
Identification of Drought-Responsive MicroRNAs from Roots and Leaves of Alfalfa by High-Throughput Sequencing

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Experiment Design
2.2. Total RNA Isolation
2.3. Small RNA Library Construction and High-Throughput Sequencing
2.4. Identification of Known and Novel MicroRNAs
2.5. MicroRNA Validation by qRT-PCR
2.6. MicroRNA Target Prediction and Function Analysis
3. Results
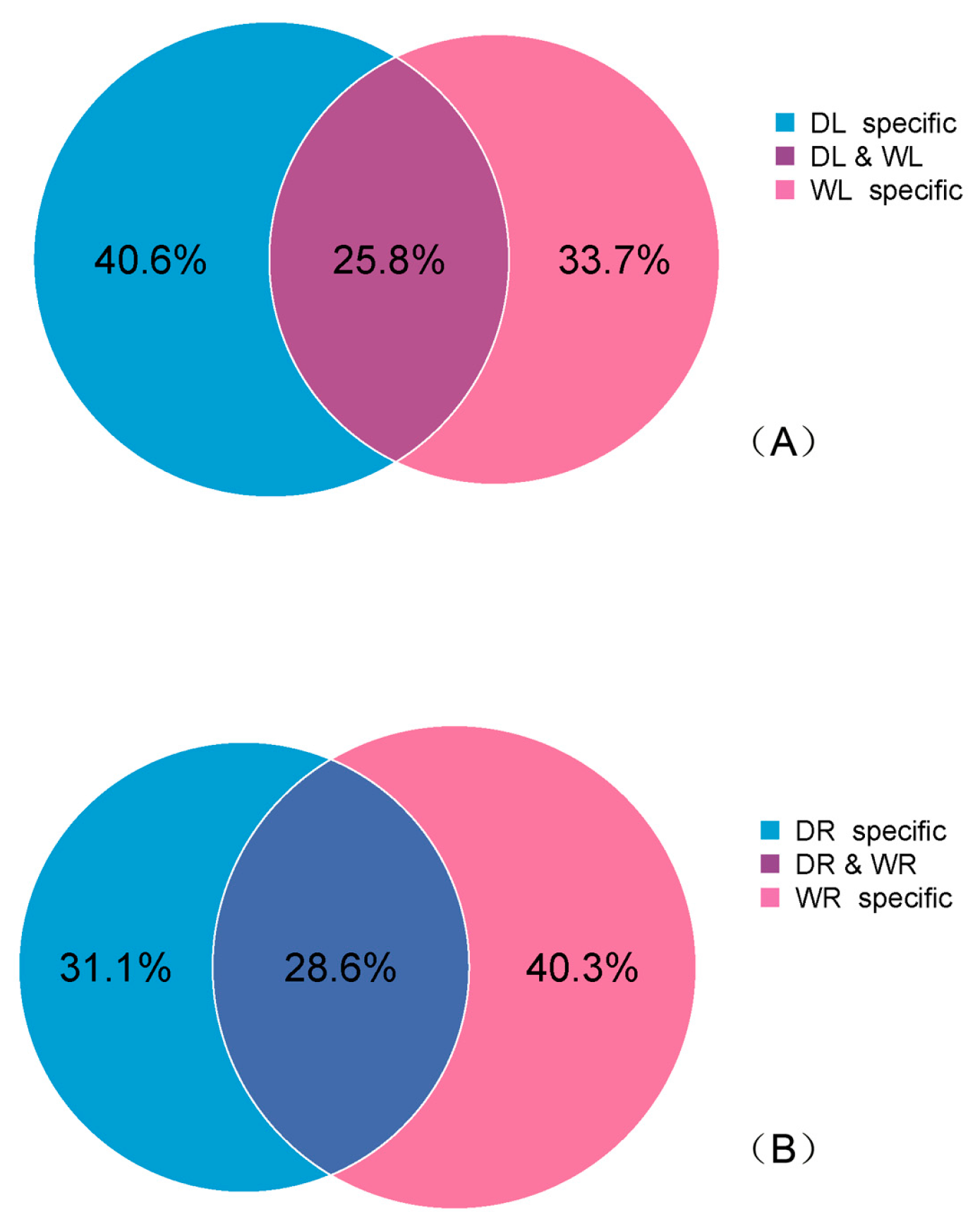
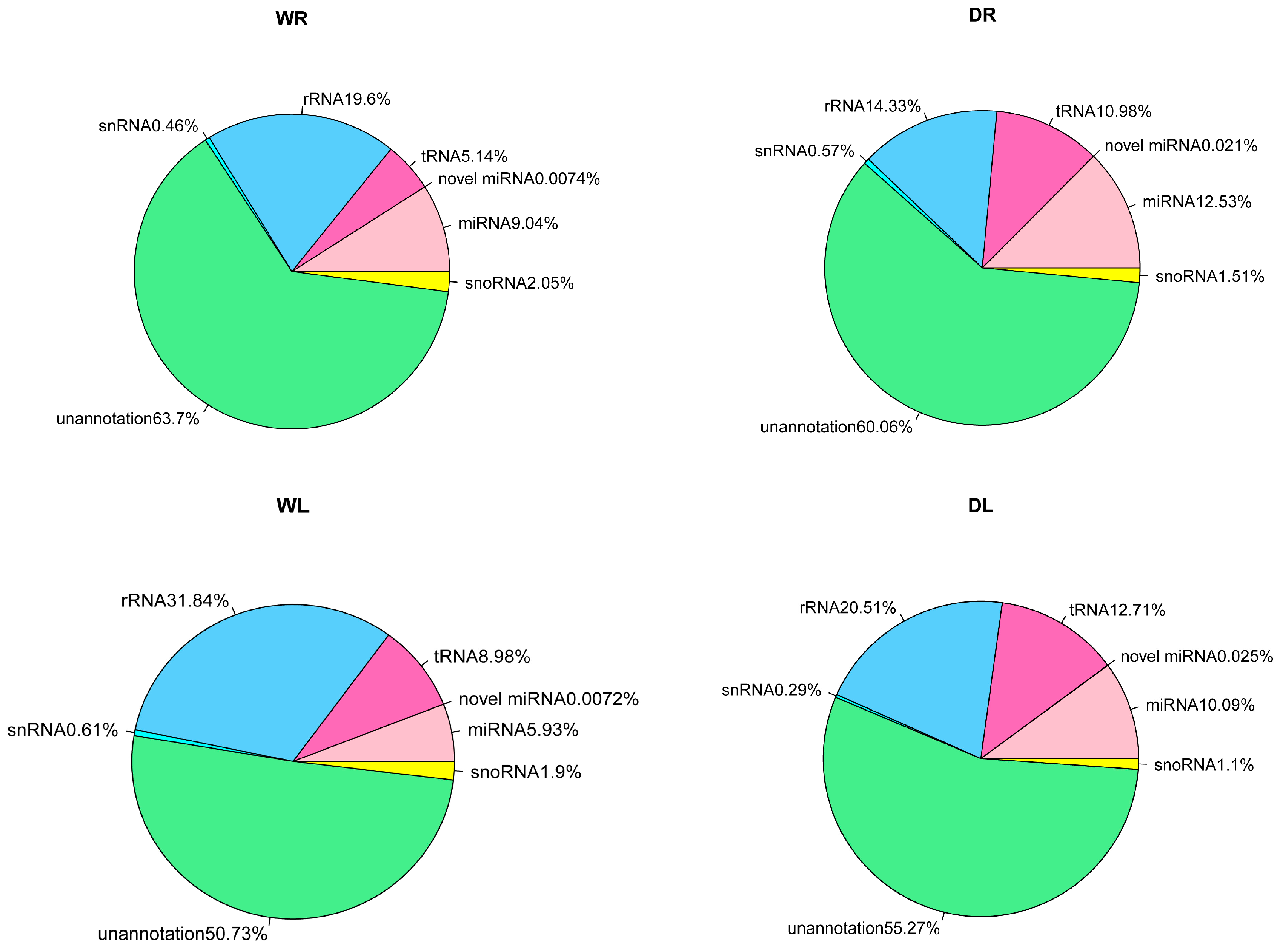
3.1. Overview of Small RNAs from Alfalfa via High-Throughput Sequencing
3.2. Identification of Known miRNAs
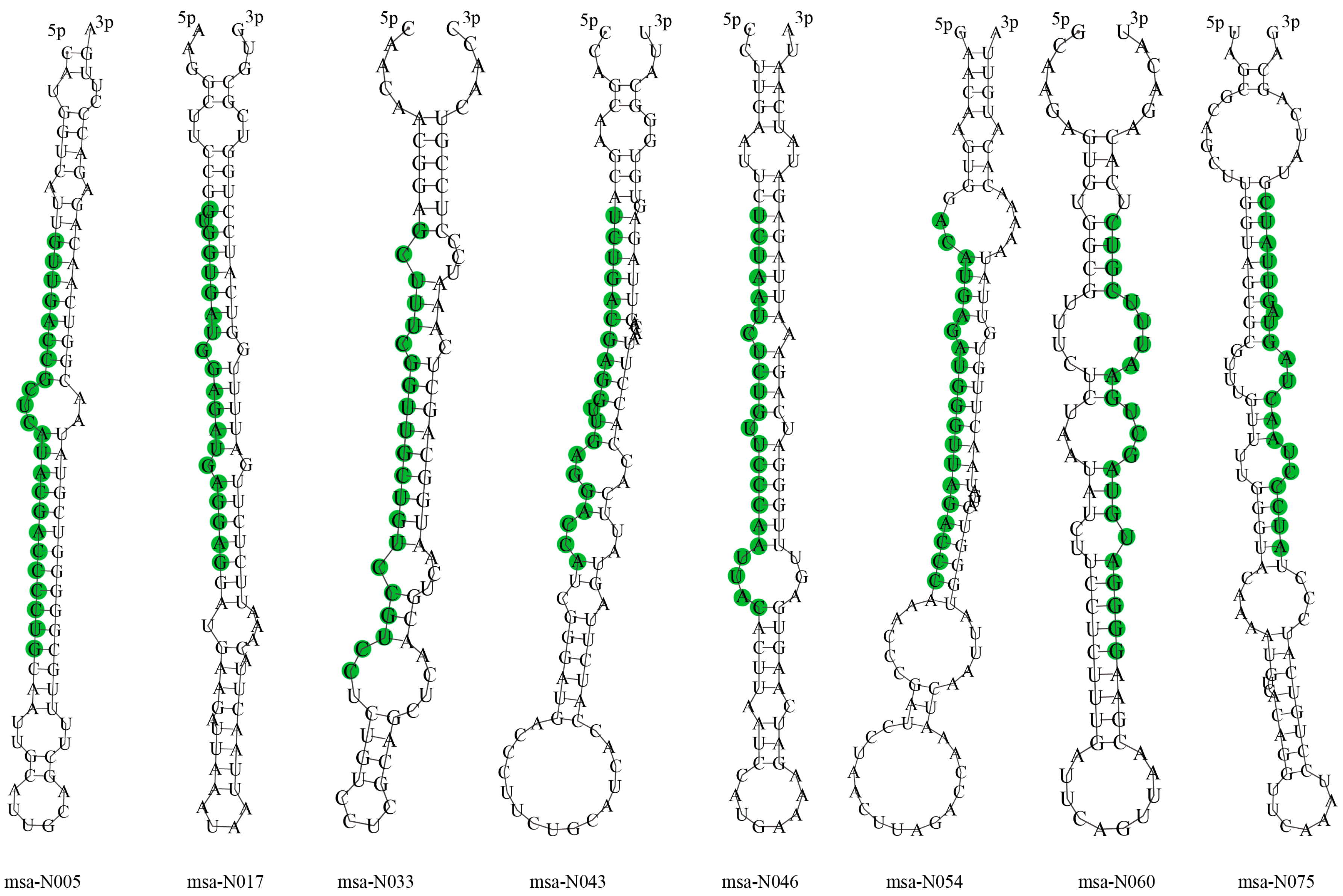
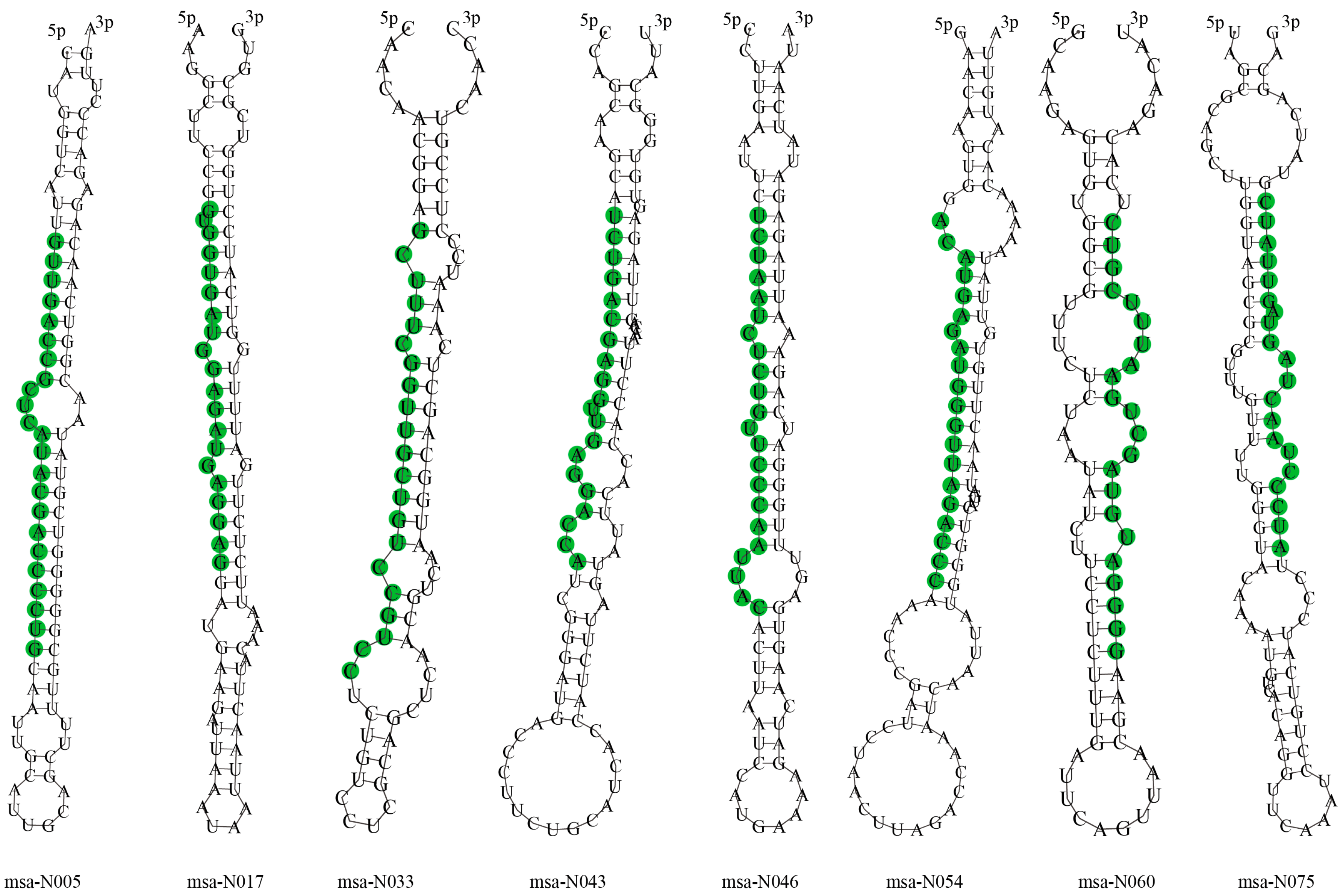
3.3. Identification of Novel miRNAs
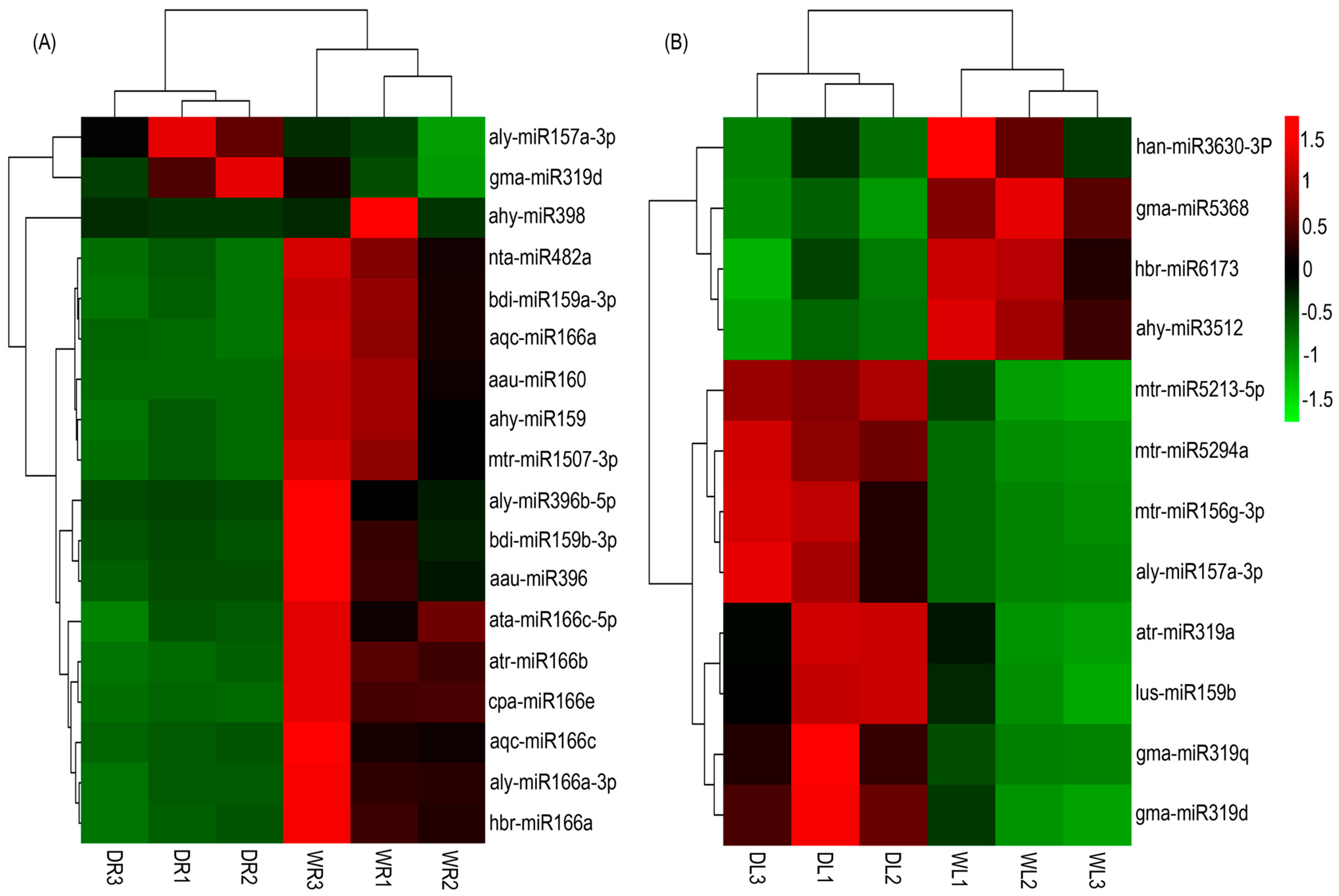
3.4. Drought-Responsive miRNAs Identified in Alfalfa
3.5. Validation of Drought-Responsive miRNAs by qRT-PCR
3.6. Targets of Drought-Responsive miRNAs and Their Functional Analysis
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fan, W.; Zhang, S.; Du, H.; Sun, X.; Shi, Y.; Wang, C. Genome-wide identification of different dormant Medicago sativa L. microRNAs in response to fall dormancy. PLoS ONE 2014, 9, e114612. [Google Scholar] [CrossRef] [PubMed]
- Barnes, D.K.; Goplen, R.P.; Baylor, J.E. Alfalfa and Alfalfa Improvement; American Society of Agronomy, Crop Science Society of America and Soil Science Society of America: Madison, WI, USA, 1988; pp. 1–24. [Google Scholar]
- Boller, B.; Posselt, U.K.; Veronesi, F.; Boller, B.; Posselt, U.K.; Veronesi, F. Fodder Crops and Amenity Grasses; Springer: New York, NY, USA, 2010; pp. 395–437. [Google Scholar]
- Akdogan, G.; Tufekci, E.D.; Uranbey, S.; Unver, T. miRNA-based drought regulation in wheat. Funct. Integr. Genom. 2016, 16, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Tao, Y.; Zhu, C. Emerging roles of microRNAs in the mediation of drought stress response in plants. J. Exp. Bot. 2013, 64, 3077–3086. [Google Scholar] [CrossRef] [PubMed]
- Cheah, B.H.; Nadarajah, K.; Divate, M.D.; Wickneswari, R. Identification of four functionally important microRNA families with contrasting differential expression profiles between drought-tolerant and susceptible rice leaf at vegetative stage. BMC Genom. 2015, 16, 692. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, K.; Yamaguchi-Shinozaki, K. Gene networks involved in drought stress response and tolerance. J. Exp. Bot. 2007, 58, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Ramanjulu, S.; Bartels, D. Drought-and desiccation-induced modulation of gene expression in plants. Plant Cell Environ. 2002, 25, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, M.; Olvera-Carrillo, Y.; Garciarrubio, A.; Campos, F.; Covarrubias, A.A. The enigmatic LEA proteins and other hydrophilins. Plant Physiol. 2008, 148, 6–24. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Zhang, Z.; Wu, H.; Huang, C.; Shuai, P.; Ye, C.Y.; Tang, S.; Wang, Y.; Yang, L.; Wang, J.; et al. Single-base-resolution methylomes of Populus trichocarpa reveal the association between DNA methylation and drought stress. BMC Genet. 2014, 15 (Suppl. 1), S9. [Google Scholar] [CrossRef] [PubMed]
- Granot, G.; Sikron-Persi, N.; Gaspan, O.; Florentin, A.; Talwara, S.; Paul, LK.; Morgenstern, Y.; Granot, Y.; Grafi, G. Histone modifications associated with drought tolerance in the desert plant Zygophyllum dumosum Boiss. Planta 2009, 231, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Ríos, G.; Gagete, A.P.; Castillo, J.; Berbel, A.; Franco, L.; Rodrigo, M.I. Abscisic acid and desiccation-dependent expression of a novel putative SNF5-type chromatin-remodeling gene in Pisum sativum. Plant Physiol. Biochem. 2007, 45, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Khraiwesh, B.; Zhu, J.K.; Zhu, J. Role of miRNAs and siRNAs in biotic and abiotic stress responses of plants. Biochim. Biophys. Acta 2012, 1819, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R. Role of microRNAs in biotic and abiotic stress responses in crop plants. Appl. Biochem. Biotechnol. 2014, 174, 93–115. [Google Scholar] [CrossRef] [PubMed]
- Chapman, E.J.; Carrington, J.C. Specialization and evolution of endogenous small RNA pathways. Nat. Rev. Genet. 2007, 8, 884–896. [Google Scholar] [CrossRef] [PubMed]
- Mallory, A.C.; Bouché, N. MicroRNA-directed regulation: To cleave or not to cleave. Trends Plant Sci. 2008, 13, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Förstemann, K.; Horwich, M.D.; Wee, L.M.; Tomari, Y.; Zamore, P.D. Drosophila microRNAs are sorted into functionally distinct Argonaute protein complexes after their production by Dicer-1. Cell 2007, 130, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Palatnik, J.F.; Allen, E.; Wu, X.; Schommer, C.; Schwab, R.; Carrington, J.C.; Weigel, D. Control of leaf morphogenesis by microRNAs. Nature 2003, 425, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Chen, X. A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Science 2004, 303, 2022–2025. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, D.; Spriggs, A.; Yang, J.; Pogson, B.J.; Dennis, E.S.; Wilson, I.W. Hypoxia-responsive microRNAs and trans-acting small interfering RNAs in Arabidopsis. J. Exp. Bot. 2010, 61, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Long, R.C.; Li, M.N.; Kang, J.M.; Zhang, T.J.; Sun, Y.; Yang, Q.C. Small RNA deep sequencing identifies novel and salt-stress-regulated microRNAs from roots of Medicago sativa and Medicago truncatula. Physiol. Plant. 2015, 154, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Ci, D.; Song, Y.P.; Tian, M.; Zhang, D.Q. Methylation of miRNA genes in the response to temperature stress in Populus simonii. Front. Plant Sci. 2015, 6, 921. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, G.; Zhang, W. UV-B responsive microRNA genes in Arabidopsis thaliana. Mol. Syst. Biol. 2007, 3, 103. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Datta, S.K.; Datta, K. miRNA regulation of nutrient homeostasis in plants. Front. Plant Sci. 2015, 6, 232. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.S.; Zeng, H.Q.; Liu, Z.P.; Yang, Z.M. Genome-wide identification of Medicago truncatula microRNAs and their targets reveals their differential regulation by heavy metal. Plant Cell Environ. 2012, 35, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Able, A.J.; Able, J.A. SMARTER De-Stressed Cereal Breeding. Trends Plant Sci. 2016, 21, 909–925. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.Z.; Chen, L.; Zhao, M.G.; Tian, Q.Y.; Zhang, W.H. Identification of drought-responsive microRNAs and their targets in Medicago truncatula by genome-wide high-throughput sequencing and degradome analysis. BMC Genom. 2011, 12, 367. [Google Scholar] [CrossRef] [PubMed]
- Shui, X.R.; Chen, Z.W.; Li, J.X. MicroRNA prediction and its function in regulating drought-related genes in cowpea. Plant Sci. 2013, 210, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Ballen-Taborda, C.; Plata, G.; Ayling, S.; Rodriguez-Zapata, F.; Becerra, L.A.; Duitama, J.; Tohme, J. Identification of cassava MicroRNAs under abiotic stress. Int. J. Genom. 2013, 2013, 857986. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Yang, J.; Wang, Z.; Wen, Y.; Wang, J.; He, W.; Liu, B.; Si, H.; Wang, D. Identification of novel and conserved microRNAs related to drought stress in potato by deep sequencing. PLoS ONE 2014, 9, e95489. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, Q.; Zhang, B. Response of miRNAs and their targets to salt and drought stresses in cotton (Gossypium hirsutum L.). Gene 2013, 30, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Gao, J.; Liu, M.; Qin, C.; Zhang, W.; Yang, A.; Xia, M.; Zhang, Z.; Shen, Y.; Lin, H.; et al. Genome-wide analysis of Water-stress-responsive microRNA expression profile in tobacco roots. Funct. Integr. Genom. 2014, 14, 319–332. [Google Scholar]
- Shuai, P.; Liang, D.; Zhang, Z.; Yin, W.; Xia, X. Identification of drought-responsive and novel Populus trichocarpa microRNAs by high-throughput sequencing and their targets using degradome analysis. BMC Genom. 2013, 14, 233. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, T.H.; Gentile, A.; Vilela, R.D.; Costa, G.G.; Dias, L.I.; Endres, L.; Menossi, M. MicroRNAs associated with drought response in the bioenergy crop sugarcane (Saccharum spp.). PLoS ONE 2012, 7, e46703. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Stewart, C.N.; Taki, F.A.; He, Q.; Liu, H.; Zhang, B. High-throughput deep sequencing shows that microRNAs play important roles in switchgrass responses to drought and salinity stress. Plant Biotechnol. J. 2014, 12, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Hackenberg, M.; Gustafson, P.; Langridge, P.; Shi, B.J. Differential expression of microRNAs and other small RNAs in barley between water and drought conditions. Plant Biotechnol. J. 2015, 13, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Searle, I.R.; Watson-Haigh, N.S.; Baumann, U.; Mather, D.E.; Able, A.J.; Able, J.A. Genome-wide identification of microRNAs in leaves and the developing head of four durum genotypes during water deficit stress. PLoS ONE 2015, 10, e0142799. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Able, A.J.; Able, J.A. Water-deficit stress-responsive microRNAs and their targets in four durum wheat genotypes. Funct. Integr. Genom. 2017, 17, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, L.; Tang, T.; Liu, J.; Zhang, H.; Zhi, H.; Jia, G.; Diao, X. Combined small RNA and degradome sequencing to identify miRNAs and their targets in response to drought in foxtail millet. BMC Genet. 2016, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- Capitão, C.; Paiva, J.A.P.; Santos, D.M.; Fevereiro, P. In Medicago truncatula, water deficit modulates the transcript accumulation of components of small RNA pathways. BMC Plant Biol. 2011, 11, 79. [Google Scholar] [CrossRef] [PubMed]
- Li, W.X.; Oono, Y.; Zhu, J.; He, X.J.; Wu, J.M.; Iida, K.; Lu, X.Y.; Cui, X.; Jin, H.; Zhu, J.K. The Arabidopsis NFYA5 transcription factor is regulated transcriptionally and posttranscriptionally to promote drought resistance. Plant Cell 2008, 20, 2238–2251. [Google Scholar] [CrossRef] [PubMed]
- Burge, S.W.; Daub, J.; Eberhardt, R.; Tate, J.; Barquist, L.; Nawrocki, E.P.; Eddy, S.R.; Gardner, P.P.; Bateman, A. Rfam 11.0: 10 years of RNA families. Nucl. Acids Res. 2013, 41, D226–D232. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucl. Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Z.; Liu, F.; Vongsangnak, W.; Jing, Q.; Shen, B. Performance comparison and evaluation of software tools for microRNA deep-sequencing data analysis. Nucl. Acids Res. 2012, 40, 4298–4305. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, T.; Zhao, M.; Tian, Q.; Zhang, W. Identification of aluminum-responsive microRNAs in Medicago truncatula by genome-wide high-throughput sequencing. Planta 2012, 235, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real time quantitative PCR and the 2−∆∆CT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Vaz, C.; Wee, C.W.; Lee, G.P. S.; Ingham, P.W.; Tanavde, V.; Mathavan, S. Deep sequencing of small RNA facilitates tissue and sex associated microRNA discovery in zebrafish. BMC Genom. 2015, 16, 950. [Google Scholar] [CrossRef] [PubMed]
- Eldem, V.; Akcay, U.C.; Ozhuner, E.; Bakir, Y.; Uranbey, S.; Unver, T. Genome-wide identification of miRNAs responsive to drought in peach (Prunus persica) by high-throughput deep sequencing. PLoS ONE 2012, 7, e50298. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.H.; Park, C.M. MIR166/165 genes exhibit dynamic expression patterns in regulating shoot apical meristem and floral development in Arabidopsis. Planta 2007, 225, 1327–1338. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, R.; Vaucheret, H.; Trejo, J.; Bartel, D.P. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev. 2006, 20, 3407–3425. [Google Scholar] [CrossRef] [PubMed]
- Fahlgren, N.; Howell, M.D.; Kasschau, K.D.; Chapman, E.J.; Sullivan, C.M.; Cumbie, J.S.; Givan, S.A.; Law, T.F.; Grant, S.R.; Dangl, J.L.; et al. High throughput sequencing of Arabidopsis microRNAs: Evidence for frequent birth and death of MIRNA genes. PLoS ONE 2007, 2, e219. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhu, X.; Chen, X.; Song, C.; Zou, Z.; Wang, Y.; Wang, M.; Fang, W.; Li, X. Identification and characterization of cold-responsive microRNAs in tea plant (Camellia sinensis) and their targets using high-throughput sequencing and degradome analysis. BMC Plant Biol. 2014, 14, 271. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Wu, Z.; Jiang, F.; Zhou, R.; Yang, Z. Identification of chilling stress-responsive tomato microRNAs and their target genes by high-throughput sequencing and degradome analysis. BMC Genom. 2014, 15, 1130. [Google Scholar] [CrossRef] [PubMed]
- Juarez, M.T.; Kui, J.S.; Thomas, J.; Heller, B.A.; Timmermans, M.C. microRNA-mediated repression of rolled leaf1 specifies maize leaf polarity. Nature 2004, 428, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Boualem, A.; Laporte, P.; Jovanovic, M.; Laffont, C.; Plet, J.; Combier, J.P.; Niebel, A.; Crespi, M.; Frugier, F. MicroRNA166 controls root and nodule development in Medicago truncatula. Plant J. 2008, 54, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Kantar, M.; Lucas, S.; Budak, H. MiRNA expression patterns of Triticum dicoccoides in response to shock drought stress. Planta 2011, 233, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Kantar, M.; Unver, T.; Budak, H. Regulation of barley miRNAs upon dehydration stress correlated with target gene expression. Funct. Integr. Genom. 2010, 10, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Trindade, I.; Capitao, C.; Dalmay, T.; Fevereiro, M.P.; Santos, D.M. miR398 and miR408 are up-regulated in response to water deficit in Medicago truncatula. Planta 2010, 231, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Abe, H.; Yamaguchi-Shinozaki, K.; Urao, T.; lwasaki, T.; Hosokawa, D.; Shinozaki, K. Role of Arabidopsis MYC and MYB homologs in drought and abscisic acid-regulated gene expression. Plant Cell 1997, 9, 1859–1868. [Google Scholar] [CrossRef] [PubMed]
- Abe, H.; Urao, T.; Ito, T.; Seki, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Arabidopsis AtMYC2 (bHLH) and AtMYB2 (MYB) function as transcriptional activators in abscisic acid signaling. Plant Cell 2003, 15, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Dai, M.; Yao, J.; Xiao, B.; Li, X.; Zhang, Q.; Xiong, L. Overexpressing a NAM, ATAF, and CUC (NAC) transcription factor enhances drought resistance and salt tolerance in rice. Proc. Natl. Acad. Sci. USA 2006, 103, 12987–12992. [Google Scholar] [CrossRef] [PubMed]
- Sunkar, R.; Kapoor, A.; Zhu, J.K. Posttranscriptional induction of two Cu/Zn superoxide dismutase genes in Arabidopsis is mediated by downregulation of miR398 and important for oxidative stress tolerance. Plant Cell 2006, 18, 2051–2065. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Zhang, D.; Xiang, F.; Zhang, Z. Differentially expressed miRNAs potentially involved in the regulation of defense mechanism to drought stress in maize seedlings. Int. J. Plant Sci. 2009, 170, 979–989. [Google Scholar] [CrossRef]
- Allen, R.S.; Li, J.Y.; Alonso-Peral, M.M.; White, R.G.; Gubler, F.; Millar, A.A. MicroR159 regulation of most conserved targets in Arabidopsis has negligible phenotypic effects. Silence 2010, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Reyes, J.; Chua, N. ABA induction of miR159 controls transcript levels of two Myb factors during Arabidopsis seed germination. Plant J. 2007, 49, 592–606. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Library | Replicates | Raw Reads | Clean Reads | Reads Mapped to the Genome | Match Known miRNAs |
|---|---|---|---|---|---|
| WL | WL1 | 16,729,829 | 14,718,375 | 13,037,667 | 1,030,262 |
| WL2 | 18,529,495 | 16,118,371 | 15,226,670 | 917,066 | |
| WL3 | 18,683,242 | 16,026,900 | 15,129,492 | 816,007 | |
| WR | WR1 | 17,888,348 | 15,351,268 | 11,164,625 | 1,360,347 |
| WR2 | 18,777,136 | 15,724,912 | 11,131,597 | 1,225,474 | |
| WR3 | 17,354,872 | 14,048,339 | 8,964,877 | 1,472,074 | |
| DL | DL1 | 15,668,523 | 12,648,491 | 10,760,915 | 1,315,694 |
| DL2 | 14,266,305 | 12,378,478 | 10,236,124 | 1,269,855 | |
| DL3 | 14,741,343 | 12,407,912 | 10,797,039 | 1,191,355 | |
| DR | DR1 | 16,484,366 | 14,594,872 | 8,586,263 | 1,814,906 |
| DR2 | 14,587,651 | 12,844,508 | 8,055,680 | 1,662,649 | |
| DR3 | 14,256,856 | 12,721,466 | 7,391,569 | 1,554,103 |
| Name | Sequences | Length | GC Contents | Loci | Minimum Free Energy (kcal/mol) |
|---|---|---|---|---|---|
| msa-N005 | GUUGACCGCUCAUACGACCCCUG | 23 | 60.87 | NC_016411.2:1711404:1711490:+ | −54.00 |
| msa-N017 | GUGGUGAUGGAGAUGAGGAG | 20 | 55 | NC_016410.2:44927035:44927123:− | −31.90 |
| msa-N033 | GCUUUCGGUUGCUGUCCGUCC | 21 | 61.90 | NC_016407.2:37807782:37807862:− | −22.80 |
| msa-N043 | UCUGACGAGGUUGAGGACCA | 20 | 55 | NC_016412.2:20603621:20603715:+ | −22.30 |
| msa-N046 | UCUAAUCUCUGUUCCCAAUUAC | 22 | 36.36 | NC_016411.2:42295745:42295832:− | −33.90 |
| msa-N054 | ACAUGAGAUGGGUUAGACCC | 20 | 50.00 | NC_016414.2:14441574:14441672:− | −20.70 |
| msa-N060 | GGGGAUGUAGCUGAAUUUCGUC | 22 | 50.00 | NC_016408.2:4907664:4907746:− | −20.50 |
| msa-N075 | AUCCCUAACUAGUAGUUAUC | 20 | 35.00 | NC_016414.2:17652459:17652554:+ | −20.30 |
| Name | Normalized Read Count | p Value | qRT-PCR | p Value | ||
|---|---|---|---|---|---|---|
| Control | Stress | Control | Stress | |||
| Root | ||||||
| gma-miR319d | 19.23 | 43.90 | 0.00 | 1.00 | 1.60 ± 1.16 | 0.63 |
| ahy-miR398 | 471.98 | 11.77 | 0.03 | 1.00 | 0.51 ± 0.11 | 0.01 |
| aau-miR396 | 107.62 | 17.07 | 0.00 | 1.00 | 0.27 ± 0.10 | 0.00 |
| mtr-miR1507-3p | 17.71 | 2.83 | 0.00 | 1.00 | 0.22 ± 0.07 | 0.00 |
| aqc-miR166a | 11.23 | 1.94 | 0.00 | 1.00 | 0.21 ± 0.03 | 0.00 |
| bdi-miR159a-3p | 5.26 | 0.98 | 0.00 | 1.00 | 0.54 ± 0.23 | 0.12 |
| aly-miR166a-3p | 1915.66 | 673.23 | 0.02 | 1.00 | 0.34 ± 0.12 | 0.01 |
| nta-miR482a | 121.66 | 37.75 | 0.01 | 1.00 | 0.86 ± 0.11 | 0.27 |
| aly-miR396b-5p | 59.07 | 12.52 | 0.03 | 1.00 | 1.32 ± 0.54 | 0.59 |
| Leaf | ||||||
| ahy-miR3512 | 7.43 | 0.96 | 0.00 | 1.00 | 0.67 ± 0.09 | 0.03 |
| gma-miR5368 | 30.07 | 9.89 | 0.00 | 1.00 | 0.63 ± 0.18 | 0.11 |
| han-miR3630-3p | 16.40 | 5.89 | 0.00 | 1.00 | 0.78 ± 0.07 | 0.04 |
| mtr-miR5294a | 6.39 | 53.70 | 0.01 | 1.00 | 1.04 ± 0.12 | 0.76 |
| gma-miR319d | 103.05 | 605.32 | 0.00 | 1.00 | 1.76 ± 0.62 | 0.28 |
| mtr-miR156g-3p | 1.04 | 17.56 | 0.01 | 1.00 | 2.49 ± 0.21 | 0.00 |
| miRNA Name | Target Accession | Expectation | UPE | Target Start | Target End | Inhibition | Target Description |
|---|---|---|---|---|---|---|---|
| han-miR3630-3p | Medtr0168s0060.1 | 2.5 | 18.13 | 35 | 56 | Translation | zein-binding protein |
| han-miR3630-3p | Medtr0021s0360.1 | 3 | 16.35 | 3893 | 3913 | Cleavage | phospholipid-transporting ATPase-like protein |
| han-miR3630-3p | Medtr1g015620.1 | 3 | 17.66 | 1832 | 1852 | Cleavage | myosin heavy chain |
| han-miR3630-3p | Medtr2g039770.1 | 3 | 17.55 | 1184 | 1203 | Cleavage | disease resistance protein (TIR-NBS-LRR class) |
| hbr-miR6173 | Medtr8g009970.1 | 3 | 15.51 | 593 | 612 | Cleavage | splicing factor 3A subunit 2 |
| ahy-miR3512 | Medtr8g063940.1 | 2 | 21.78 | 980 | 999 | Cleavage | spermidine synthase |
| ahy-miR3512 | Medtr3g058220.1 | 2.5 | 15.17 | 1732 | 1751 | Cleavage | cytochrome P450 family 71 protein |
| ahy-miR3512 | Medtr7g028160.1 | 3 | 13.87 | 1624 | 1643 | Cleavage | TCP family transcription factor |
| aly-miR157a-3p | Medtr8g101880.1 | 3 | 15.82 | 23 | 42 | Cleavage | ATP synthase G subunit family protein |
| mtr-miR5213-5p | Medtr3g025460.1 | 0 | 14.58 | 73 | 94 | Cleavage | neutral/alkaline invertase |
| mtr-miR5213-5p | Medtr4g014580.1 | 1.5 | 13.06 | 121 | 142 | Cleavage | TIR-NBS-LRR class disease resistance protein |
| mtr-miR1507-3p | Medtr7g091550.1 | 2 | 20.51 | 883 | 904 | Cleavage | NBS-LRR disease resistance protein |
| mtr-miR1507-3p | Medtr7g078630.1 | 2 | 22.30 | 655 | 676 | Cleavage | cysteine proteinase superfamily protein |
| mtr-miR1507-3p | Medtr5g015600.1 | 3 | 15.75 | 1400 | 1419 | Cleavage | condensin-2 complex subunit G2, putative |
| nta-miR482a | Medtr6g463480.1 | 1 | 17.17 | 329 | 350 | Cleavage | kinesin KIF2A-like protein |
| nta-miR482a | Medtr6g477950.1 | 1 | 13.93 | 39 | 60 | Cleavage | B3 DNA-binding domain protein |
| nta-miR482a | Medtr7g088640.1 | 2 | 17.84 | 550 | 569 | Cleavage | NBS-LRR type disease resistance protein |
| nta-miR482a | Medtr7g026400.1 | 3 | 14.25 | 1263 | 1283 | Cleavage | response regulator receiver domain protein |
| nta-miR482a | Medtr5g099160.1 | 3 | 14.68 | 67 | 86 | Cleavage | cation/calcium exchanger, putative |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Wan, L.; Bi, S.; Wan, X.; Li, Z.; Cao, J.; Tong, Z.; Xu, H.; He, F.; Li, X. Identification of Drought-Responsive MicroRNAs from Roots and Leaves of Alfalfa by High-Throughput Sequencing. Genes 2017, 8, 119. https://doi.org/10.3390/genes8040119
Li Y, Wan L, Bi S, Wan X, Li Z, Cao J, Tong Z, Xu H, He F, Li X. Identification of Drought-Responsive MicroRNAs from Roots and Leaves of Alfalfa by High-Throughput Sequencing. Genes. 2017; 8(4):119. https://doi.org/10.3390/genes8040119
Chicago/Turabian StyleLi, Yue, Liqiang Wan, Shuyi Bi, Xiufu Wan, Zhenyi Li, Jing Cao, Zongyong Tong, Hongyu Xu, Feng He, and Xianglin Li. 2017. "Identification of Drought-Responsive MicroRNAs from Roots and Leaves of Alfalfa by High-Throughput Sequencing" Genes 8, no. 4: 119. https://doi.org/10.3390/genes8040119
APA StyleLi, Y., Wan, L., Bi, S., Wan, X., Li, Z., Cao, J., Tong, Z., Xu, H., He, F., & Li, X. (2017). Identification of Drought-Responsive MicroRNAs from Roots and Leaves of Alfalfa by High-Throughput Sequencing. Genes, 8(4), 119. https://doi.org/10.3390/genes8040119