
Role of MYC in B Cell Lymphomagenesis



Abstract
:1. Introduction
2. MYC in B Cell Development and Maturation
3. MYC in B Cell Lymphoma Classification
4. Aberrant MYC Signaling Pathways in B Cell Lymphomagenesis
4.1. BL
4.2. DLBCL
4.3. BCLU
4.4. Other B Cell Lymphomas
5. MYC Overexpression as a Prognostic Marker
6. MYC-Based Therapy and Future Perspective
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardiman, J.W. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; IARC Press: Lyon, France, 2008. [Google Scholar]
- Ott, G.; Rosenwald, A; Campo, E. Understanding MYC-driven aggressive B-cell lymphomas: Pathogenesis and classification. Blood 2013, 122, 3884–3891. [Google Scholar] [CrossRef] [PubMed]
- Karube, K.; Campo, E. MYC alterations in diffuse large B-cell lymphomas. Semin. Hematol. 2015, 52, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef] [PubMed]
- Sander, S.; Calado, D.P.; Srinivasan, L.; Köchert, K.; Zhang, B.; Rosolowski, M.; Rodig, S.J.; Holzmann, K.; Stilgenbauer, S.; Siebert, R.; et al. Synergy between PI3K signaling and MYC in Burkitt lymphomagenesis. Cancer Cell 2012, 22, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Nie, Z.; Hu, G.; Wei, G.; Cui, K.; Yamane, A.; Resch, W.; Wang, R.; Green, D.R.; Tessarollo, L.; Casellas, R.; et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 2012, 151, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Lovén, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.D.; Ansel, K.M.; Low, C.; Lesley, R.; Tamamura, H.; Fujii, N.; Cyster, J.G. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat. Immunol. 2004, 5, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.D.; Okada, T.; Cyster, J.G. Germinal-center organization and cellular dynamics. Immunity 2007, 27, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.D.; Okada, T.; Tang, H.L.; Cyster, J.G. Imaging of germinal center selection events during affinity maturation. Science 2007, 315, 528–531. [Google Scholar] [CrossRef] [PubMed]
- Victora, G.D.; Nussenzweig, M.C. Germinal centers. Annu. Rev. Immunol. 2012, 30, 429–457. [Google Scholar] [CrossRef] [PubMed]
- Bannard, O.; Horton, R.M.; Allen, C.D.; An, J.; Nagasawa, T.; Cyster, J.G. Germinal center centroblasts transition to a centrocyte phenotype according to a timed program and depend on the dark zone for effective selection. Immunity 2013, 39, 912–924. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Sola, D.; Victora, G.D.; Ying, C.Y.; Phan, R.T.; Saito, M.; Nussenzweig, M.C.; Dalla-Favera, R. The proto-oncogene MYC is required for selection in the germinal center and cyclic reentry. Nat. Immunol. 2012, 13, 1083–1091. [Google Scholar] [CrossRef]
- Calado, D.P.; Sasaki, Y.; Godinho, S.A.; Pellerin, A.; Köchert, K.; Sleckman, B.P.; de Alborán, I.M.; Janz, M.; Rodig, S.; Rajewsky, K. The cell-cycle regulator c-Myc is essential for the formation and maintenance of germinal centers. Nat. Immunol. 2012, 13, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Campo, E. New pathogenic mechanisms in Burkitt lymphoma. Nat. Genet. 2012, 44, 1288–1289. [Google Scholar] [CrossRef] [PubMed]
- Basso, K.; Dalla-Favera, R. Germinal centres and B cell lymphomagenesis. Nat. Rev. Immunol. 2015, 15, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.; Schlesner, M.; Hoffmann, S.; Kreuz, M.; Leich, E.; Burkhardt, B.; Rosolowski, M.; Ammerpohl, O.; Wagener, R.; Bernhart, S.H.; et al. Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat. Genet. 2012, 44, 1316–1320. [Google Scholar] [CrossRef] [PubMed]
- Ott, G. Impact of MYC on malignant behavior. Hematol. Am. Soc. Hematol. Educ. Program 2014, 2014, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Korać, P.; Dotlić, S.; Dominis, M. “Double hit” lymphoma or secondary MYC translocation lymphoma? Period. Biologor. 2012, 114, 527–537. [Google Scholar]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed]
- Lester, J.F.; Dojcinov, S.D.; Attanoos, R.L.; O’Brien, C.J.; Maughan, T.S.; Toy, E.T.; Poynton, C.H. The clinical impact of expert pathological review on lymphoma management: A regional experience. Br. J. Haematol. 2003, 123, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Coiffier, B.; Lepage, E.; Briere, J.; Herbrecht, R.; Tilly, H.; Bouabdallah, R.; Morel, P.; Van Den Neste, E.; Salles, G.; Gaulard, P.; et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N. Engl. J. Med. 2002, 346, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Lenz, G.; Wright, G.; Dave, S.S.; Xiao, W.; Powell, J.; Zhao, H.; Xu, W.; Tan, B.; Goldschmidt, N.; Iqbal, J.; et al. Stromal gene signatures in large-B-cell lymphomas. N. Engl. J. Med. 2008, 359, 2313–2323. [Google Scholar] [CrossRef] [PubMed]
- Green, T.M.; Young, K.H.; Visco, C.; Xu-Monette, Z.Y.; Orazi, A.; Go, R.S.; Nielsen, O.; Gadeberg, O.V.; Mourits-Andersen, T.; Frederiksen, M.; et al. Immunohistochemical double-hit score is a strong predictor of outcome in patients with diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J. Clin. Oncol. 2012, 30, 3460–3467. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.A.; Slack, G.W.; Savage, K.J.; Connors, J.M.; Ben-Neriah, S.; Rogic, S.; Scott, D.W.; Tan, K.L.; Steidl, C.; Sehn, L.H.; et al. Concurrent expression of MYC and BCL2 in diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J. Clin. Oncol. 2012, 30, 3452–3459. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, M.Ø.; Gang, A.O.; Poulsen, T.S.; Knudsen, H.; Lauritzen, A.F.; Nielsen, S.L.; Klausen, T.W.; Nørgaard, P. MYC translocation partner gene determines survival of patients with large B-cell lymphoma with MYC- or double-hit MYC/BCL2 translocations. Eur. J. Haematol. 2014, 92, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Barrans, S.; Crouch, S.; Smith, A.; Turner, K.; Owen, R.; Patmore, R.; Roman, E.; Jack, A. Rearrangement of MYC is associated with poor prognosis in patients with diffuse large B-cell lymphoma treated in the era of rituximab. J. Clin. Oncol. 2010, 28, 3360–3365. [Google Scholar] [CrossRef]
- Savage, K.J.; Johnson, N.A.; Ben-Neriah, S.; Connors, J.M.; Sehn, L.H.; Farinha, P.; Horsman, D.E.; Gascoyne, R.D. MYC gene rearrangements are associated with a poor prognosis in diffuse large B-cell lymphoma patients treated with R-CHOP chemotherapy. Blood 2009, 114, 3533–3537. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Xu-Monette, Z.Y.; Tzankov, A.; Green, T.; Wu, L.; Balasubramanyam, A.; Liu, W.M.; Visco, C.; Li, Y.; Miranda, R.N.; et al. MYC/BCL2 protein coexpression contributes to the inferior survival of activated B-cell subtype of diffuse large B-cell lymphoma and demonstrates high-risk gene expression signatures: A report from The International DLBCL Rituximab-CHOP Consortium Program. Blood 2013, 121, 4021–4031. [Google Scholar] [CrossRef] [PubMed]
- Valera, A.; López-Guillermo, A.; Cardesa-Salzmann, T.; Climent, F.; González-Barca, E.; Mercadal, S.; Espinosa, I.; Novelli, S.; Briones, J.; Mate, J.L.; et al. MYC protein expression and genetic alterations have prognostic impact in patients with diffuse large B-cell lymphoma treated with immunochemotherapy. Haematologica 2013, 98, 1554–1562. [Google Scholar] [CrossRef] [PubMed]
- Boerma, E.G.; Siebert, R.; Kluin, P.M.; Baudis, M. Translocations involving 8q24 in Burkitt lymphoma and other malignant lymphomas: A historical review of cytogenetics in the light of todays knowledge. Leukemia 2009, 23, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Salaverria, I.; Martin-Guerrero, I.; Wagener, R.; Kreuz, M.; Kohler, C.W.; Richter, J.; Pienkowska-Grela, B.; Adam, P.; Burkhardt, B.; Claviez, A.; et al. A recurrent 11q aberration pattern characterizes a subset of MYC-negative high-grade B-cell lymphomas resembling Burkitt lymphoma. Blood 2014, 123, 1187–1198. [Google Scholar] [CrossRef] [PubMed]
- Ferreiro, J.F.; Morscio, J.; Dierickx, D.; Marcelis, L.; Verhoef, G.; Vandenberghe, P.; Tousseyn, T.; Wlodarska, I. Post-transplant molecularly defined Burkitt lymphomas are frequently MYC-negative and characterized by the 11q-gain/loss pattern. Haematologica 2015, 100, e275–e279. [Google Scholar] [CrossRef] [PubMed]
- Leucci, E.; Cocco, M.; Onnis, A.; De Falco, G.; van Cleef, P.; Bellan, C.; van Rijk, A.; Nyagol, J.; Byakika, B.; Lazzi, S.; et al. MYC translocation-negative classical Burkitt lymphoma cases: An alternative pathogenetic mechanism involving miRNA deregulation. J. Pathol. 2008, 216, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Eischen, C.M.; Weber, J.D.; Roussel, M.F.; Sherr, C.J.; Cleveland, J.L. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999, 13, 2658–2669. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Medeiros, L.J.; Xu, X.; Young, K.H. MYC-driven aggressive B-cell lymphomas: Biology, entity, differential diagnosis and clinical management. Oncotarget 2015, 6, 38591–38616. [Google Scholar] [CrossRef] [PubMed]
- Dalla-Favera, R.; Bregni, M.; Erikson, J.; Patterson, D.; Gallo, R.C.; Croce, C.M. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7824–7827. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.; Ceribelli, M.; Pittaluga, S.; Wright, G.; Staudt, L.M. Oncogenic mechanisms in Burkitt lymphoma. Cold Spring Harb. Perspect. Med. 2014, 4, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Spender, L.C.; Inman, G. Developments in Burkitt’s lymphoma: Novel cooperations in oncogenic MYC signaling. J. Cancer Manag. Res. 2014, 6, 27–38. [Google Scholar] [CrossRef]
- Ryan, R.J.; Drier, Y.; Whitton, H.; Cotton, M.J.; Kaur, J.; Issner, R.; Gillespie, S.; Epstein, C.B.; Nardi, V.; Sohani, A.R.; et al. Detection of Enhancer-Associated Rearrangements Reveals Mechanisms of Oncogene Dysregulation in B-cell Lymphoma. Cancer Discov. 2015, 5, 1058–1071. [Google Scholar] [CrossRef] [PubMed]
- Sears, R.; Nuckolls, F.; Haura, E.; Taya, Y.; Tamai, K.; Nevins, J.R. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000, 14, 2501–2514. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Lwin, T.; Zhang, X.; Huang, A.; Wang, J.; Marquez, V.E.; Chen-Kiang, S.; Dalton, W.S.; Sotomayor, E.; Tao, J. Disruption of the MYC-miRNA-EZH2 loop to suppress aggressive B-cell lymphoma survival and clonogenicity. Leukemia 2013, 27, 2341–2350. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G. Genome sequencing of lymphoid malignancies. Blood 2013, 122, 3899–3907. [Google Scholar] [CrossRef] [PubMed]
- Béguelin, W.; Popovic, R.; Teater, M.; Jiang, Y.; Bunting, K.L.; Rosen, M.; Shen, H.; Yang, S.N.; Wang, L.; Ezponda, T.; et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 2013, 23, 677–692. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.; Young, R.M.; Ceribelli, M.; Jhavar, S.; Xiao, W.; Zhang, M.; Wright, G.; Shaffer, A.L.; Hodson, D.J.; Buras, E.; et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 2012, 490, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Dorsett, Y.; Robbiani, D.F.; Jankovic, M.; Reina-San-Martin, B.; Eisenreich, T.R.; Nussenzweig, M.C. A role for AID in chromosome translocations between c-myc and the IgH variable region. J. Exp. Med. 2007, 204, 2225–2232. [Google Scholar] [CrossRef] [PubMed]
- Pasqualucci, L.; Bhagat, G.; Jankovic, M.; Compagno, M.; Smith, P.; Muramatsu, M.; Honjo, T.; Morse, H.C., 3rd; Nussenzweig, M.C.; Dalla-Favera, R. AID is required for germinal center-derived lymphomagenesis. Nat. Genet. 2008, 40, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Robbiani, D.F.; Deroubaix, S.; Feldhahn, N.; Oliveira, T.Y.; Callen, E.; Wang, Q.; Jankovic, M.; Silva, I.T.; Rommel, P.C.; Bosque, D.; et al. Plasmodium Infection Promotes Genomic Instability and AID-Dependent B Cell Lymphoma. Cell 2015, 162, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Kwon, K.; Hutter, C.; Sun, Q.; Bilic, I.; Cobaleda, C.; Malin, S.; Busslinger, M. Instructive role of the transcription factor E2A in early B lymphopoiesis and germinal center B cell development. Immunity 2008, 28, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Peled, J.U.; Yu, J.J.; Venkatesh, J.; Bi, E.; Ding, B.B.; Krupski-Downs, M.; Shaknovich, R.; Sicinski, P.; Diamond, B.; Scharff, M.D.; et al. Requirement for cyclin D3 in germinal center formation and function. Cell Res. 2010, 20, 631–646. [Google Scholar] [CrossRef] [PubMed]
- Pasqualucci, L.; Trifonov, V.; Fabbri, G.; Ma, J.; Rossi, D.; Chiarenza, A.; Wells, V.A.; Grunn, A.; Messina, M.; Elliot, O.; et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat. Genet. 2011, 43, 830–837. [Google Scholar] [CrossRef] [PubMed]
- Basso, K.; Schneider, C.; Shen, Q.; Holmes, A.B.; Setty, M.; Leslie, C.; Dalla-Favera, R. BCL6 positively regulates AID and germinal center gene expression via repression of miR-155. J. Exp. Med. 2012, 209, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Ying, C.Y.; Dominguez-Sola, D.; Fabi, M.; Lorenz, I.C.; Hussein, S.; Bansal, M.; Califano, A.; Pasqualucci, L.; Basso, K.; Dalla-Favera, R. MEF2B mutations lead to deregulated expression of the oncogene BCL6 in diffuse large B cell lymphoma. Nat. Immunol. 2013, 14, 1084–1092. [Google Scholar] [CrossRef] [PubMed]
- Hatzi, K.; Melnick, A. Breaking bad in the germinal center: how deregulation of BCL6 contributes to lymphomagenesis. Trends Mol. Med. 2014, 20, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Ci, W.; Polo, J.M.; Cerchietti, L.; Shaknovich, R.; Wang, L.; Yang, S.N.; Ye, K.; Farinha, P.; Horsman, D.E.; Gascoyne, R.D.; et al. The BCL6 transcriptional program features repression of multiple oncogenes in primary B cells and is deregulated in DLBCL. Blood 2009, 113, 5536–5548. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Iida, S.; Louie, D.C.; Dalla-Favera, R.; Chaganti, R.S. Heterologous promoters fused to BCL6 by chromosomal translocations affecting band 3q27 cause its deregulated expression during B-cell differentiation. Blood 1998, 91, 603–607. [Google Scholar] [PubMed]
- Duan, S.; Cermak, L.; Pagan, J.K.; Rossi, M.; Martinengo, C.; di Celle, P.F.; Chapuy, B.; Shipp, M.; Chiarle, R.; Pagano, M. FBXO11 targets BCL6 for degradation and is inactivated in diffuse large B-cell lymphomas. Nature 2012, 481, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Tam, W.; Gomez, M.; Chadburn, A.; Lee, J.W.; Chan, W.C.; Knowles, D.M. Mutational analysis of PRDM1 indicates a tumor-suppressor role in diffuse large B-cell lymphomas. Blood 2006, 107, 4090–4100. [Google Scholar] [CrossRef] [PubMed]
- Mandelbaum, J.; Bhagat, G.; Tang, H.; Mo, T.; Brahmachary, M.; Shen, Q.; Chadburn, A.; Rajewsky, K.; Tarakhovsky, A.; Pasqualucci, L.; et al. BLIMP1 is a tumor suppressor gene frequently disrupted in activated B cell-like diffuse large B cell lymphoma. Cancer Cell 2010, 18, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Aukema, S.M.; Kreuz, M.; Kohler, C.W.; Rosolowski, M.; Hasenclever, D.; Hummel, M.; Küppers, R.; Lenze, D.; Ott, G.; Pott, C.; et al. Molecular Mechanisms in Malignant Lymphomas Network Project. Biological characterization of adult MYC-translocation-positive mature B-cell lymphomas other than molecular Burkitt lymphoma. Haematologica 2014, 99, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Chng, W.J.; Huang, G.F.; Chung, T.H.; Ng, S.B.; Gonzalez-Paz, N.; Troska-Price, T.; Mulligan, G.; Chesi, M.; Bergsagel, P.L.; Fonseca, R. Clinical and biological implications of MYC activation: A common difference between MGUS and newly diagnosed multiple myeloma. Leukemia 2011, 25, 1026–1035. [Google Scholar] [CrossRef] [PubMed]
- Hart, L.S.; Cunningham, J.T.; Datta, T.; Dey, S.; Tameire, F.; Lehman, S.L.; Qiu, B.; Zhang, H.; Cerniglia, G.; Bi, M.; et al. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J. Clin. Investig. 2012, 122, 4621–4634. [Google Scholar] [CrossRef] [PubMed]
- Momose, S.; Tamaru, J.; Kishi, H.; Mikata, I.; Mori, M.; Toyozumi, Y.; Itoyama, S. Hyperactivated STAT3 in ALK-positive diffuse large B-cell lymphoma with clathrin-ALK fusion. Hum. Pathol. 2009, 40, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Snuderl, M.; Kolman, O.K.; Chen, Y.B.; Hsu, J.J.; Ackerman, A.M.; Dal Cin, P.; Ferry, J.A.; Harris, N.L.; Hasserjian, R.P.; Zukerberg, L.R.; et al. B-cell lymphomas with concurrent IGH-BCL2 and MYC rearrangements are aggressive neoplasms with clinical and pathologic features distinct from Burkitt lymphoma and diffuse large B-cell lymphoma. Am. J. Surg. Pathol. 2010, 34, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, L.J.; Hai, S.; Thomazy, V.A.; Estalilla, O.C.; Romaguera, J.; Luthra, R. Real-time RT-PCR assay for quantifying cyclin D1 mRNA in B-cell non-Hodgkin’s lymphomas. Mod. Pathol. 2002, 15, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Woroniecka, R.; Rymkiewicz, G.; Grygalewicz, B.; Błachnio, K.; Rygier, J.; Jarmuż-Szymczak, M.; Ratajczak, B.; Pieńkowska-Grela, B. Cytogenetic and flow cytometry evaluation of Richter syndrome reveals MYC, CDKN2A, IGH alterations with loss of CD52, CD62L and increase of CD71 antigen expression as the most frequent recurrent abnormalities. Am. J. Clin. Pathol. 2015, 143, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Fonseka, L.N.; Tirado, C.A. C-MYC Involvement in Chronic Lymphocytic Leukemia (CLL): A Molecular and Cytogenetic Update. J. Assoc. Genet. Technol. 2015, 41, 176–183. [Google Scholar] [PubMed]
- Haberl, S.; Haferlach, T.; Stengel, A.; Jeromin, S.; Kern, W.; Haferlach, C. MYC rearranged B-cell neoplasms: Impact of genetics on classification. Cancer Genet. 2016, 209, 431–439. [Google Scholar] [CrossRef] [PubMed]
- God, J.M.; Cameron, C.; Figueroa, J.; Amria, S.; Hossain, A.; Kempkes, B.; Bornkamm, G.W.; Stuart, R.K.; Blum, J.S.; Haque, A. Elevation of c-MYC disrupts HLA class II-mediated immune recognition of human B cell tumors. J. Immunol. 2015, 194, 1434–1445. [Google Scholar] [CrossRef] [PubMed]
- Perry, A.M.; Alvarado-Bernal, Y.; Laurini, J.A.; Smith, L.M.; Slack, G.W.; Tan, K.L.; Sehn, L.H.; Fu, K.; Aoun, P.; Greiner, T.C.; et al. MYC and BCL2 protein expression predicts survival in patients with diffuse large B-cell lymphoma treated with rituximab. Br. J. Haematol. 2014, 165, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Xu, D.; Cao, Y.; Wang, J.; Yang, Y.; Huang, M. C-MYC aberrations as prognostic factors in diffuse large B-cell lymphoma: A meta-analysis of epidemiological studies. PLoS ONE 2014, 9, e95020. [Google Scholar] [CrossRef] [PubMed]
- Kluk, M.J.; Chapuy, B.; Sinha, P.; Roy, A.; Dal Cin, P.; Neuberg, D.S.; Monti, S.; Pinkus, G.S.; Shipp, M.A.; Rodig, S.J. Immunohistochemical detection of MYC-driven diffuse large B-cell lymphomas. PLoS ONE 2012, 7, e33813. [Google Scholar] [CrossRef] [PubMed]
- Horn, H.; Ziepert, M.; Becher, C.; Barth, T.F.; Bernd, H.; Feller, A.C.; Klapper, W.; Hummel, M.; Stein, H.; Hansmann, M.; et al. MYC status in concert with BCL2 and BCL6 expression predicts outcome in diffuse large B-cell lymphoma. Blood 2013, 121, 2253–2263. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.X.; Liu, Y.H.; Luo, D.L.; Zhang, F.; Cheng, Y.; Luo, X.L.; Xu, J.; Cheng, J.; Zhuang, H.G. MYC Expression in Concert with BCL2 and BCL6 Expression Predicts Outcome in Chinese Patients with Diffuse Large B-Cell Lymphoma Not Otherwise Specified. PLoS ONE 2014, 9, e104068. [Google Scholar] [CrossRef] [PubMed]
- Molina, T.J.; Canioni, D.; Copie-Bergman, C.; Recher, C.; Brière, J.; Haioun, C.; Berger, F.; Fermé, C.; Copin, M.C.; Casasnovas, O.; et al. Young patients with non-germinal center B-cell-like diffuse large B-cell lymphoma benefit from intensified chemotherapy with ACVBP plus rituximab compared with CHOP plus rituximab: analysis of data from the Groupe d’Etudes des Lymphomes de l’Adulte/Lymphoma Study Association phase III trial LNH 03–2B. J. Clin. Oncol. 2014, 32, 3996–4003. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Guo, L.; Liu, H.; Zheng, B.; Ying, J.; Lv, N. C-MYC overexpression predicts aggressive transformation and a poor outcome in mucosa-associated lymphoid tissue lymphomas. Int. J. Clin. Exp. Pathol. 2014, 7, 5634–5644. [Google Scholar] [PubMed]
- Gill, K.Z.; Iwamoto, F.; Allen, A.; Hoehn, D.; Murty, V.V.; Alobeid, B.; Bhagat, G. MYC protein expression in primary diffuse large B-cell lymphoma of the central nervous system. PLoS ONE 2014, 9, e114398. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.W.; Mottok, A.; Ennishi, D.; Wright, G.W.; Farinha, P.; Ben-Neriah, S.; Kridel, R.; Barry, G.S.; Hother, C.; Abrisqueta, P.; et al. Prognostic significance of diffuse large B-cell lymphoma cell of origin determined by digital gene expression in formalin-fixed paraffin-embedded tissue biopsies. J. Clin. Oncol. 2015, 33, 2848–2856. [Google Scholar] [CrossRef] [PubMed]
- Choe, J.Y.; Park, M.; Yun, J.Y.; Na, H.Y.; Go, H.; Kim, H.J.; Oh, S.; Kim, J.E. PELI1 expression is correlated with MYC and BCL6 expression and associated with poor prognosis in diffuse large B-cell lymphoma. Mod. Pathol. 2016, 29, 1313–1323. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Y.; Medeiros, L.J.; Bueso-Ramos, C.E.; Tang, G.; Wang, S.; Oki, Y.; Desai, P.; Khoury, J.D.; Miranda, R.N.; Tang, Z.; et al. P53 expression correlates with poorer survival and augments the negative prognostic effect of MYC rearrangement; expression or concurrent MYC/BCL2 expression in diffuse large B-cell lymphoma. Mod. Pathol. 2016. [Google Scholar] [CrossRef]
- Kawamoto, K.; Miyoshi, H.; Yoshida, N.; Nakamura, N.; Ohshima, K.; Sone, H.; Takizawa, J. MYC translocation and/or BCL 2 protein expression are associated with poor prognosis in diffuse large B-cell lymphoma. Cancer Sci. 2016, 107, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Xu-Monette, Z.Y.; Tzankov, A.; Deng, L.; Wang, X.; Manyam, G.C.; Visco, C.; Montes-Moreno, S.; Zhang, L.; Dybkær, K.; et al. Prognostic impact of concurrent MYC and BCL6 rearrangements and expression in de novo diffuse large B-cell lymphoma. Oncotarget 2016, 7, 2401–2416. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Nam, S.J.; Kwon, D.; Kim, H.; Lee, E.; Kim, T.M.; Heo, D.S.S.; Park, S.H.; Kim, C.W.; Jeon, K.Y. MYC and BCL2 overexpression is associated with a higher class of Memorial Sloan-Kettering Cancer Center prognostic model and poor clinical outcome in primary diffuse large B-cell lymphoma of the central nervous system. BMC Cancer 2016, 16, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Son, S.M.; Ha, S.Y.; Yoo, H.Y.; Oh, D.; Kim, S.J.; Kim, W.S.; Ko, Y.H. Prognostic impact of MYC protein expression in central nervous system diffuse large B-cell lymphoma: Comparison with MYC rearrangement and MYC mRNA expression. Mod. Pathol. 2017, 30, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; PRISMA Group. Preferred reporting items for systemic reviews and meta-analyses: The PRISMA statement. PLoS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef] [PubMed]
- Walhout, A.J.; Gubbels, J.M.; Bernards, R.; van der Vliet, P.C.; Timmers, H.T. c-Myc/Max heterodimers bind cooperatively to the E-box sequences located in the first intron of the rat ornithine decarboxylase (ODC) gene. Nucleic Acids Res. 1997, 25, 1493–1501. [Google Scholar] [CrossRef] [PubMed]
- Berg, T.; Cohen, S.B.; Desharnais, J.; Sonderegger, C.; Maslyar, D.J.; Goldberg, J.; Boger, D.L.; Vogt, P.K. Small-molecule antagonists of Myc/Max dimerization inhibit Myc-induced transformation of chicken embryo fibroblasts. Proc. Natl. Acad. Sci. USA 2002, 99, 3830–3835. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Giap, C.; Lazo, J.S.; Prochownik, E.V. Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene 2003, 22, 6151–6159. [Google Scholar] [CrossRef] [PubMed]
- Kiessling, A.; Sperl, B.; Hollis, A.; Eick, D.; Berg, T. Selective inhibition of c-Myc/Max dimerization and DNA binding by small molecules. Chem. Biol. 2006, 13, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Vogt, P.K.; Boger, D.L.; Lunec, J. Disruption of the MYC transcriptional function by a small-molecule antagonist of MYC/MAXdimerization. Oncol. Rep. 2008, 19, 825–830. [Google Scholar] [PubMed]
- Wang, H.; Hammoudeh, D.I.; Follis, A.V.; Reese, B.E.; Lazo, J.S.; Metallo, S.J.; Prochownik, E.V. Improved low molecular weight Myc-Max inhibitors. Mol. Cancer Ther. 2007, 6, 2399–2408. [Google Scholar] [CrossRef] [PubMed]
- Hammoudeh, D.I.; Follis, A.V.; Prochownik, E.V.; Metallo, S.J. Multiple independent binding sites for small-molecule inhibitors on the oncoprotein c-Myc. J. Am. Chem. Soc. 2009, 131, 7390–7401. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Stover, J.S.; Whitby, L.R.; Vogt, P.K.; Boger, D.L. Small molecule inhibitors of Myc/Max dimerization and Myc-induced cell transformation. Bioorg. Med. Chem. Lett. 2009, 19, 6038–6041. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Parise, R.A.; Joseph, E.; Egorin, M.J.; Lazo, J.S.; Prochownik, E.V.; Eiseman, J.L. Efficacy, pharmacokinetics, tisssue distribution, and metabolism of the Myc-Max disruptor, 10058-F4 [Z,E]-5-[4-ethylbenzylidine]-2-thioxothiazolidin-4-one, in mice. Cancer Chemother. Pharmacol. 2009, 63, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S.; Prochownik, E.V. Small-molecule inhibitors of the Myc oncoprotein. Biochim. Biophys. Acta 2015, 1849, 525–543. [Google Scholar] [CrossRef] [PubMed]
- Mo, H.; Vita, M.; Crespin, M.; Henriksson, M. Myc overexpression enhances apoptosis induced by small molecules. Cell Cycle 2006, 5, 2191–2194. [Google Scholar] [CrossRef] [PubMed]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Filippakopoulos, P. Beating the odds: BETs in disease. Trends Biochem. Sci. 2015, 40, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Loven, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [PubMed]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-André, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Boi, M.; Gaudio, E.; Bonetti, P.; Kwee, I.; Bernasconi, E.; Tarantelli, C.; Rinaldi, A.; Testoni, M.; Cascione, L.; Ponzoni, M.; et al. The BET Bromodomain inhibitor OTX015 affects pathogenetic pathways in pre-clinical B-cell tumor models and synergizes with targeted drugs. Clin. Cancer Res. 2015, 21, 1628–1638. [Google Scholar] [CrossRef] [PubMed]
- Chapuy, B.; McKeown, M.R.; Lin, C.Y.; Monti, S.; Roemer, M.G.; Qi, J.; Rahl, P.B.; Sun, H.H.; Yeda, K.T.; Doench, J.G.; et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell 2013, 24, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Trabucco, S.E.; Gerstein, R.M.; Evens, A.M.; Bradner, J.E.; Shultz, L.D.; Greiner, D.L.; Zhang, H. Inhibition of bromodomain proteins for the treatment of human diffuse large B-cell lymphoma. Clin. Cancer Res. 2015, 21, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Abedin, S.M.; Boddy, C.S.; Munshi, H.G. BET inhibitors in the treatment of hematologic malignancies: Current insights and future prospects. OncoTargets Ther. 2016, 9, 5943–5953. [Google Scholar] [CrossRef]
- Cortiguera, M.G.; Batlle-López, A.; Albajar, M.; Delgado, M.D.; León, J. MYC as therapeutic target in leukemia and lymphoma. Blood Lymphat. Cancer 2015, 5, 75–91. [Google Scholar] [CrossRef]
- Cermelli, S.; Jang, I.S.; Bernard, B.; Grandori, C. Synthetic lethal screens as a means to understand and treat MYC-driven cancers. Cold Spring Harb. Perspect. Med. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.C.; Wims, M.; Spotts, G.D.; Hann, S.R.; Bradley, A. A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev. 1993, 7, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.G.; Cha, J.; Chandrasegaran, S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc. Natl. Acad. Sci. USA 1996, 93, 1156–1160. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Huang, S.; Zhao, X.; Wright, D.A.; Carpenter, S.; Spalding, M.H.; Weeks, D.P.; Yang, B. Modularly assembled designer TAL effector nucleases for targeted gene knockout and gene replacement in eukaryotes. Nucleic Acids Res. 2011, 39, 6315–6325. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Perez-Pinera, P.; Kocak, D.D.; Vockley, C.M.; Adler, A.F.; Kabadi, A.M.; Polstein, L.R.; Thakore, P.I.; Glass, K.A.; Ousterout, D.G.; Leong, K.W.; et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat. Methods 2013, 10, 973–976. [Google Scholar] [CrossRef] [PubMed]
- Thakore, P.I.; D’Ippolito, A.M.; Song, L.; Safi, A.; Shivakumar, N.K.; Kabadi, A.M.; Reddy, T.E.; Crawford, G.E.; Gersbach, C.A. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat. Methods 2015, 12, 1143–1149. [Google Scholar] [CrossRef] [PubMed]
- Hilton, I.B.; D’Ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Vojta, A.; Dobrinić, P.; Tadić, V.; Bočkor, L.; Korać, P.; Julg, B.; Klasić, M.; Zoldoš, V. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016, 44, 5615–5628. [Google Scholar] [CrossRef] [PubMed]
- Maddalo, D.; Manchado, E.; Concepcion, C.P.; Bonetti, C.; Vidigal, J.A.; Han, Y.C.; Ogrodowski, P.; Crippa, A.; Rekhtman, N.; de Stanchina, E.; et al. In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature 2014, 516, 423–427. [Google Scholar] [CrossRef] [PubMed]
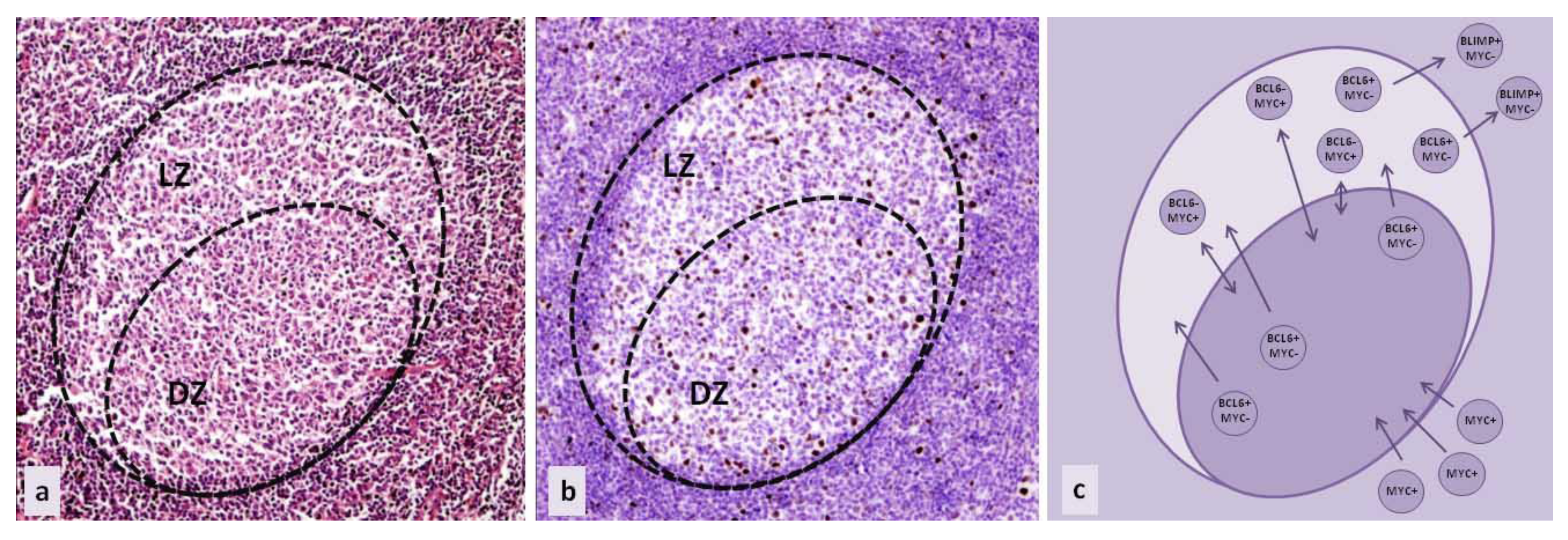

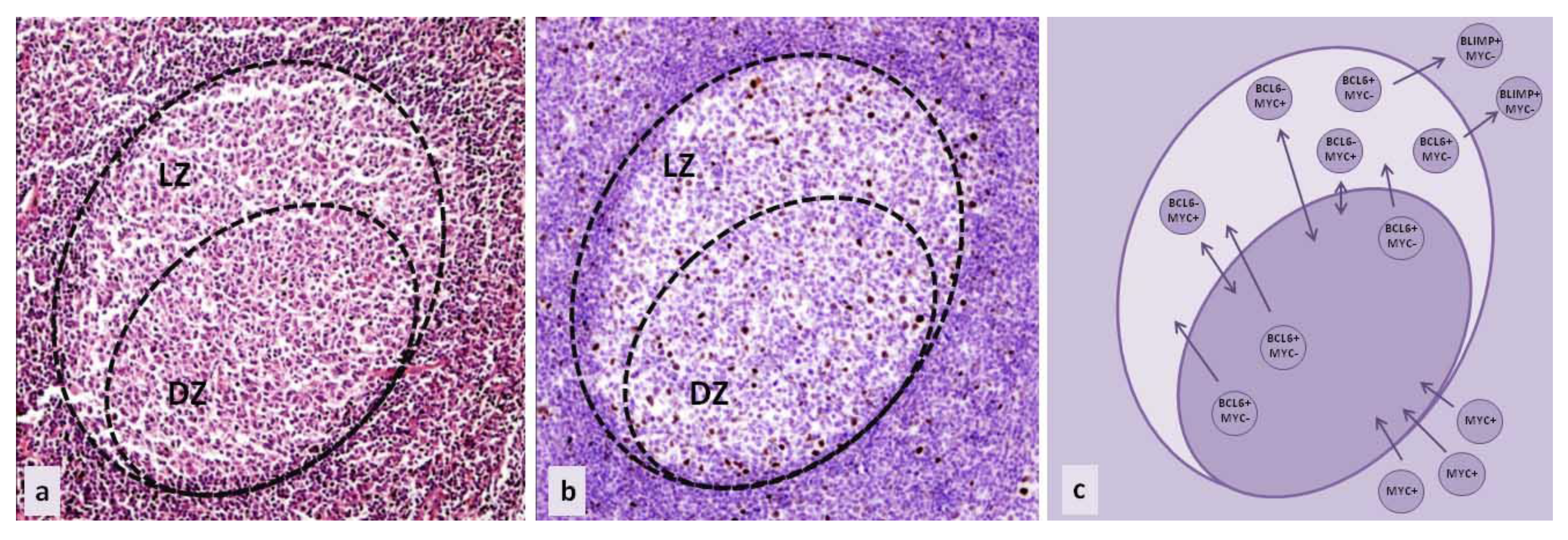{kind=link}
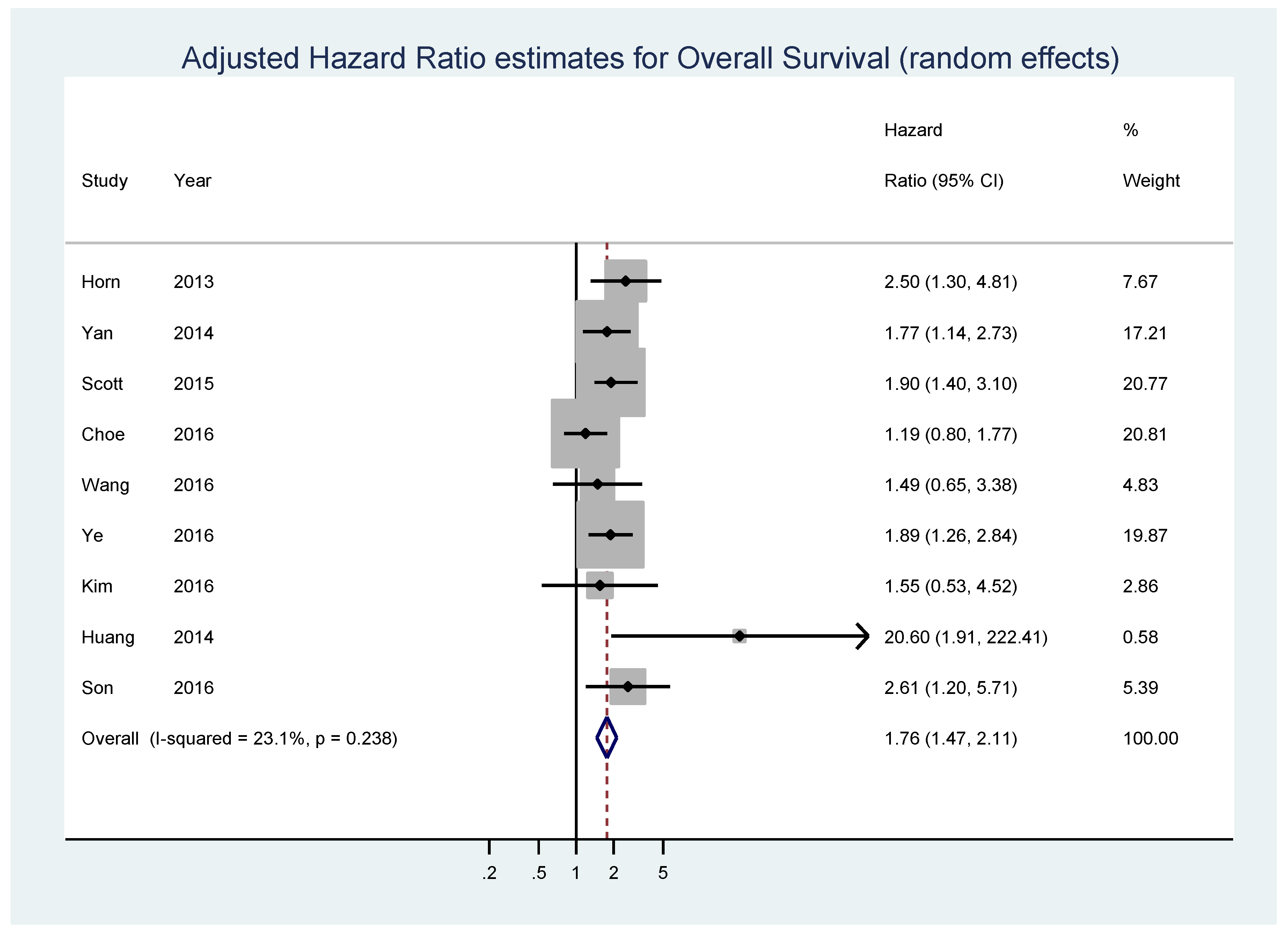{kind=link}
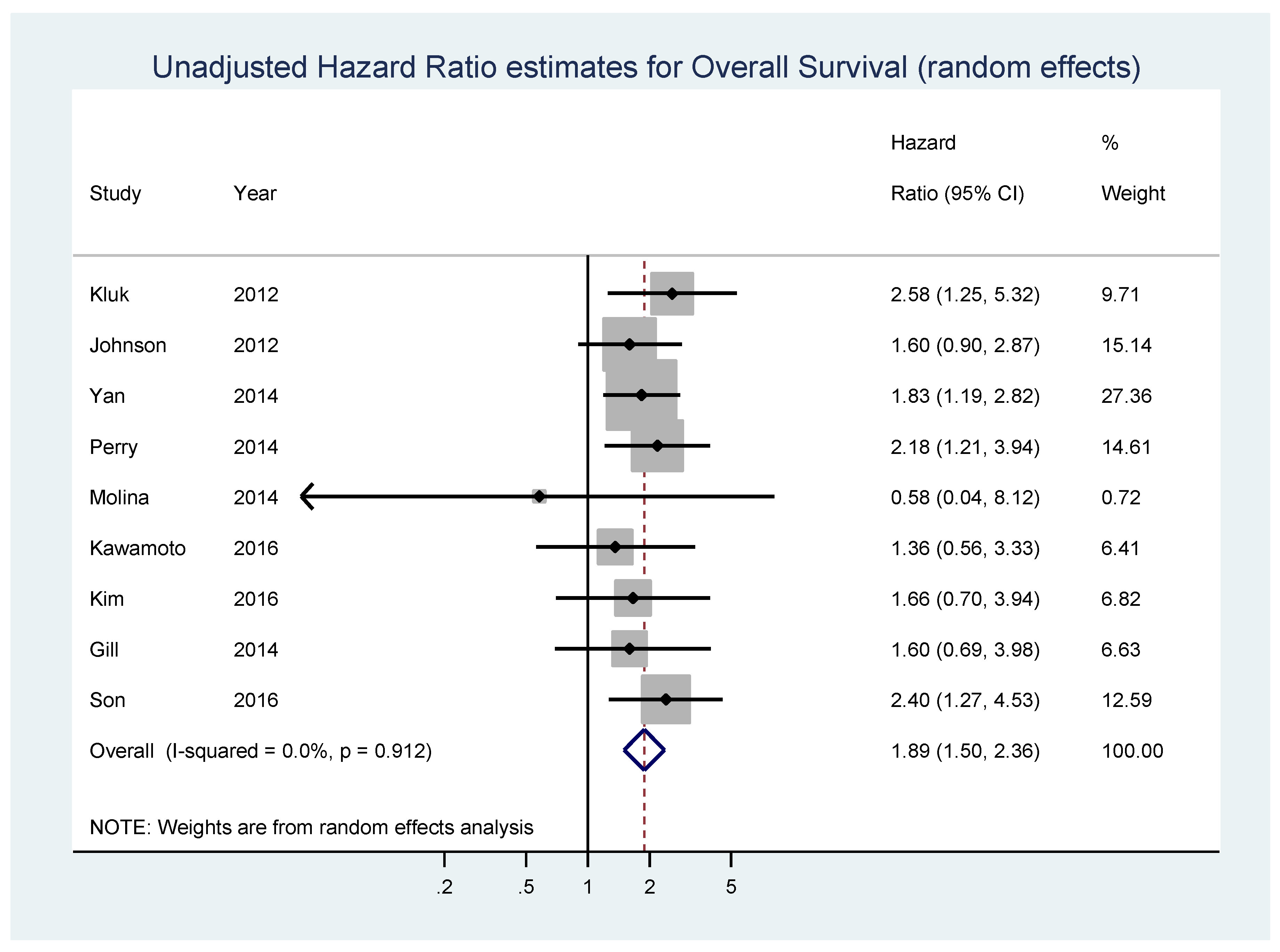{kind=link}
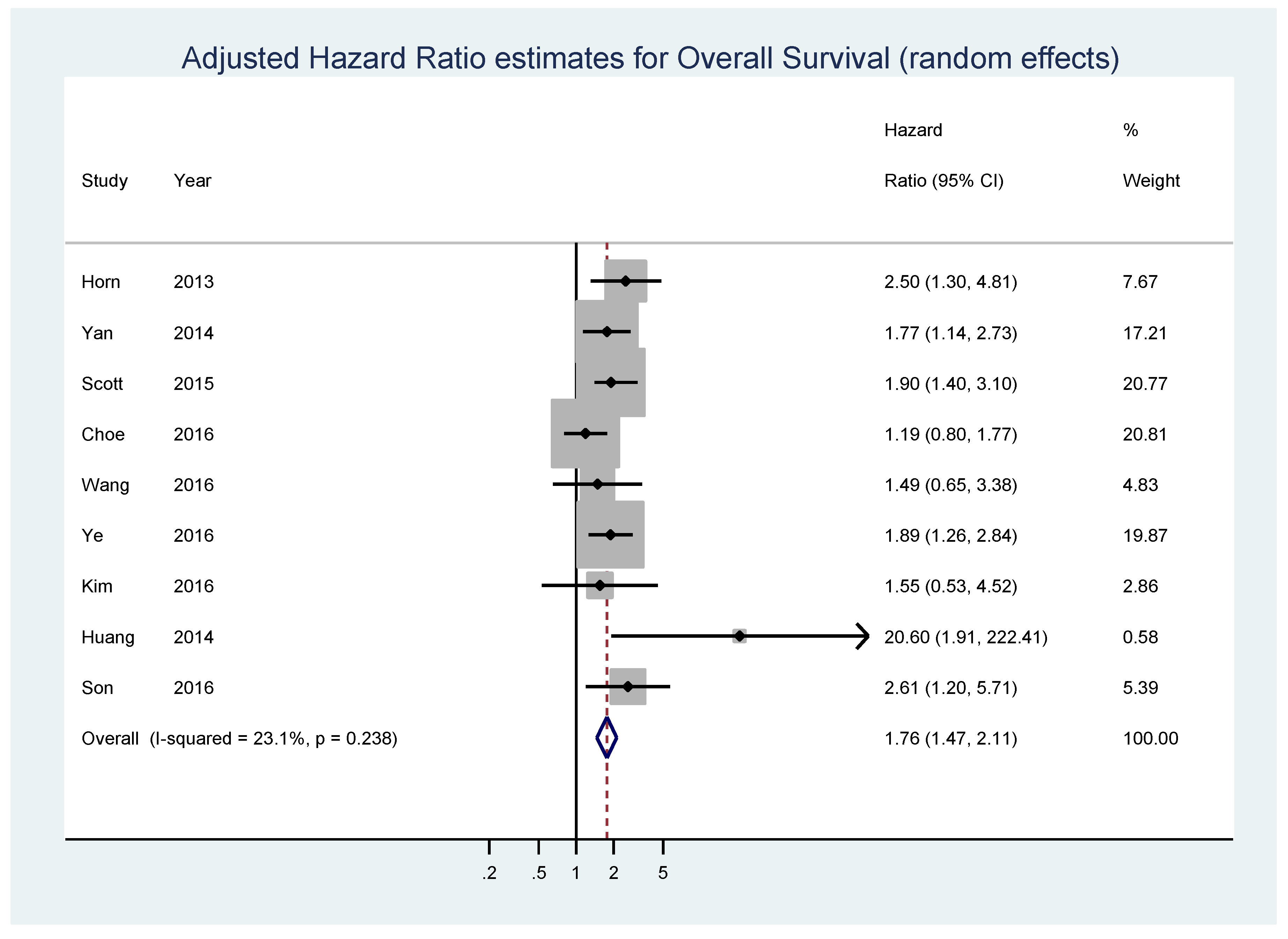
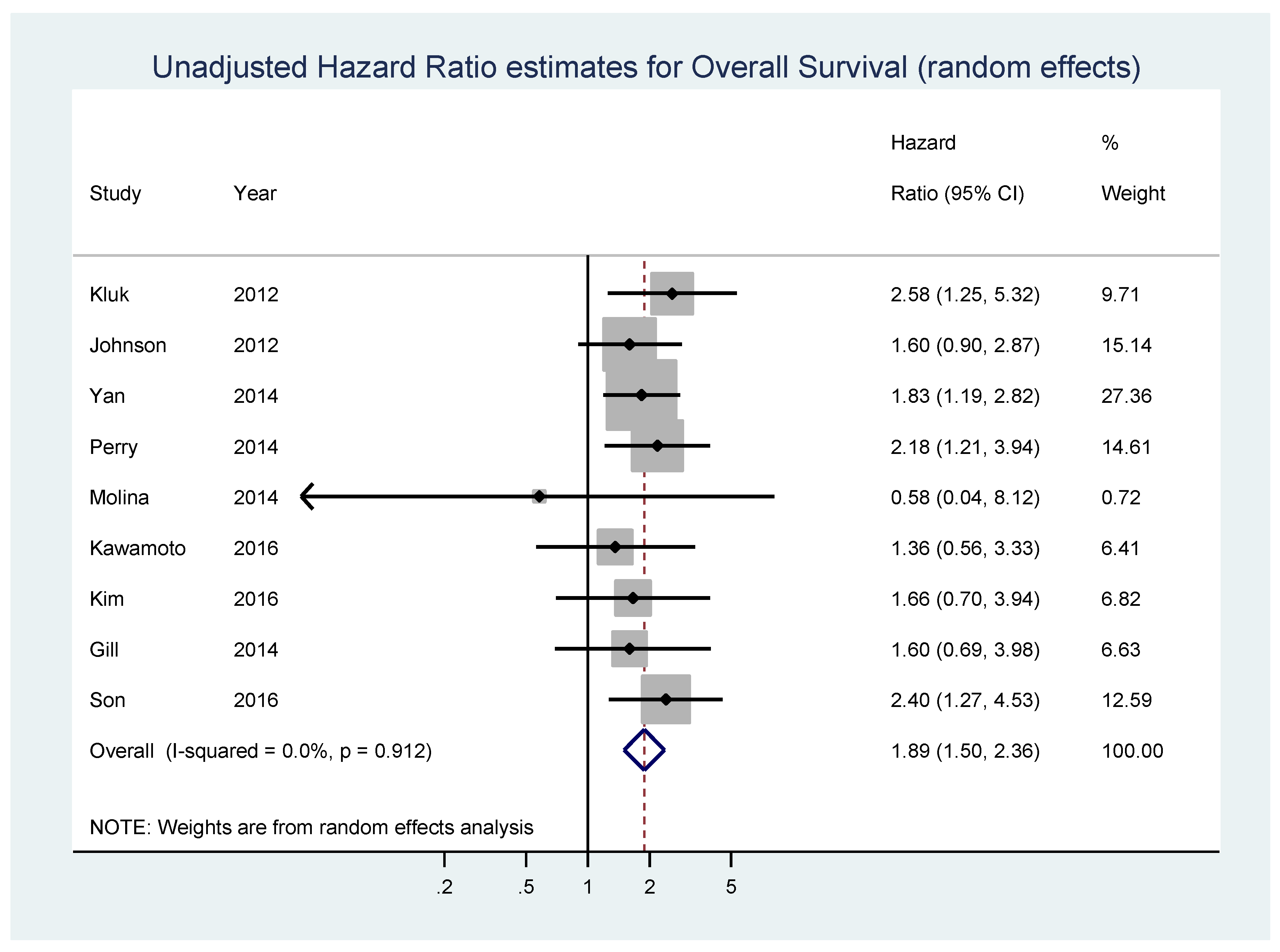
| Study | Year | Population | Disease | Number of Patients | Detection Method | Available Data | Median, Follow-Up, Months (Minimum,Maximum) | Threshold for MYC Expression | N myc_high | N myc_low | Origin of Data |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Kluk [69] | 2012 | USA (Boston, Massachusetts) | diffuse large B-cell lymphoma | 38 | IHC | OS-uni | 42 (2,87) | >50% | 6 | 32 | extrapolated |
| Johnson [25] | 2012 | not specified (from 10 international institutions) | de novo diagnosed diffuse large B-cell lymphoma | 307 | IHC/Microarray | OS-uni | 49 (6,136) | ≥40% | 100 | 207 | reported in text |
| Horn [70] | 2013 | Germany (samples taken from the RICOVER study of the German High-Grade Non-Hodgkin Lymphoma Study Group (DSHNHL) | diffuse large B-cell lymphoma | 283 | IHC/FISH | OS-multi, EFS-multi | 29 (4,64) | ≥40% | 43 | 98 | reported in text |
| Yan [71] | 2014 | China | diffuse large B-cell lymphoma | 203 | IHC | OS-uni, PFS-uni; OS-multi, PFS-multi | 37 (1,145) | ≥40% | 108 | 95 | reported in text |
| Perry [67] | 2014 | USA (patients from the Nebraska Lymphoma Study Group cohort) | diffuse large B-cell lymphoma | 106 | IHC | OS-uni | 54 (7,145) | >50% | 69 | 37 | extrapolated |
| Molina [72] | 2014 | France (participants in the Groupe d’Etudes des Lymphomes de l’Adulte/Lymphoma Study Association (LYSA) LNH 03-2B trial) | previously untreated de novo CD20+ difuse large B-cell lymphoma | 174 | IHC | OS-uni, PFS-uni | not specified | ≥40% | 47 | 127 | reported in text |
| Huang [73] | 2014 | China (samples collected at the Cancer Institute and Hospital, Chinese Academy of Medical Sciences (CICAMS) in Beijing) | MALT lymphoma without LTCs patients, 20 cases of MALT lymphoma with LTCs and 7 cases of DLBCL with a MALT lymphoma component | 62 | IHC | OS-multi | 43 (1,84) | ≥20% | 16 | 46 | reported in text |
| Gill [74] | 2014 | USA | primary diffuse large B-cell lymphoma of the central nervous system (CNS DLBCL) | 59 | IHC | OS-uni | 22 (<1,128) | ≥40% | 43 | 16 | calculated from raw data in supplemental table |
| Scott [75] | 2015 | Pretreatment tumor biopsies of patients diagnosed with de novo DLBCL, treated at the British Columbia Cancer Agency | de novo untreated diffuse large B-cell lymphoma | 339 | IHC | OS-multi, PFS-multi | 78 (9,158) | ≥40% | 105 | 234 | reported in text |
| Choe [76] | 2016 | Korea | diffuse large B-cell lymphoma | 173 | IHC | OS-multi | not specified | ≥20% | not specified | not specified | reported in text |
| Wang [77] | 2016 | USA (Nashville, Tennessee and Houston, Texas) | de novo untreated diffuse large B-cell lymphoma | 192 | IHC | OS-multi | not specified | ≥40% | 106 | 86 | reported in text |
| Kawamoto [78] | 2016 | Japan (Nigata) | diffuse large B-cell lymphoma | 52 | IHC | OS-uni, PFS | 76 (4,127) | ≥30% | 32 | 29 | reported in text |
| Ye [79] | 2016 | USA (participants in the The International DLBCL Rituximab-CHOP Consortium Program Study) | de novo untreated diffuse large B-cell lymphoma | 825 | IHC | OS-multi | 59 (1,187) | ≥70% | 249 | 576 | reported in text |
| Kim [80] | 2016 | Korea (patients from the Seoul National University Hospital) | primary diffuse large B-cell lymphoma of the central nervous system | 114 | IHC | OS-uni, PFS-uni; OS-multi, PFS-multi | 83 (0.2,118) | ≥40% | 21 | 93 | calculated from raw data in supplemental table |
| Son [81] | 2016 | Korea (Department of Pathology of the Samsung Medical Center in Seoul) | pretreatment tumor biopsies of patients diagnosed with de novo central nervous system diffuse large B-cell lymphoma | 74 | IHC | OS-uni ; OS-multi | 35.2 (1.8,148.8) | ≥44% | 49 | 25 | reported in text |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korać, P.; Dotlić, S.; Matulić, M.; Zajc Petranović, M.; Dominis, M. Role of MYC in B Cell Lymphomagenesis. Genes 2017, 8, 115. https://doi.org/10.3390/genes8040115
Korać P, Dotlić S, Matulić M, Zajc Petranović M, Dominis M. Role of MYC in B Cell Lymphomagenesis. Genes. 2017; 8(4):115. https://doi.org/10.3390/genes8040115
Chicago/Turabian StyleKorać, Petra, Snježana Dotlić, Maja Matulić, Matea Zajc Petranović, and Mara Dominis. 2017. "Role of MYC in B Cell Lymphomagenesis" Genes 8, no. 4: 115. https://doi.org/10.3390/genes8040115
APA StyleKorać, P., Dotlić, S., Matulić, M., Zajc Petranović, M., & Dominis, M. (2017). Role of MYC in B Cell Lymphomagenesis. Genes, 8(4), 115. https://doi.org/10.3390/genes8040115