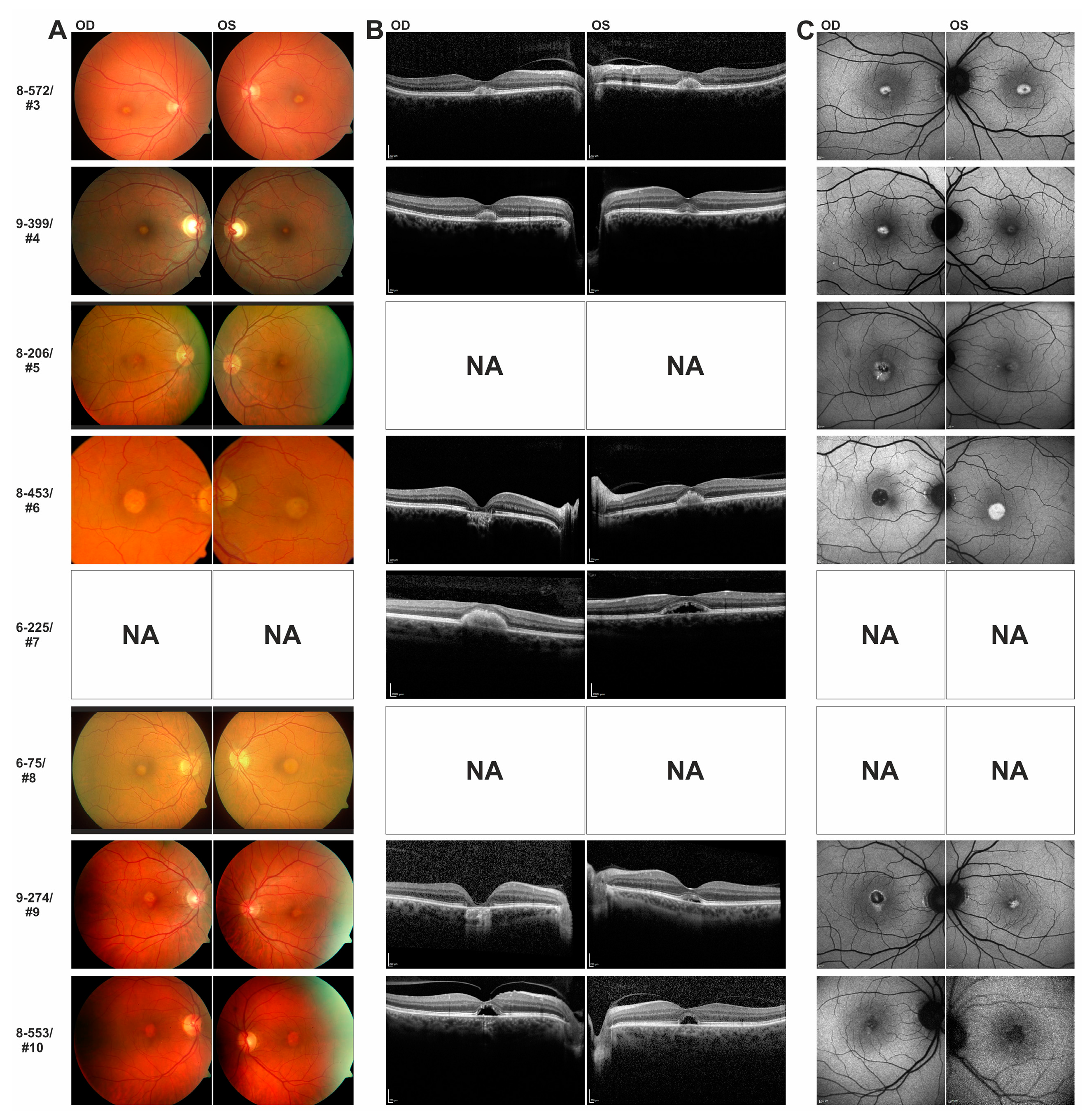
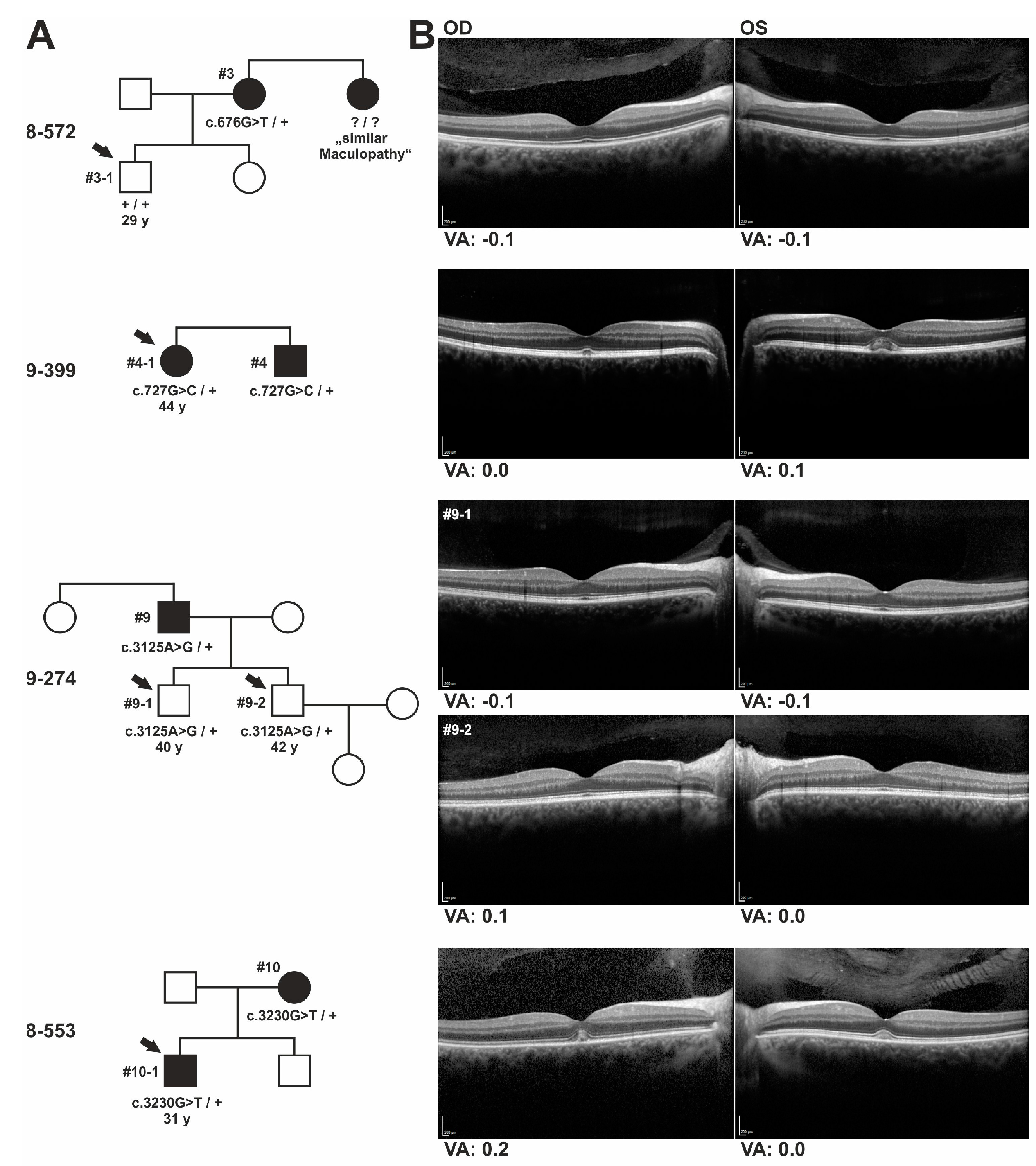
The same c.3230G > T/p.(Cys1077Phe)
IMPG2 mutation detected in the 70-year old female index patient of family 8-553 has previously been reported in a 44-year old male with progressive bilateral vitelliform lesions and his asymptomatic 22-year old son [
8]. SD-OCT scans of both of the son’s eyes revealed an abnormal thin hyperreflective line between the ellipsoid and outer segment-RPE interdigitation lines which was interpreted as a preclinical stage of
IMPG2-VMD [
8]. In support of an autosomal-dominant inheritance of the p.(Cys1077Phe) mutation, we observed distinct foveal lesions in the 31-year old son of patient #10 consisting with an accumulation of material above the RPE and mild visual impairment. Similarly, the sister of index patient #4 who is a heterozygous carrier of the familial c.727G > C/p.(Ala243Pro) mutation in
IMPG2 exon 7 and showed subclinical deposits above the RPE in the foveal zone and slightly decreased visual acuity.
Both sons of family 9-274, who inherited the heterozygous c.3125A > G/p.(Tyr1042Cys) mutation in exon 15 of the IMPG2 gene from their father, were 40 and 42 years of age when ophthalmologically examined. They were then asymptomatic and revealed regular retinal structures in SD-OCT. Their father was diagnosed with VMD not before the age of 62 and they may develop retinal disease later in life consistent with a rather late disease onset for symptomatic carriers of known C-terminal IMPG2 missense mutations.
Four of the five VMD-associated missense mutations in the IMPG2 gene are located within a stretch of 80 amino acids in the two epidermal growth factor (EGF)-like motifs at the N-terminal vicinity of a highly hydrophobic putative transmembrane spanning domain. The p.(Gly1008Asp) amino acid exchange affects one of the four consensus sequences of glycosaminoglycan (GAG) attachment sites in IMPG2, namely Ser1007-Gly-(acidic) and may influence protein interaction that is important for the establishment and stability of the IPM matrix.
Six conserved cysteine residues in each of the two tandem EGF-like motifs of IMPG2 likely form the characteristic EGF-like disulfide bridges required for stability of EGF-like motifs [
20]. IMPG2 mutations p.(Tyr1042Cys) and p.(Cys1077Phe) add or replace a cysteine residue in the EGF-like domain and may thus interfere with interchain disulfide bonding. Disruption of the disulfide bond pattern of EGF-like domains plays a major role in the pathogenesis of several inherited diseases [
21,
22]. Unravelling the molecular processes linking mutations in the
IMPG1 and
IMPG2 genes to retinal degeneration will be an important task for future experiments.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}