
CpG and Non-CpG Methylation in Epigenetic Gene Regulation and Brain Function


Abstract
:1. Introduction
2. CpG and Non-CpG Methylation in Epigenetic Gene Regulation
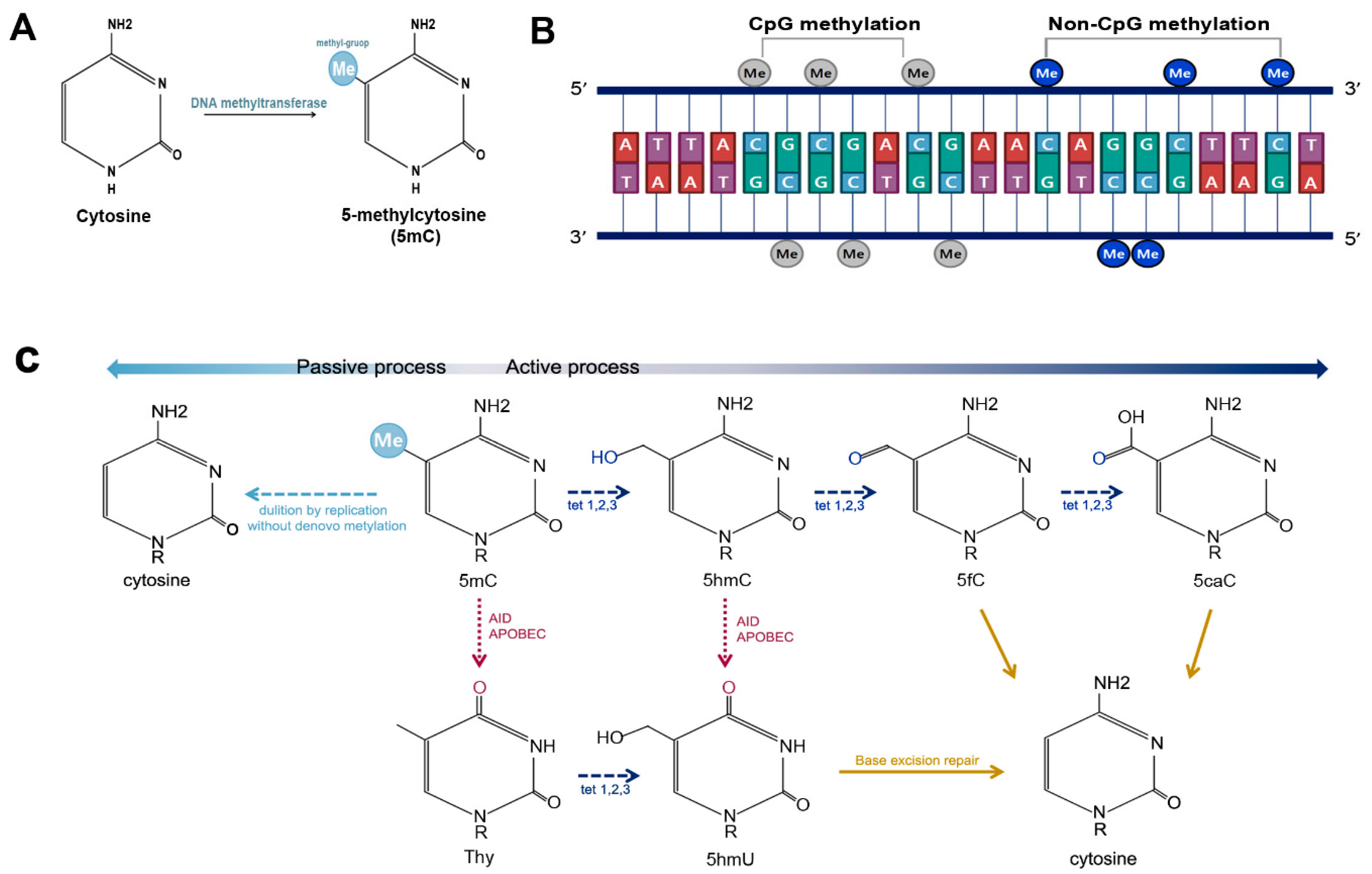
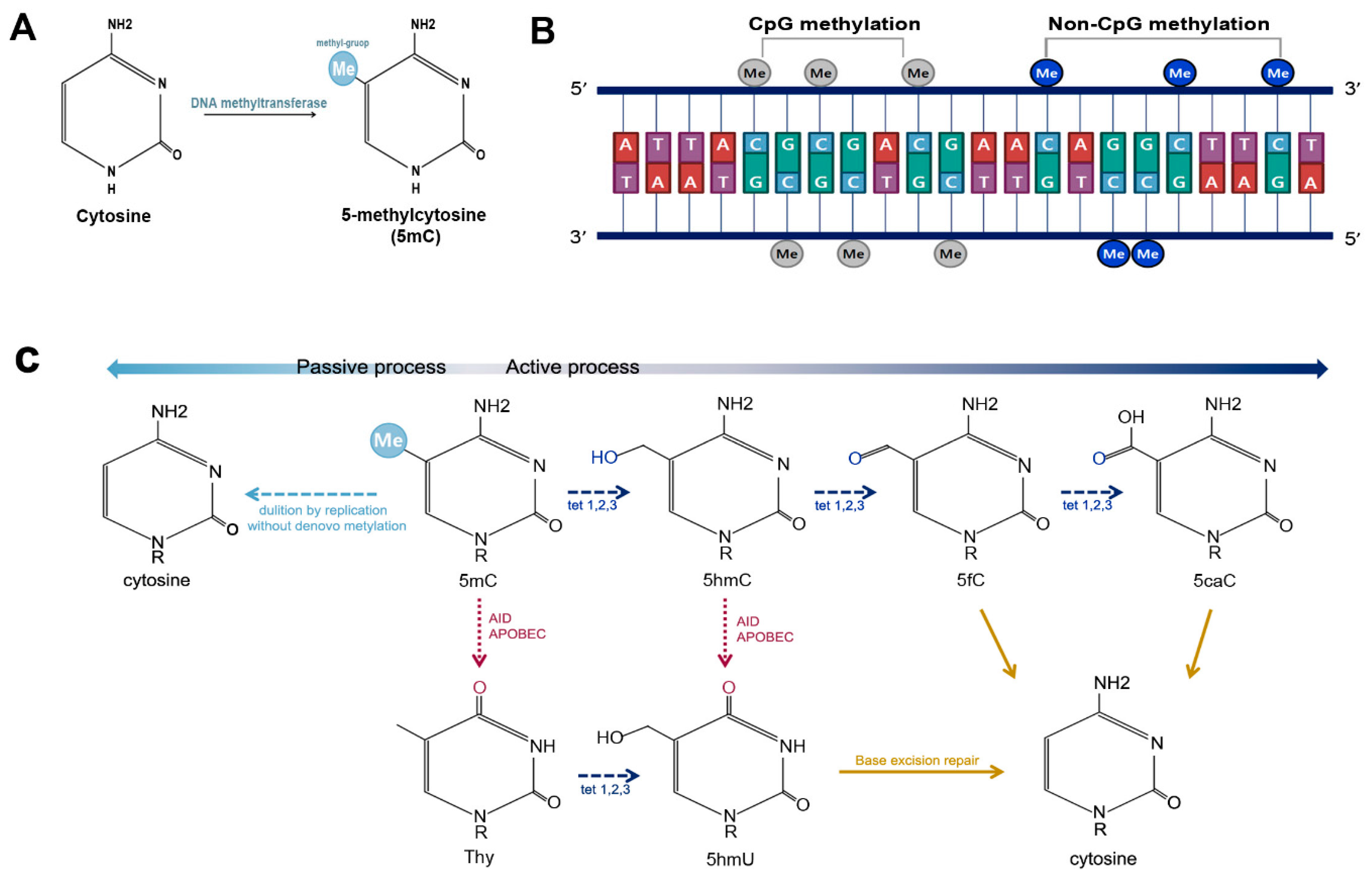
2.1. DNA Methylation
2.2. Non-CpG Methylation
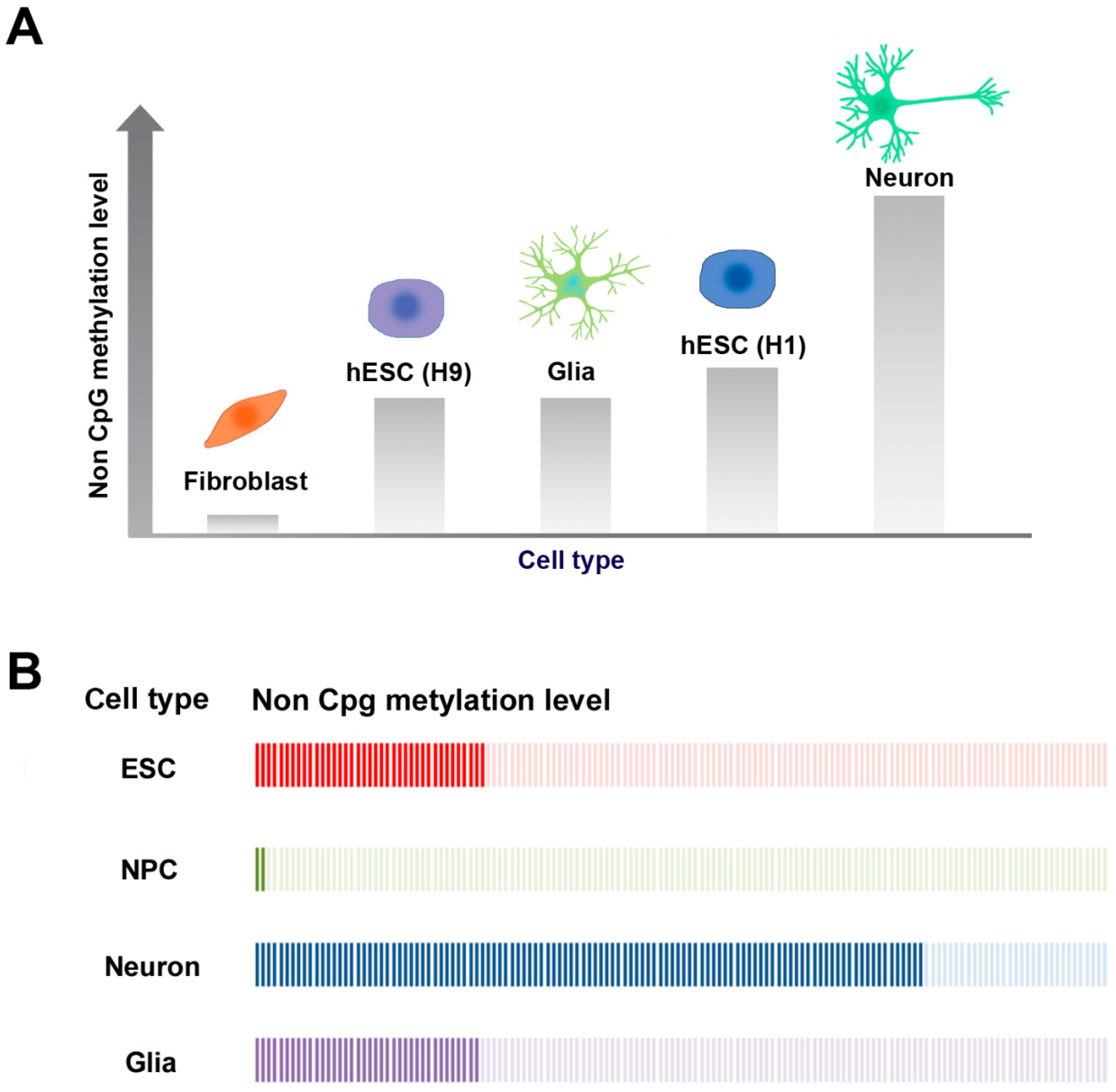
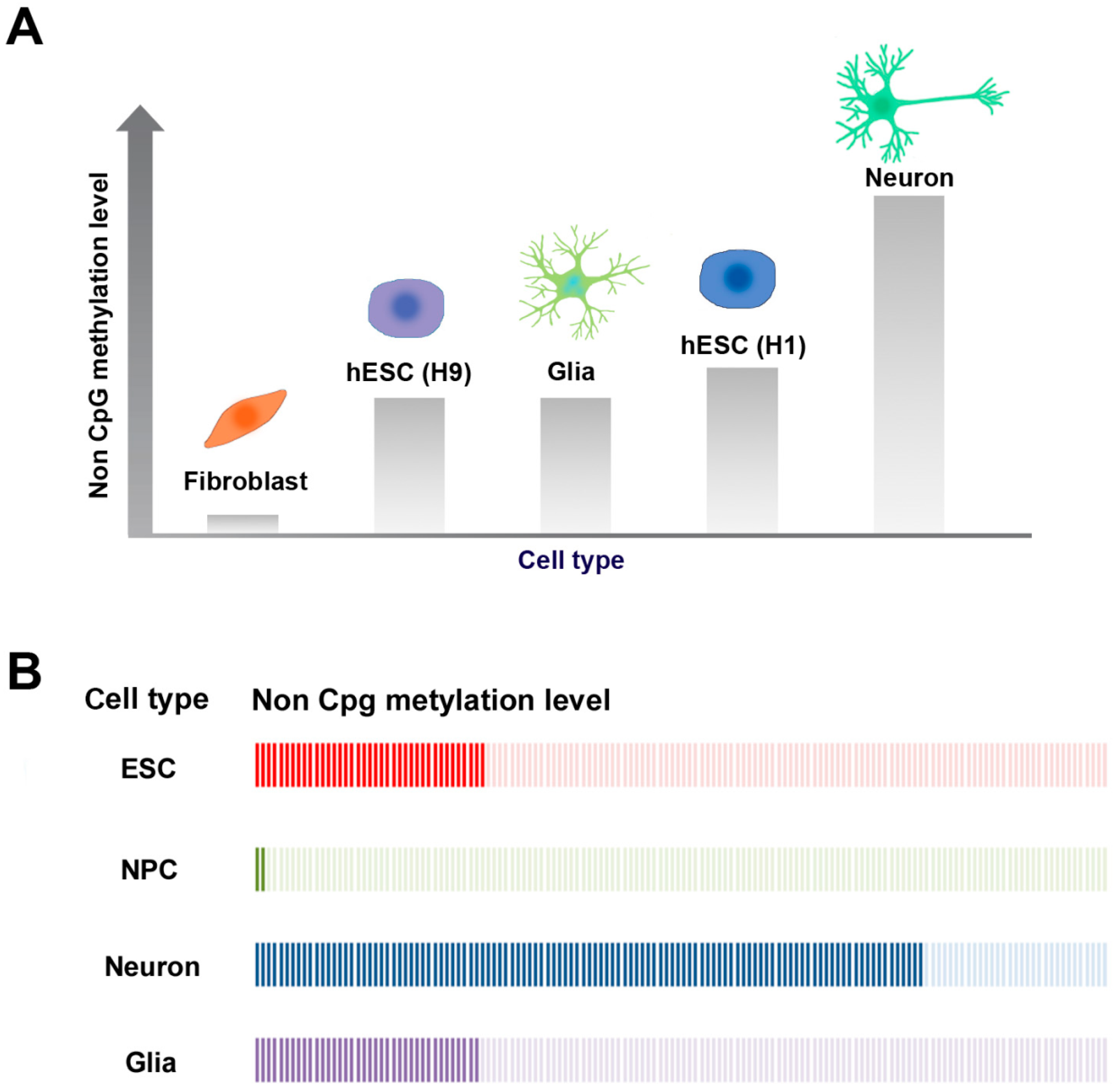
2.3. CpG and Non-CpG Methylation in Brain
2.4. 5hmC in Brain and Neural Development
3. Neurological Disorders Associated with DNA Methylation
3.1. Alzheimer’s Disease
3.2. Rett Syndrome
3.3. Fragile X Syndrome (FXS)
3.4. Parkinson’s Disease (PD)
3.5. Huntington’s Disease
3.6. Amyotrophic Lateral Sclerosis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Russo, V.E.; Martienssen, R.A.; Riggs, A.D. Epigenetic Mechanisms of Gene Regulation; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1996. [Google Scholar]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Riggs, A.D. X inactivation, differentiation, and DNA methylation. Cytogenet. Genome Res. 1975, 14, 9–25. [Google Scholar] [CrossRef]
- Holliday, R.; Pugh, J.E. DNA modification mechanisms and gene activity during development. Science 1975, 187, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Christman, J.K. 5-azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: Mechanistic studies and their implications for cancer therapy. Oncogene 2002, 21, 5483–5495. [Google Scholar] [CrossRef] [PubMed]
- Lindroth, A.M.; Cao, X.; Jackson, J.P.; Zilberman, D.; McCallum, C.M.; Henikoff, S.; Jacobsen, S.E. Requirement of chromomethylase3 for maintenance of cpxpg methylation. Science 2001, 292, 2077–2080. [Google Scholar] [CrossRef] [PubMed]
- Laurent, L.; Wong, E.; Li, G.; Huynh, T.; Tsirigos, A.; Ong, C.T.; Low, H.M.; Kin Sung, K.W.; Rigoutsos, I.; Loring, J.; et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010, 20, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global epigenomic reconfiguration during mammalian brain development. Science 2013, 341, 1237905. [Google Scholar] [CrossRef] [PubMed]
- Gowher, H.; Jeltsch, A. Enzymatic properties of recombinant Dnmt3a DNA methyltransferase from mouse: The enzyme modifies DNA in a non-processive manner and also methylates non-CpG [correction of non-CpA] sites. J. Mol. Biol. 2001, 309, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Ramsahoye, B.H.; Biniszkiewicz, D.; Lyko, F.; Clark, V.; Bird, A.P.; Jaenisch, R. Non-cpg methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc. Natl. Acad. Sci. USA 2000, 97, 5237–5242. [Google Scholar] [CrossRef] [PubMed]
- Schofield, M.J.; Hsieh, P. DNA mismatch repair: Molecular mechanisms and biological function. Ann. Rev. Microbiol. 2003, 57, 579–608. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [PubMed]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by tdg in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef] [PubMed]
- Mellen, M.; Ayata, P.; Dewell, S.; Kriaucionis, S.; Heintz, N. MeCP2 binds to 5hmc enriched within active genes and accessible chromatin in the nervous system. Cell 2012, 151, 1417–1430. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef] [PubMed]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Illingworth, R.; Kerr, A.; Desousa, D.; Jorgensen, H.; Ellis, P.; Stalker, J.; Jackson, D.; Clee, C.; Plumb, R.; Rogers, J.; et al. A novel CpG island set identifies tissue-specific methylation at developmental gene loci. PLoS Biol. 2008, 6, e22. [Google Scholar] [CrossRef] [PubMed]
- Felsenfeld, G. A brief history of epigenetics. Cold Spring Harb. Perspect. Biol. 2014, 6, a018200. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Han, H.; De Carvalho, D.D.; Lay, F.D.; Jones, P.A.; Liang, G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 2014, 26, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Chodavarapu, R.K.; Feng, S.; Bernatavichute, Y.V.; Chen, P.Y.; Stroud, H.; Yu, Y.; Hetzel, J.A.; Kuo, F.; Kim, J.; Cokus, S.J.; et al. Relationship between nucleosome positioning and DNA methylation. Nature 2010, 466, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Kavak, E.; Gregory, M.; Imashimizu, M.; Shutinoski, B.; Kashlev, M.; Oberdoerffer, P.; Sandberg, R.; Oberdoerffer, S. Ctcf-promoted rna polymerase ii pausing links DNA methylation to splicing. Nature 2011, 479, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, R.A.; Ladd-Acosta, C.; Wen, B.; Wu, Z.; Montano, C.; Onyango, P.; Cui, H.; Gabo, K.; Rongione, M.; Webster, M.; et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009, 41, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Alisch, R.S.; Barwick, B.G.; Chopra, P.; Myrick, L.K.; Satten, G.A.; Conneely, K.N.; Warren, S.T. Age-associated DNA methylation in pediatric populations. Genome Res. 2012, 22, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Pirazzini, C.; Giuliani, C.; Bacalini, M.G.; Boattini, A.; Capri, M.; Fontanesi, E.; Marasco, E.; Mantovani, V.; Pierini, M.; Pini, E.; et al. Space/population and time/age in DNA methylation variability in humans: A study on igf2/h19 locus in different italian populations and in mono- and di-zygotic twins of different age. Aging 2012, 4, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Heyn, H.; Li, N.; Ferreira, H.J.; Moran, S.; Pisano, D.G.; Gomez, A.; Diez, J.; Sanchez-Mut, J.V.; Setien, F.; Carmona, F.J.; et al. Distinct DNA methylomes of newborns and centenarians. Proc. Natl. Acad. Sci. USA 2012, 109, 10522–10527. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, N.; Izawa, T.; Yoshizawa, A.; Yokoi, H.; Kikuchi, Y.; Hashimoto, N. Decrease in cytosine methylation at CpG island shores and increase in DNA fragmentation during zebrafish aging. Age 2014, 36, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Yen, R.W.; Vertino, P.M.; Nelkin, B.D.; Yu, J.J.; el-Deiry, W.; Cumaraswamy, A.; Lennon, G.G.; Trask, B.J.; Celano, P.; Baylin, S.B. Isolation and characterization of the cdna encoding human DNA methyltransferase. Nucleic Acids Res. 1992, 20, 2287–2291. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Fan, G. The role of DNA methylation in the central nervous system and neuropsychiatric disorders. Int. Rev. Neurobiol. 2009, 89, 67–84. [Google Scholar] [PubMed]
- Chen, W.G.; Chang, Q.; Lin, Y.; Meissner, A.; West, A.E.; Griffith, E.C.; Jaenisch, R.; Greenberg, M.E. Derepression of Bdnf transcription involves calcium-dependent phosphorylation of MeCP2. Science 2003, 302, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Tohgi, H.; Utsugisawa, K.; Nagane, Y.; Yoshimura, M.; Genda, Y.; Ukitsu, M. Reduction with age in methylcytosine in the promoter region -224 approximately -101 of the amyloid precursor protein gene in autopsy human cortex. Mol. Brain Res. 1999, 70, 288–292. [Google Scholar] [CrossRef]
- Okano, M.; Xie, S.; Li, E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat. Genet. 1998, 19, 219–220. [Google Scholar] [PubMed]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases DNMT3A and DNMT3B are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Klein, C.J.; Botuyan, M.V.; Wu, Y.; Ward, C.J.; Nicholson, G.A.; Hammans, S.; Hojo, K.; Yamanishi, H.; Karpf, A.R.; Wallace, D.C.; et al. Mutations in dnmt1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat. Genet. 2011, 43, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Jurkowska, R.Z.; Jurkowski, T.P.; Jeltsch, A. Structure and function of mammalian DNA methyltransferases. ChemBioChem 2011, 12, 206–222. [Google Scholar] [CrossRef] [PubMed]
- Goll, M.G.; Bestor, T.H. Eukaryotic cytosine methyltransferases. Ann. Rev. Biochem. 2005, 74, 481–514. [Google Scholar] [CrossRef] [PubMed]
- Reik, W.; Dean, W.; Walter, J. Epigenetic reprogramming in mammalian development. Science 2001, 293, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Reik, W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 2007, 447, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Patil, V.; Ward, R.L.; Hesson, L.B. The evidence for functional non-CpG methylation in mammalian cells. Epigenetics 2014, 9, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Ziller, M.J.; Muller, F.; Liao, J.; Zhang, Y.; Gu, H.; Bock, C.; Boyle, P.; Epstein, C.B.; Bernstein, B.E.; Lengauer, T.; et al. Genomic distribution and inter-sample variation of non-CpG methylation across human cell types. PLoS Genet. 2011, 7, e1002389. [Google Scholar] [CrossRef] [PubMed]
- Barres, R.; Osler, M.E.; Yan, J.; Rune, A.; Fritz, T.; Caidahl, K.; Krook, A.; Zierath, J.R. Non-cpg methylation of the PGC-1alpha promoter through dnmt3b controls mitochondrial density. Cell Metabol. 2009, 10, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Kida, Y.S.; Hawkins, R.D.; Nery, J.R.; Hon, G.; Antosiewicz-Bourget, J.; O′Malley, R.; Castanon, R.; Klugman, S.; et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature 2011, 471, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Morey, R.; O′Neil, R.C.; He, Y.; Daughtry, B.; Schultz, M.D.; Hariharan, M.; Nery, J.R.; Castanon, R.; Sabatini, K.; et al. Abnormalities in human pluripotent cells due to reprogramming mechanisms. Nature 2014, 511, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.U.; Su, Y.; Shin, J.H.; Shin, J.; Li, H.; Xie, B.; Zhong, C.; Hu, S.; Le, T.; Fan, G.; et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci. 2014, 17, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Barres, R.; Kirchner, H.; Rasmussen, M.; Yan, J.; Kantor, F.R.; Krook, A.; Naslund, E.; Zierath, J.R. Weight loss after gastric bypass surgery in human obesity remodels promoter methylation. Cell Rep. 2013, 3, 1020–1027. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Barr, C.L.; Kim, A.; Yue, F.; Lee, A.Y.; Eubanks, J.; Dempster, E.L.; Ren, B. Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell 2012, 148, 816–831. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Oishi, M. Effects of methylation of non-CpG sequence in the promoter region on the expression of human synaptotagmin XI (syt11). Gene 2005, 348, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Zhu, P.; Yan, L.; Li, R.; Hu, B.; Lian, Y.; Yan, J.; Ren, X.; Lin, S.; Li, J.; et al. The DNA methylation landscape of human early embryos. Nature 2014, 511, 606–610. [Google Scholar] [CrossRef] [PubMed]
- Tomizawa, S.; Kobayashi, H.; Watanabe, T.; Andrews, S.; Hata, K.; Kelsey, G.; Sasaki, H. Dynamic stage-specific changes in imprinted differentially methylated regions during early mammalian development and prevalence of non-CpG methylation in oocytes. Development 2011, 138, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Varley, K.E.; Gertz, J.; Bowling, K.M.; Parker, S.L.; Reddy, T.E.; Pauli-Behn, F.; Cross, M.K.; Williams, B.A.; Stamatoyannopoulos, J.A.; Crawford, G.E.; et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 2013, 23, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Zvetkova, I.; Apedaile, A.; Ramsahoye, B.; Mermoud, J.E.; Crompton, L.A.; John, R.; Feil, R.; Brockdorff, N. Global hypomethylation of the genome in XX embryonic stem cells. Nat. Genet. 2005, 37, 1274–1279. [Google Scholar] [CrossRef] [PubMed]
- Arand, J.; Spieler, D.; Karius, T.; Branco, M.R.; Meilinger, D.; Meissner, A.; Jenuwein, T.; Xu, G.; Leonhardt, H.; Wolf, V.; et al. In vivo control of CpG and non-CpG DNA methylation by DNA methyltransferases. PLoS Genet. 2012, 8, e1002750. [Google Scholar] [CrossRef] [PubMed]
- Ichiyanagi, T.; Ichiyanagi, K.; Miyake, M.; Sasaki, H. Accumulation and loss of asymmetric non-CpG methylation during male germ-cell development. Nucleic Acids Res. 2013, 41, 738–745. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Karnik, R.; Gu, H.; Ziller, M.J.; Clement, K.; Tsankov, A.M.; Akopian, V.; Gifford, C.A.; Donaghey, J.; Galonska, C.; et al. Targeted disruption of DNMT1, DNMT3A and DNMT3B in human embryonic stem cells. Nat. Genet. 2015, 47, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Ladd-Acosta, C.; Pevsner, J.; Sabunciyan, S.; Yolken, R.H.; Webster, M.J.; Dinkins, T.; Callinan, P.A.; Fan, J.B.; Potash, J.B.; Feinberg, A.P. DNA methylation signatures within the human brain. Am. J. Hum. Genet. 2007, 81, 1304–1315. [Google Scholar] [CrossRef] [PubMed]
- Kozlenkov, A.; Wang, M.; Roussos, P.; Rudchenko, S.; Barbu, M.; Bibikova, M.; Klotzle, B.; Dwork, A.J.; Zhang, B.; Hurd, Y.L.; et al. Substantial DNA methylation differences between two major neuronal subtypes in human brain. Nucleic acids Res. 2016, 44, 2593–2612. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.U.; Ma, D.K.; Mo, H.; Ball, M.P.; Jang, M.H.; Bonaguidi, M.A.; Balazer, J.A.; Eaves, H.L.; Xie, B.; Ford, E.; et al. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat. Neurosci. 2011, 14, 1345–1351. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Schultz, M.D.; Lister, R.; Hou, Z.; Rajagopal, N.; Ray, P.; Whitaker, J.W.; Tian, S.; Hawkins, R.D.; Leung, D.; et al. Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell 2013, 153, 1134–1148. [Google Scholar] [CrossRef] [PubMed]
- Reubinoff, B.E.; Pera, M.F.; Fong, C.Y.; Trounson, A.; Bongso, A. Embryonic stem cell lines from human blastocysts: Somatic differentiation in vitro. Nat. Biotechnol. 2000, 18, 399–404. [Google Scholar] [PubMed]
- Zhang, S.C.; Wernig, M.; Duncan, I.D.; Brustle, O.; Thomson, J.A. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat. Biotechnol. 2001, 19, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Ecker, J.R. Non-CG methylation in the human genome. Ann. Rev. Genom. Hum. Genet. 2015, 16, 55–77. [Google Scholar] [CrossRef] [PubMed]
- Kozlenkov, A.; Roussos, P.; Timashpolsky, A.; Barbu, M.; Rudchenko, S.; Bibikova, M.; Klotzle, B.; Byne, W.; Lyddon, R.; Di Narzo, A.F.; et al. Differences in DNA methylation between human neuronal and glial cells are concentrated in enhancers and non-CpG sites. Nucleic Acids Res. 2014, 42, 109–127. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Bird, A.P. Genomic DNA methylation: The mark and its mediators. Trends Biochem. Sci. 2006, 31, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Shahbazian, M.D.; Antalffy, B.; Armstrong, D.L.; Zoghbi, H.Y. Insight into rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum. Mol. Genet. 2002, 11, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Guy, J.; Cheval, H.; Selfridge, J.; Bird, A. The role of MeCP2 in the brain. Ann. Rev. Cell Dev. Biol. 2011, 27, 631–652. [Google Scholar] [CrossRef] [PubMed]
- Kishi, N.; Macklis, J.D. MeCP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Mol. Cell. Neurosci. 2004, 27, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Skene, P.J.; Illingworth, R.S.; Webb, S.; Kerr, A.R.; James, K.D.; Turner, D.J.; Andrews, R.; Bird, A.P. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol. Cell 2010, 37, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Nan, X.; Ng, H.H.; Johnson, C.A.; Laherty, C.D.; Turner, B.M.; Eisenman, R.N.; Bird, A. Transcriptional repression by the methyl-cpg-binding protein MeCP2 involves a histone deacetylase complex. Nature 1998, 393, 386–389. [Google Scholar] [PubMed]
- Jones, P.L.; Veenstra, G.J.; Wade, P.A.; Vermaak, D.; Kass, S.U.; Landsberger, N.; Strouboulis, J.; Wolffe, A.P. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998, 19, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Kriaucionis, S.; Heintz, N. The nuclear DNA base 5-hydroxymethylcytosine is present in purkinje neurons and the brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef] [PubMed]
- Globisch, D.; Munzel, M.; Muller, M.; Michalakis, S.; Wagner, M.; Koch, S.; Bruckl, T.; Biel, M.; Carell, T. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS ONE 2010, 5, e15367. [Google Scholar] [CrossRef] [PubMed]
- Munzel, M.; Globisch, D.; Bruckl, T.; Wagner, M.; Welzmiller, V.; Michalakis, S.; Muller, M.; Biel, M.; Carell, T. Quantification of the sixth DNA base hydroxymethylcytosine in the brain. Angew. Chem. 2010, 49, 5375–5377. [Google Scholar] [CrossRef] [PubMed]
- Song, C.X.; Szulwach, K.E.; Fu, Y.; Dai, Q.; Yi, C.; Li, X.; Li, Y.; Chen, C.H.; Zhang, W.; Jian, X.; et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotechnol. 2011, 29, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Dawlaty, M.M.; Ganz, K.; Powell, B.E.; Hu, Y.C.; Markoulaki, S.; Cheng, A.W.; Gao, Q.; Kim, J.; Choi, S.W.; Page, D.C.; et al. TET1 is dispensable for maintaining pluripotency and its loss is compatible with embryonic and postnatal development. Cell Stem Cell 2011, 9, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Hahn, M.A.; Qiu, R.; Wu, X.; Li, A.X.; Zhang, H.; Wang, J.; Jui, J.; Jin, S.G.; Jiang, Y.; Pfeifer, G.P.; et al. Dynamics of 5-hydroxymethylcytosine and chromatin marks in mammalian neurogenesis. Cell Rep. 2013, 3, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.R.; Cui, Q.Y.; Murai, K.; Lim, Y.C.; Smith, Z.D.; Jin, S.; Ye, P.; Rosa, L.; Lee, Y.K.; Wu, H.P.; et al. TET1 regulates adult hippocampal neurogenesis and cognition. Cell Stem Cell 2013, 13, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Yang, D.; Li, J.; Tang, Y.; Yang, J.; Le, W. Critical role of tet3 in neural progenitor cell maintenance and terminal differentiation. Mol. Neurobiol. 2015, 51, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Szwagierczak, A.; Bultmann, S.; Schmidt, C.S.; Spada, F.; Leonhardt, H. Sensitive enzymatic quantification of 5-hydroxymethylcytosine in genomic DNA. Nucleic Acids Res. 2010, 38, e181. [Google Scholar] [CrossRef] [PubMed]
- Szulwach, K.E.; Li, X.; Li, Y.; Song, C.X.; Han, J.W.; Kim, S.; Namburi, S.; Hermetz, K.; Kim, J.J.; Rudd, M.K.; et al. Integrating 5-hydroxymethylcytosine into the epigenomic landscape of human embryonic stem cells. PLoS Genet. 2011, 7, e1002154. [Google Scholar] [CrossRef] [PubMed]
- Ruzov, A.; Tsenkina, Y.; Serio, A.; Dudnakova, T.; Fletcher, J.; Bai, Y.; Chebotareva, T.; Pells, S.; Hannoun, Z.; Sullivan, G.; et al. Lineage-specific distribution of high levels of genomic 5-hydroxymethylcytosine in mammalian development. Cell Res. 2011, 21, 1332–1342. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Xiong, L.; Xu, W.; Wu, F.; Huang, N.; Xu, Y.; Kong, L.; Zheng, L.; Schwartz, L.; Shi, Y.; et al. Genome-wide comparison of DNA hydroxymethylation in mouse embryonic stem cells and neural progenitor cells by a new comparative hmedip-seq method. Nucleic Acids Res. 2013, 41, e84. [Google Scholar] [CrossRef] [PubMed]
- Colquitt, B.M.; Allen, W.E.; Barnea, G.; Lomvardas, S. Alteration of genic 5-hydroxymethylcytosine patterning in olfactory neurons correlates with changes in gene expression and cell identity. Proc. Natl. Acad. Sci. USA 2013, 110, 14682–14687. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.; Wen, L.; Liao, S.; Lin, X.; Ma, T.; Liu, J.; Song, C.X.; Wang, M.; He, C.; Han, C.; et al. Dynamics of 5-hydroxymethylcytosine during mouse spermatogenesis. Nat. Commun. 2013, 4, 1995. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Li, X.; Yan, L.; Tan, Y.; Li, R.; Zhao, Y.; Wang, Y.; Xie, J.; Zhang, Y.; Song, C.; et al. Whole-genome analysis of 5-hydroxymethylcytosine and 5-methylcytosine at base resolution in the human brain. Genome Biol. 2014, 15, R49. [Google Scholar] [CrossRef] [PubMed]
- Gabel, H.W.; Kinde, B.; Stroud, H.; Gilbert, C.S.; Harmin, D.A.; Kastan, N.R.; Hemberg, M.; Ebert, D.H.; Greenberg, M.E. Disruption of DNA-methylation-dependent long gene repression in rett syndrome. Nature 2015, 522, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Spruijt, C.G.; Gnerlich, F.; Smits, A.H.; Pfaffeneder, T.; Jansen, P.W.; Bauer, C.; Munzel, M.; Wagner, M.; Muller, M.; Khan, F.; et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell 2013, 152, 1146–1159. [Google Scholar] [CrossRef] [PubMed]
- Minor, E.A.; Court, B.L.; Young, J.I.; Wang, G. Ascorbate induces ten-eleven translocation (TET) methylcytosine dioxygenase-mediated generation of 5-hydroxymethylcytosine. J. Biol. Chem. 2013, 288, 13669–13674. [Google Scholar] [CrossRef] [PubMed]
- Spector, R.; Johanson, C.E. The nexus of vitamin homeostasis and DNA synthesis and modification in mammalian brain. Mol. Brain 2014, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Sherwani, S.I.; Khan, H.A. Role of 5-hydroxymethylcytosine in neurodegeneration. Gene 2015, 570, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Wheldon, L.M.; Abakir, A.; Ferjentsik, Z.; Dudnakova, T.; Strohbuecker, S.; Christie, D.; Dai, N.; Guan, S.; Foster, J.M.; Correa, I.R., Jr.; et al. Transient accumulation of 5-carboxylcytosine indicates involvement of active demethylation in lineage specification of neural stem cells. Cell Rep. 2014, 7, 1353–1361. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.A.; Sweatt, J.D. Covalent modification of DNA regulates memory formation. Neuron 2007, 53, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Schanen, N.C. Epigenetics of autism spectrum disorders. Hum. Mol. Genet. 2006, 15, R138–R150. [Google Scholar] [CrossRef] [PubMed]
- LaSalle, J.M.; Yasui, D.H. Evolving role of MeCP2 in rett syndrome and autism. Epigenomics 2009, 1, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Nicolia, V.; Cavallaro, R.A.; Lopez-Gonzalez, I.; Maccarrone, M.; Scarpa, S.; Ferrer, I.; Fuso, A. DNA methylation profiles of selected pro-inflammatory cytokines in alzheimer disease. J. Neuropathol. Exp. Neurol. 2017, 76, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D.; Wolffe, A.P. DNA methylation in health and disease. Nat. Rev. Genet. 2000, 1, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Urdinguio, R.G.; Sanchez-Mut, J.V.; Esteller, M. Epigenetic mechanisms in neurological diseases: Genes, syndromes, and therapies. Lancet Neurol. 2009, 8, 1056–1072. [Google Scholar] [CrossRef]
- Iqbal, K.; Grundke-Iqbal, I. Alzheimer neurofibrillary degeneration: Significance, etiopathogenesis, therapeutics and prevention. J. Cell. Mol. Med. 2008, 12, 38–55. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Mut, J.V.; Aso, E.; Panayotis, N.; Lott, I.; Dierssen, M.; Rabano, A.; Urdinguio, R.G.; Fernandez, A.F.; Astudillo, A.; Martin-Subero, J.I.; et al. DNA methylation map of mouse and human brain identifies target genes in Alzheimer’s disease. Brain 2013, 136, 3018–3027. [Google Scholar] [CrossRef] [PubMed]
- Blanch, M.; Mosquera, J.L.; Ansoleaga, B.; Ferrer, I.; Barrachina, M. Altered mitochondrial DNA methylation pattern in Alzheimer disease-related pathology and in parkinson disease. Am. J. Pathol. 2016, 186, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Du, Q.; Chen, L.; Fu, G.; Li, S.; Fu, L.; Zhang, X.; Ma, C.; Bin, C. Cpg methylation patterns of human mitochondrial DNA. Sci. Rep. 2016, 6, 23421. [Google Scholar] [CrossRef] [PubMed]
- Chouliaras, L.; Mastroeni, D.; Delvaux, E.; Grover, A.; Kenis, G.; Hof, P.R.; Steinbusch, H.W.; Coleman, P.D.; Rutten, B.P.; van den Hove, D.L. Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer’s disease patients. Neurobiol. Aging 2013, 34, 2091–2099. [Google Scholar] [CrossRef] [PubMed]
- Condliffe, D.; Wong, A.; Troakes, C.; Proitsi, P.; Patel, Y.; Chouliaras, L.; Fernandes, C.; Cooper, J.; Lovestone, S.; Schalkwyk, L. Cross-region reduction in 5-hydroxymethylcytosine in alzheimer’s disease brain. Neurobiol. Aging 2014, 35, 1850–1854. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, N.; Dieriks, B.V.; Lill, C.; Faull, R.L.; Curtis, M.A.; Dragunow, M. Global changes in DNA methylation and hydroxymethylation in Alzheimer’s disease human brain. Neurobiol. Aging 2014, 35, 1334–1344. [Google Scholar] [CrossRef] [PubMed]
- Bradley-Whitman, M.; Lovell, M. Epigenetic changes in the progression of Alzheimer’s disease. Mech. Ageing Dev. 2013, 134, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, B. Rett’s syndrome: Prevalence and impact on progressive severe mental retardation in girls. Acta Paediatr. Scand. 1985, 74, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Rett, A. On a unusual brain atrophy syndrome in hyperammonemia in childhood. In Wiener Medizinische Wochenschrift; Springer: Vienna, Austria, 1966; Volume 116, pp. 723–726. [Google Scholar]
- Martinowich, K.; Hattori, D.; Wu, H.; Fouse, S.; He, F.; Hu, Y.; Fan, G.; Sun, Y.E. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 2003, 302, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Nuber, U.A.; Kriaucionis, S.; Roloff, T.C.; Guy, J.; Selfridge, J.; Steinhoff, C.; Schulz, R.; Lipkowitz, B.; Ropers, H.H.; Holmes, M.C.; et al. Up-regulation of glucocorticoid-regulated genes in a mouse model of Rett syndrome. Hum. Mol. Genet. 2005, 14, 2247–2256. [Google Scholar] [CrossRef] [PubMed]
- Horike, S.; Cai, S.; Miyano, M.; Cheng, J.F.; Kohwi-Shigematsu, T. Loss of silent-chromatin looping and impaired imprinting of Dlx5 in Rett syndrome. Nat. Genet. 2005, 37, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Sarraf, S.A.; Schmiedeberg, L.; McDermott, S.M.; Stancheva, I.; Bird, A.P. DNA binding selectivity of MeCP2 due to a requirement for A/T sequences adjacent to methyl-CpG. Mol. Cell 2005, 19, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Makedonski, K.; Abuhatzira, L.; Kaufman, Y.; Razin, A.; Shemer, R. MeCP2 deficiency in Rett syndrome causes epigenetic aberrations at the PWS/AS imprinting center that affects UBE3A expression. Hum. Mol. Genet. 2005, 14, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MeCP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [PubMed]
- Chen, L.; Chen, K.; Lavery, L.A.; Baker, S.A.; Shaw, C.A.; Li, W.; Zoghbi, H.Y. MeCP2 binds to non-CG methylated DNA as neurons mature, influencing transcription and the timing of onset for Rett syndrome. Proc. Natl. Acad. Sci. USA 2015, 112, 5509–5514. [Google Scholar] [CrossRef] [PubMed]
- Irwin, S.A.; Galvez, R.; Greenough, W.T. Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb. Cortex 2000, 10, 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Huber, K.M.; Gallagher, S.M.; Warren, S.T.; Bear, M.F. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. USA 2002, 99, 7746–7750. [Google Scholar] [CrossRef] [PubMed]
- Garber, K.B.; Visootsak, J.; Warren, S.T. Fragile X syndrome. Eur. J. Hum. Genet. 2008, 16, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Mostofsky, S.H.; Mazzocco, M.M.; Aakalu, G.; Warsofsky, I.S.; Denckla, M.B.; Reiss, A.L. Decreased cerebellar posterior vermis size in fragile X syndrome: Correlation with neurocognitive performance. Neurology 1998, 50, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Nolin, S.L.; Lewis, F.A., 3rd; Ye, L.L.; Houck, G.E., Jr.; Glicksman, A.E.; Limprasert, P.; Li, S.Y.; Zhong, N.; Ashley, A.E.; Feingold, E.; et al. Familial transmission of the FMR1 CgG repeat. Am. J. Hum. Genet. 1996, 59, 1252–1261. [Google Scholar] [PubMed]
- Kremer, E.; Pritchard, M.; Lynch, M.; Yu, S.; Holman, K.; Baker, E.; Warren, S.; Schlessinger, D.; Sutherland, G.; Richards, R. Mapping of DNA instability at the fragile x to a trinucleotide repeat sequence p (CcG) n. Science 1991, 252, 1711–1714. [Google Scholar] [CrossRef] [PubMed]
- Oberle, I.; Rousseau, F.; Heitz, D.; Kretz, C.; Devys, D.; Hanauer, A.; Boue, J.; Bertheas, M.F.; Mandel, J.L. Instability of a 550-base pair DNA segment and abnormal methylation in fragile x syndrome. Science 1991, 252, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Coffee, B.; Zhang, F.; Warren, S.T.; Reines, D. Acetylated histones are associated with fmr1 in normal but not fragile X-syndrome cells. Nat. Genet. 1999, 22, 98–101. [Google Scholar] [PubMed]
- Tabolacci, E.; Palumbo, F.; Nobile, V.; Neri, G. Transcriptional reactivation of the fmr1 gene. A possible approach to the treatment of the fragile X syndrome. Genes 2016, 7, 49. [Google Scholar] [CrossRef] [PubMed]
- Lees, A.J.; Hardy, J.; Revesz, T. Parkinson’s disease. Lancet 2009, 373, 2055–2066. [Google Scholar] [CrossRef]
- Volta, M.; Milnerwood, A.J.; Farrer, M.J. Insights from late-onset familial parkinsonism on the pathogenesis of idiopathic parkinson’s disease. Lancet Neurol. 2015, 14, 1054–1064. [Google Scholar] [CrossRef]
- Grundemann, J.; Schlaudraff, F.; Haeckel, O.; Liss, B. Elevated alpha-synuclein mrna levels in individual UV-laser-microdissected dopaminergic substantia nigra neurons in idiopathic parkinson’s disease. Nucleic Acids Res. 2008, 36, e38. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, I.; Kaut, O.; Khazneh, H.; deBoni, L.; Ahmad, A.; Berg, D.; Klein, C.; Frohlich, H.; Wullner, U. L-dopa increases alpha-synuclein DNA methylation in parkinson’s disease patients in vivo and in vitro. Mov. Disord. 2015, 30, 1794–1801. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yuan, Y.; Tong, Q.; Jiang, S.; Xu, Q.; Ding, J.; Zhang, L.; Zhang, R.; Zhang, K. Reduced plasma taurine level in parkinson’s disease: Association with motor severity and levodopa treatment. Int. J. Neurosci. 2016, 126, 630–636. [Google Scholar] [PubMed]
- Desplats, P.; Spencer, B.; Coffee, E.; Patel, P.; Michael, S.; Patrick, C.; Adame, A.; Rockenstein, E.; Masliah, E. Alpha-synuclein sequesters DNMT1 from the nucleus: A novel mechanism for epigenetic alterations in lewy body diseases. J. Biol. Chem. 2011, 286, 9031–9037. [Google Scholar] [CrossRef] [PubMed]
- Carvey, P.M.; Chen, E.Y.; Lipton, J.W.; Tong, C.W.; Chang, Q.A.; Ling, Z.D. Intra-parenchymal injection of tumor necrosis factor-alpha and interleukin 1-beta produces dopamine neuron loss in the rat. J. Neural Transm. 2005, 112, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Pieper, H.C.; Evert, B.O.; Kaut, O.; Riederer, P.F.; Waha, A.; Wullner, U. Different methylation of the tnf-alpha promoter in cortex and substantia nigra: Implications for selective neuronal vulnerability. Neurobiol. Dis. 2008, 32, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Pringsheim, T.; Wiltshire, K.; Day, L.; Dykeman, J.; Steeves, T.; Jette, N. The incidence and prevalence of Huntington’s disease: A systematic review and meta-analysis. Mov. Disord. 2012, 27, 1083–1091. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N. A novel gene containing a trinucleotide repeat that is expanded and unstable on huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Kremer, B.; Goldberg, P.; Andrew, S.E.; Theilmann, J.; Telenius, H.; Zeisler, J.; Squitieri, F.; Lin, B.; Bassett, A.; Almqvist, E. A worldwide study of the Huntington’s disease mutation: The sensitivity and specificity of measuring cag repeats. N. Engl. J. Med. 1994, 330, 1401–1406. [Google Scholar] [CrossRef] [PubMed]
- De Souza, R.A.; Islam, S.A.; McEwen, L.M.; Mathelier, A.; Hill, A.; Mah, S.M.; Wasserman, W.W.; Kobor, M.S.; Leavitt, B.R. DNA methylation profiling in human Huntington’s disease brain. Hum. Mol. Genet. 2016, 25, 2013–2030. [Google Scholar] [CrossRef] [PubMed]
- Villar-Menéndez, I.; Blanch, M.; Tyebji, S.; Pereira-Veiga, T.; Albasanz, J.L.; Martín, M.; Ferrer, I.; Pérez-Navarro, E.; Barrachina, M. Increased 5-methylcytosine and decreased 5-hydroxymethylcytosine levels are associated with reduced striatal A2AR levels in Huntington’s disease. Neuromol. Med. 2013, 15, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Thomas, B.; Matson, S.; Chopra, V.; Sun, L.; Sharma, S.; Hersch, S.; Rosas, H.D.; Scherzer, C.; Ferrante, R.; Matson, W. A novel method for detecting 7-methyl guanine reveals aberrant methylation levels in Huntington disease. Anal. Biochem. 2013, 436, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Yang, Y.; Lin, X.; Wang, J.-Q.; Wu, Y.-S.; Xie, W.; Wang, D.; Zhu, S.; Liao, Y.-Q.; Sun, Q. Genome-wide loss of 5-hmC is a novel epigenetic feature of Huntington’s disease. Hum. Mol. Genet. 2013, 22, 3641–3653. [Google Scholar] [CrossRef] [PubMed]
- Bai, G.; Cheung, I.; Shulha, H.P.; Coelho, J.E.; Li, P.; Dong, X.; Jakovcevski, M.; Wang, Y.; Grigorenko, A.; Jiang, Y. Epigenetic dysregulation of hairy and enhancer of split 4 (HES4) is associated with striatal degeneration in postmortem Huntington brains. Hum. Mol. Genet. 2014, 24, 1441–1456. [Google Scholar] [CrossRef] [PubMed]
- McFarland, K.N.; Huizenga, M.N.; Darnell, S.B.; Sangrey, G.R.; Berezovska, O.; Cha, J.-H.J.; Outeiro, T.F.; Sadri-Vakili, G. MeCP2: A novel huntingtin interactor. Hum. Mol. Genet. 2014, 23, 1036–1044. [Google Scholar] [CrossRef] [PubMed]
- Rowland, L.P.; Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Heath, P.R.; Shaw, P.J. Update on the glutamatergic neurotransmitter system and the role of excitotoxicity in amyotrophic lateral sclerosis. Muscle Nerv. 2002, 26, 438–458. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J. Mitochondrial and cell death mechanisms in neurodegenerative diseases. Pharmaceuticals 2010, 3, 839–915. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Wong, M. Aberrant regulation of DNA methylation in amyotrophic lateral sclerosis: A new target of disease mechanisms. Neurotherapeutics 2013, 10, 722–733. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho, M.; Swash, M. Amyotrophic lateral sclerosis: An update. Curr. Opin. Neurol. 2011, 24, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Romero, C.; Hur, J.; Bender, D.E.; Delaney, C.E.; Cataldo, M.D.; Smith, A.L.; Yung, R.; Ruden, D.M.; Callaghan, B.C.; Feldman, E.L. Identification of epigenetically altered genes in sporadic amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e52672. [Google Scholar] [CrossRef] [PubMed]
- Chestnut, B.A.; Chang, Q.; Price, A.; Lesuisse, C.; Wong, M.; Martin, L.J. Epigenetic regulation of motor neuron cell death through DNA methylation. J. Neurosci. 2011, 31, 16619–16636. [Google Scholar] [CrossRef] [PubMed]
- Bollati, V.; Galimberti, D.; Pergoli, L.; Dalla Valle, E.; Barretta, F.; Cortini, F.; Scarpini, E.; Bertazzi, P.; Baccarelli, A. DNA methylation in repetitive elements and alzheimer disease. Brain Behav. Immun. 2011, 25, 1078–1083. [Google Scholar] [CrossRef] [PubMed]
- Paez-Colasante, X.; Figueroa-Romero, C.; Sakowski, S.A.; Goutman, S.A.; Feldman, E.L. Amyotrophic lateral sclerosis: Mechanisms and therapeutics in the epigenomic era. Nat. Rev. Neurol. 2015, 11, 266–279. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9orf72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Xi, Z.; Zinman, L.; Moreno, D.; Schymick, J.; Liang, Y.; Sato, C.; Zheng, Y.; Ghani, M.; Dib, S.; Keith, J. Hypermethylation of the CpG island near the G 4 C 2 repeat in ALS with A C9orf72 expansion. Am. J. Hum. Genet. 2013, 92, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Xi, Z.; Zhang, M.; Bruni, A.C.; Maletta, R.G.; Colao, R.; Fratta, P.; Polke, J.M.; Sweeney, M.G.; Mudanohwo, E.; Nacmias, B.; et al. The C9orf72 repeat expansion itself is methylated in ALS and FTLD patients. Acta Neuropathol. 2015, 129, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Ciura, S.; Lattante, S.; Le Ber, I.; Latouche, M.; Tostivint, H.; Brice, A.; Kabashi, E. Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis. Ann. Neurol. 2013, 74, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Bauer, P.O. Methylation of C9orf72 expansion reduces rna foci formation and dipeptide-repeat proteins expression in cells. Neurosci. Lett. 2016, 612, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Day, J.J.; Roberson, E.D. DNA methylation slows effects of C9orf72 mutations an epigenetic brake on genetic inheritance. Neurology 2015, 84, 1616–1617. [Google Scholar] [CrossRef] [PubMed]
- Pihlstrom, L.; Berge, V.; Rengmark, A.; Toft, M. Parkinson’s disease correlates with promoter methylation in the alpha-synuclein gene. Mov. Disord. 2015, 30, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Dumaop, W.; Galasko, D.; Desplats, P. Distinctive patterns of DNA methylation associated with parkinson disease: Identification of concordant epigenetic changes in brain and peripheral blood leukocytes. Epigenetics 2013, 8, 1030–1038. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Mariani, J.; Coppola, G.; Zhang, P.; Abyzov, A.; Provini, L.; Tomasini, L.; Amenduni, M.; Szekely, A.; Palejev, D.; Wilson, M.; et al. Foxg1-dependent dysregulation of gaba/glutamate neuron differentiation in autism spectrum disorders. Cell 2015, 162, 375–390. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Disease | Patterns | Main Factors | References |
|---|---|---|---|
| Alzheimer’s disease | CpG methylation | Methylated interleukin-6 | [97] |
| CpG methylation | Hypermethylated TXBA2R, SORBS3, and SPTBN4 | [102] | |
| CpG/non-CpG methylation | Hypermethylated mitochondrial DNA | [103] | |
| Rett syndrome | MeCp2 mutation | Mutations in MeCP2 target genes (Bdnf, Fkbp5, Dlx5, Dlx6, UBE3A, and Sgk) | [111,112,113,114,115] |
| Non-CpG methylation | Binding of MeCP2 to methylated non-CpG sites | [117] | |
| MeCP2-5hmC interaction | Inhibition of conversion from 5mC to 5hmC | [89] | |
| Fragile X syndrome | Histone deacetylation and DNA hypermethylation | Abnormal expansion of CGG repeat in 5’ UTR of FMR1 gene | [121] |
| Parkinson’s disease | CpG methylation | Hypomethylation of SNCA | [130] |
| CpG methylation | Hypomethylation of TNF-αpromoter | [134] | |
| CpG/non-CpG methylation | Hypomethylation of mitochondrial DNA | [102] | |
| Huntington’s disease | CpG methylation | Neurotrophic factors (Bdnf and A2A) | [139] |
| CpG methylation | Interaction of mutant Htt with MeCP2 | [143] | |
| 5hmC | Regulation of Adora2a expression by 5hmC levels | [139] | |
| Amyotrophic lateral sclerosis | CpG methylation | 5mC accumulation in motor neurons | [150] |
| DMNT1 transcription | Increased levels of Dnmt3a lead to apoptosis | [150] | |
| Genetic mutation | Gene mutations in SOD1, C9orf72, TARDBP, and FUS genes | [152] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, H.S.; Shin, W.J.; Lee, J.E.; Do, J.T. CpG and Non-CpG Methylation in Epigenetic Gene Regulation and Brain Function. Genes 2017, 8, 148. https://doi.org/10.3390/genes8060148
Jang HS, Shin WJ, Lee JE, Do JT. CpG and Non-CpG Methylation in Epigenetic Gene Regulation and Brain Function. Genes. 2017; 8(6):148. https://doi.org/10.3390/genes8060148
Chicago/Turabian StyleJang, Hyun Sik, Woo Jung Shin, Jeong Eon Lee, and Jeong Tae Do. 2017. "CpG and Non-CpG Methylation in Epigenetic Gene Regulation and Brain Function" Genes 8, no. 6: 148. https://doi.org/10.3390/genes8060148
APA StyleJang, H. S., Shin, W. J., Lee, J. E., & Do, J. T. (2017). CpG and Non-CpG Methylation in Epigenetic Gene Regulation and Brain Function. Genes, 8(6), 148. https://doi.org/10.3390/genes8060148