
Differences in the Bacteriome of Smokeless Tobacco Products with Different Oral Carcinogenicity: Compositional and Predicted Functional Analysis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Smokeless Tobacco Products—DNA Extraction
2.2. Determination of Bacterial Load
2.3. Amplicon Library Preparation and Sequencing
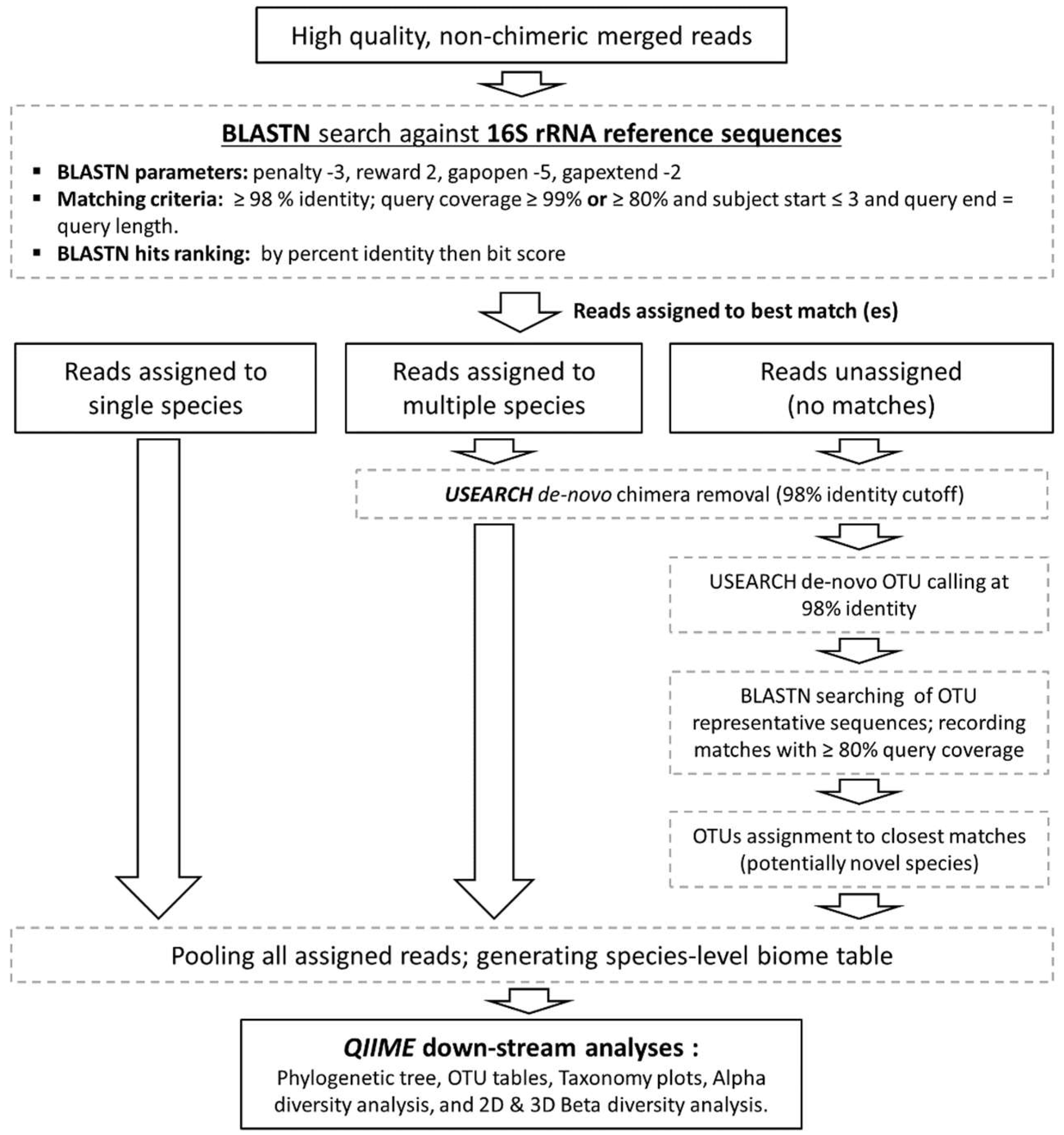
2.4. Preprocessing of Sequencing Data
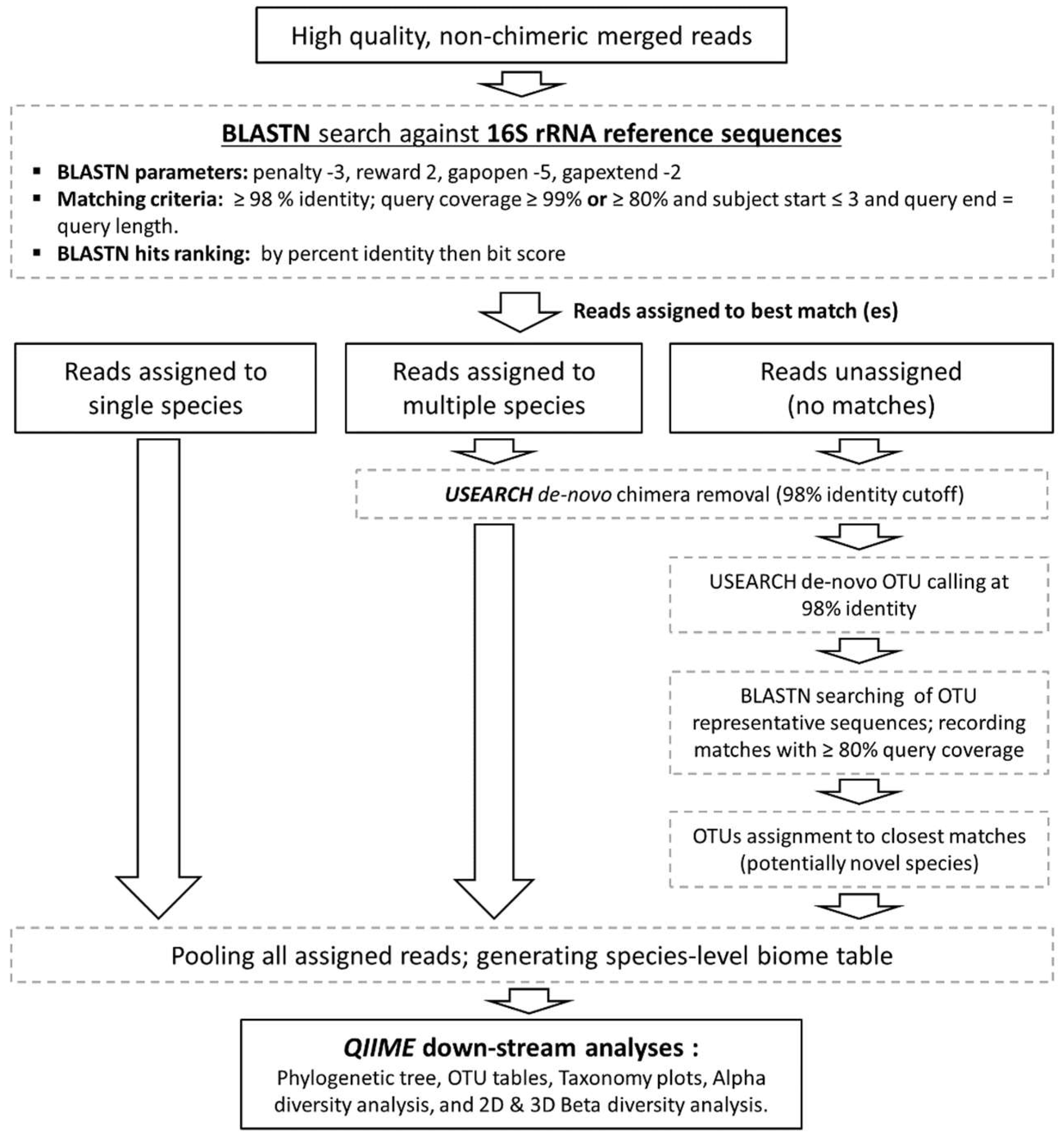
2.5. Taxonomy Assignment Algorithm
2.6. Down-Stream Biological Observation Matrix Analysis
2.7. Imputed Functional Predictions
3. Results
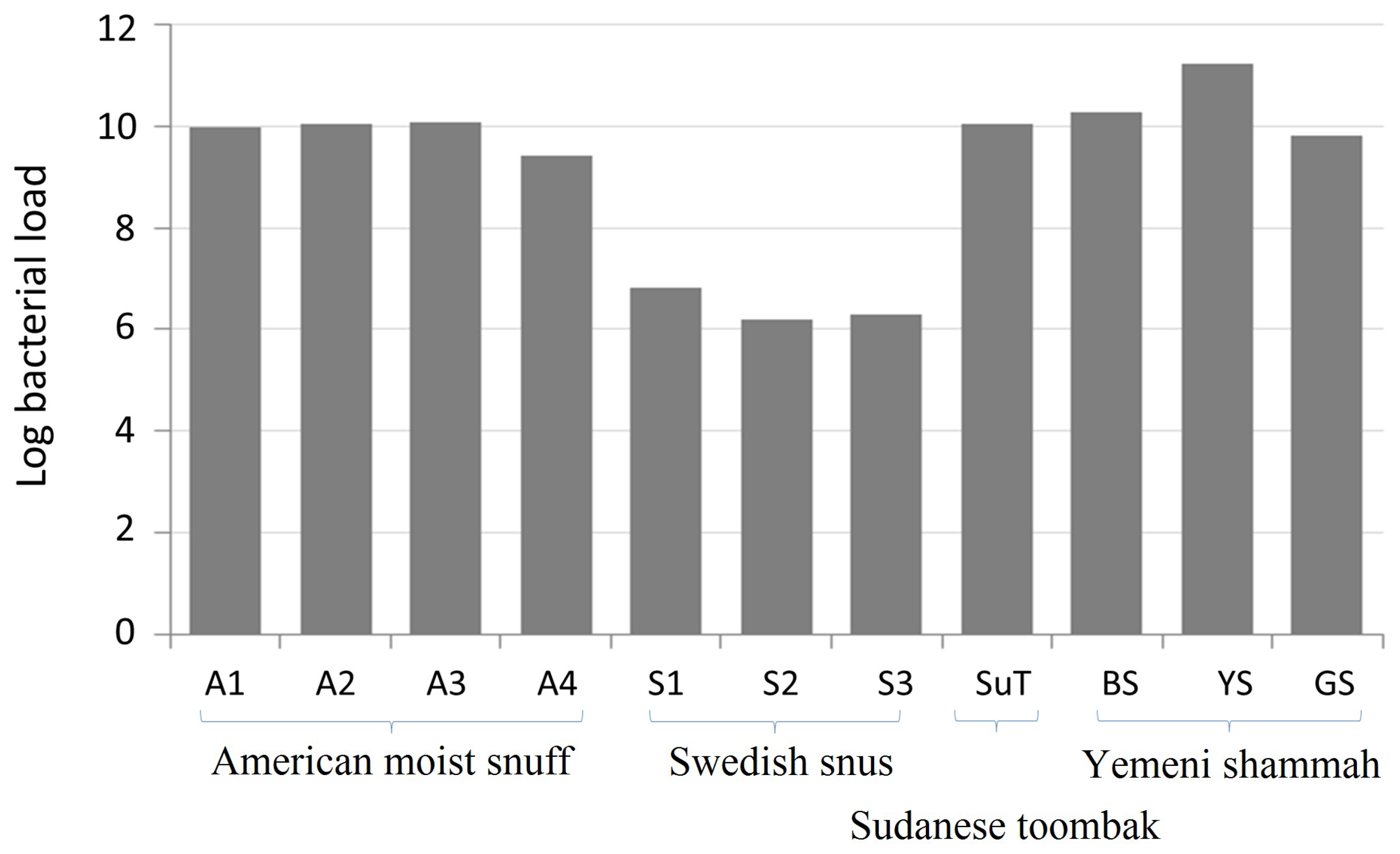
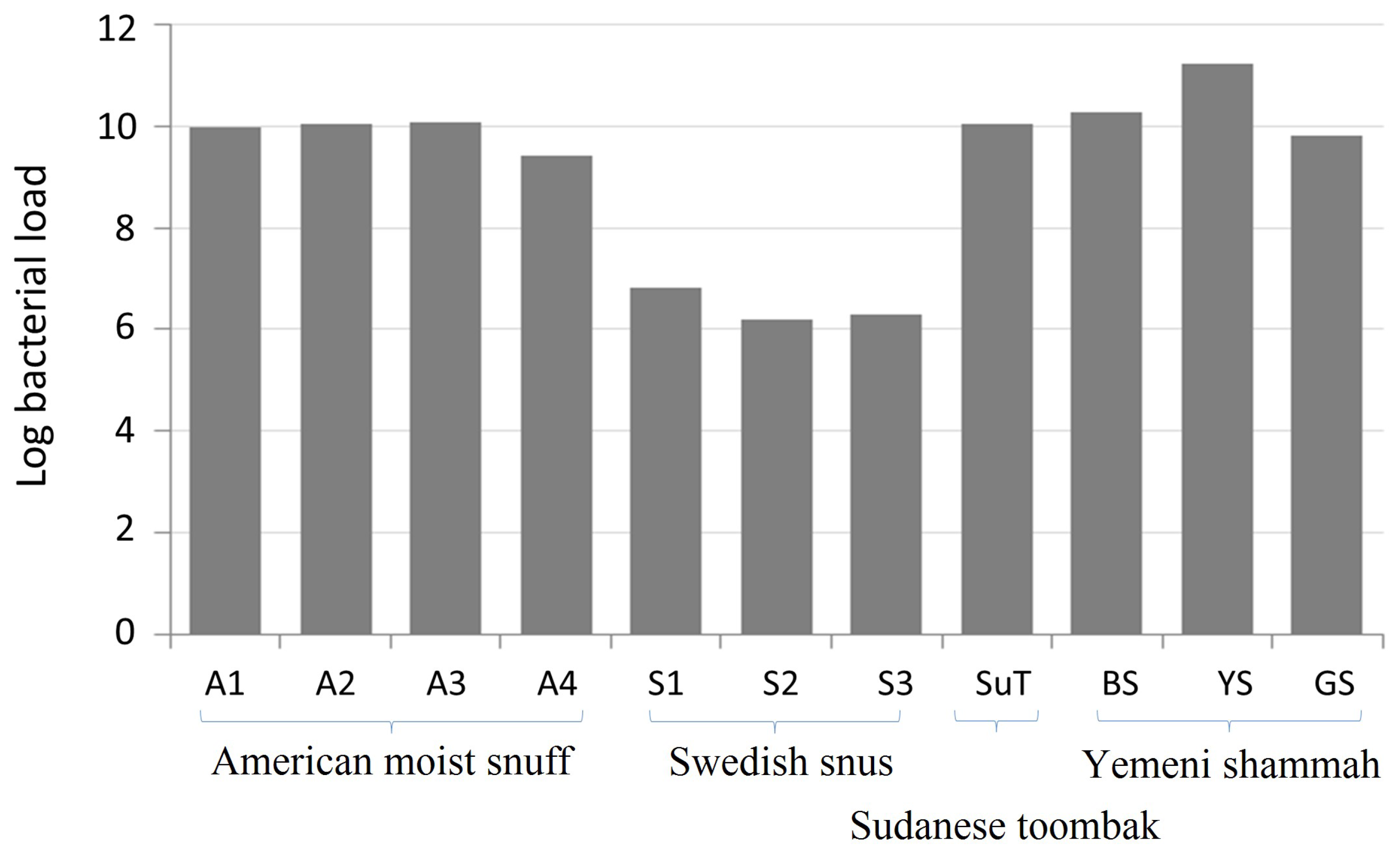
3.1. Bacterial Load of the ST Products
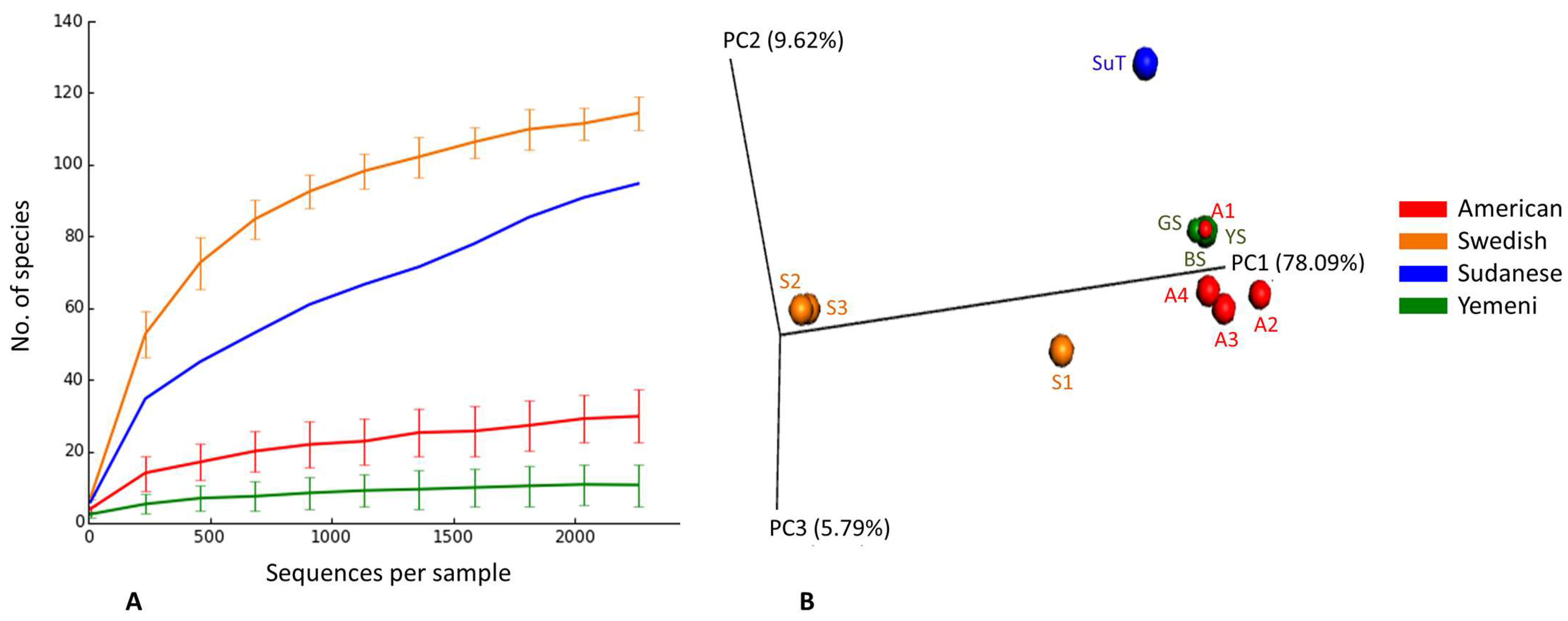
3.2. Sequencing and Data Processing Statistics
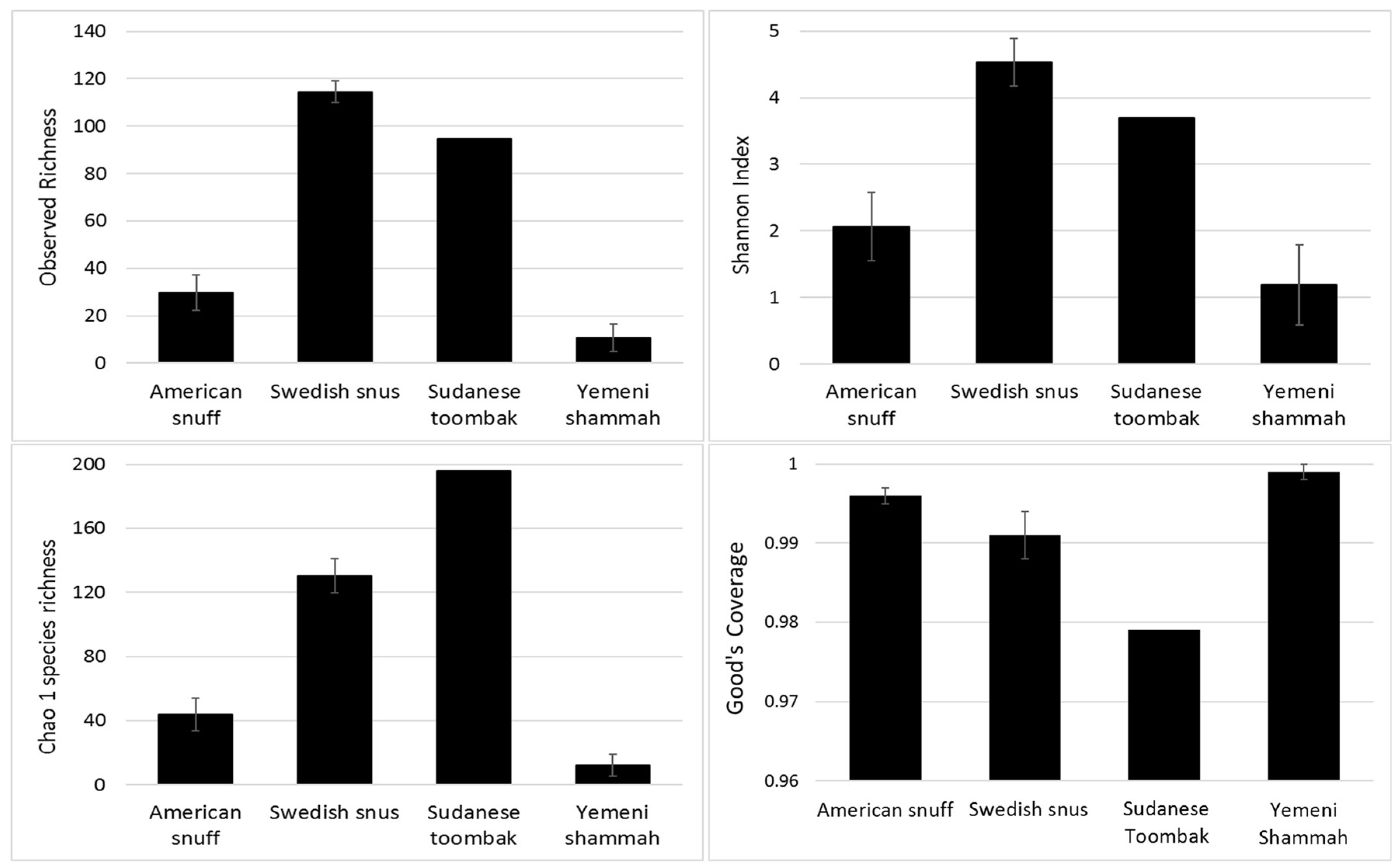
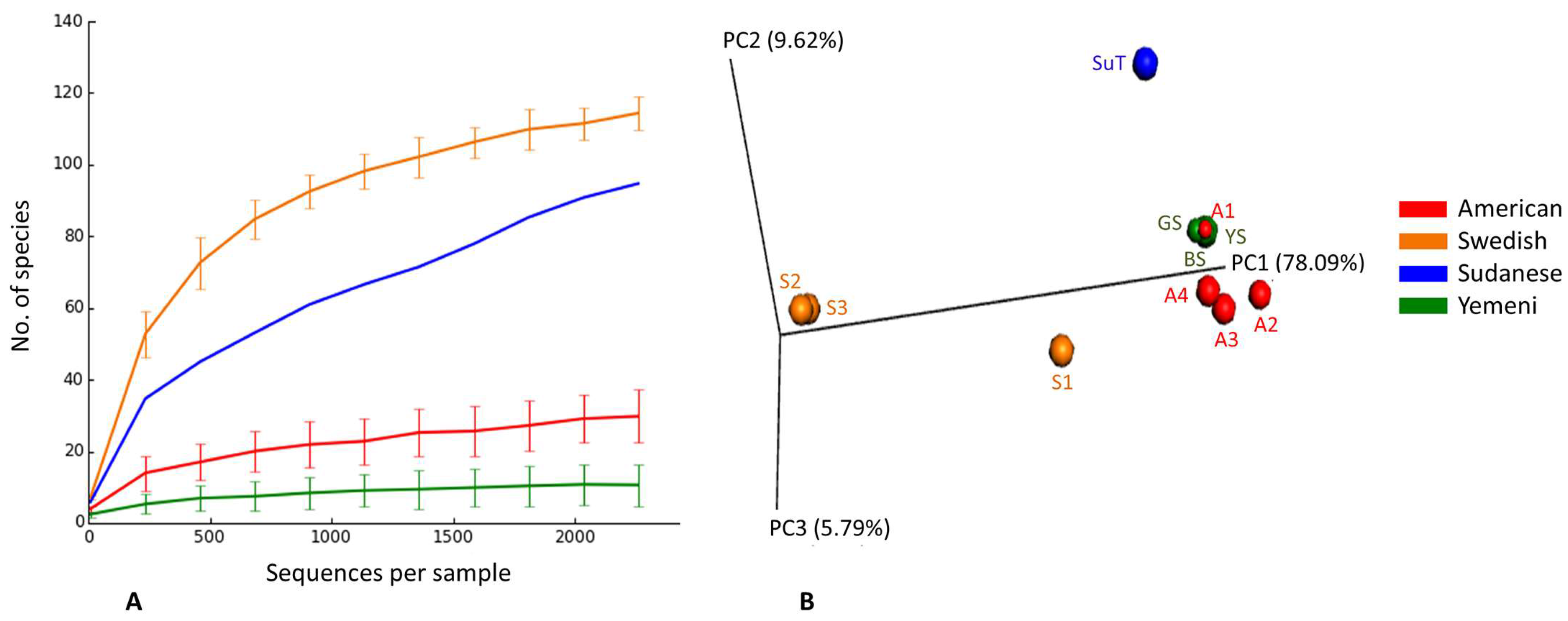
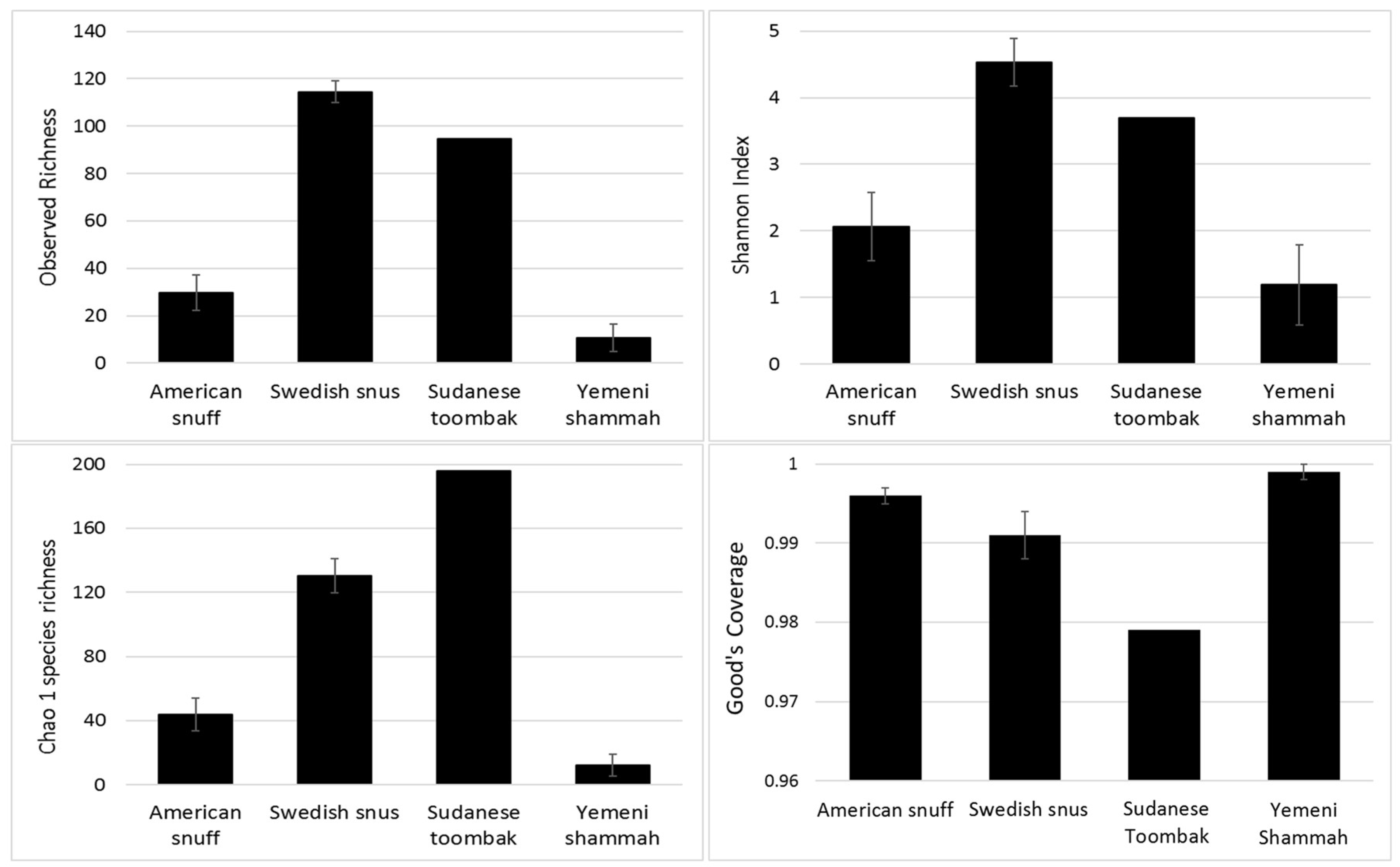
3.3. Species Richness, Diversity, and Coverage
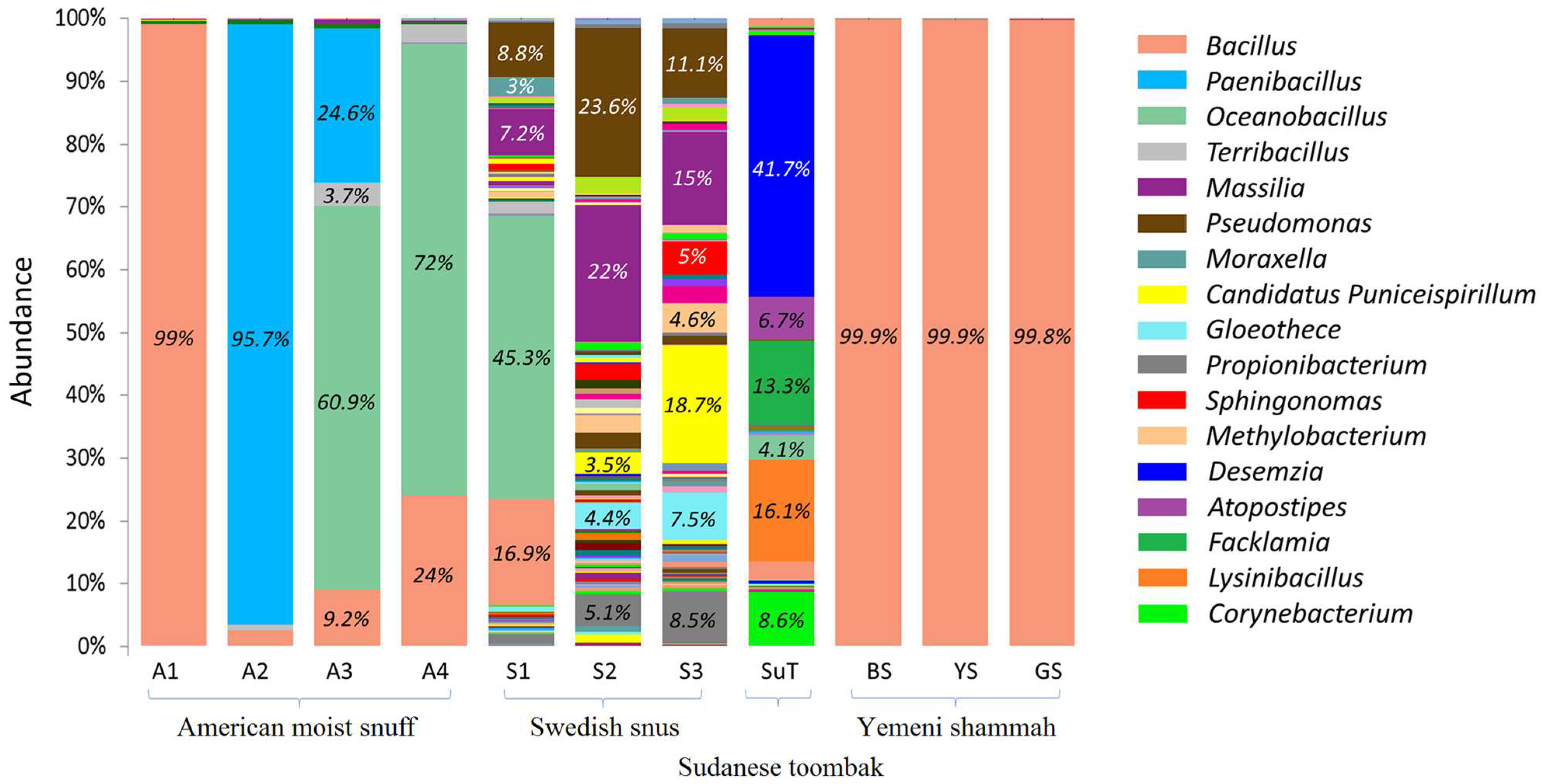
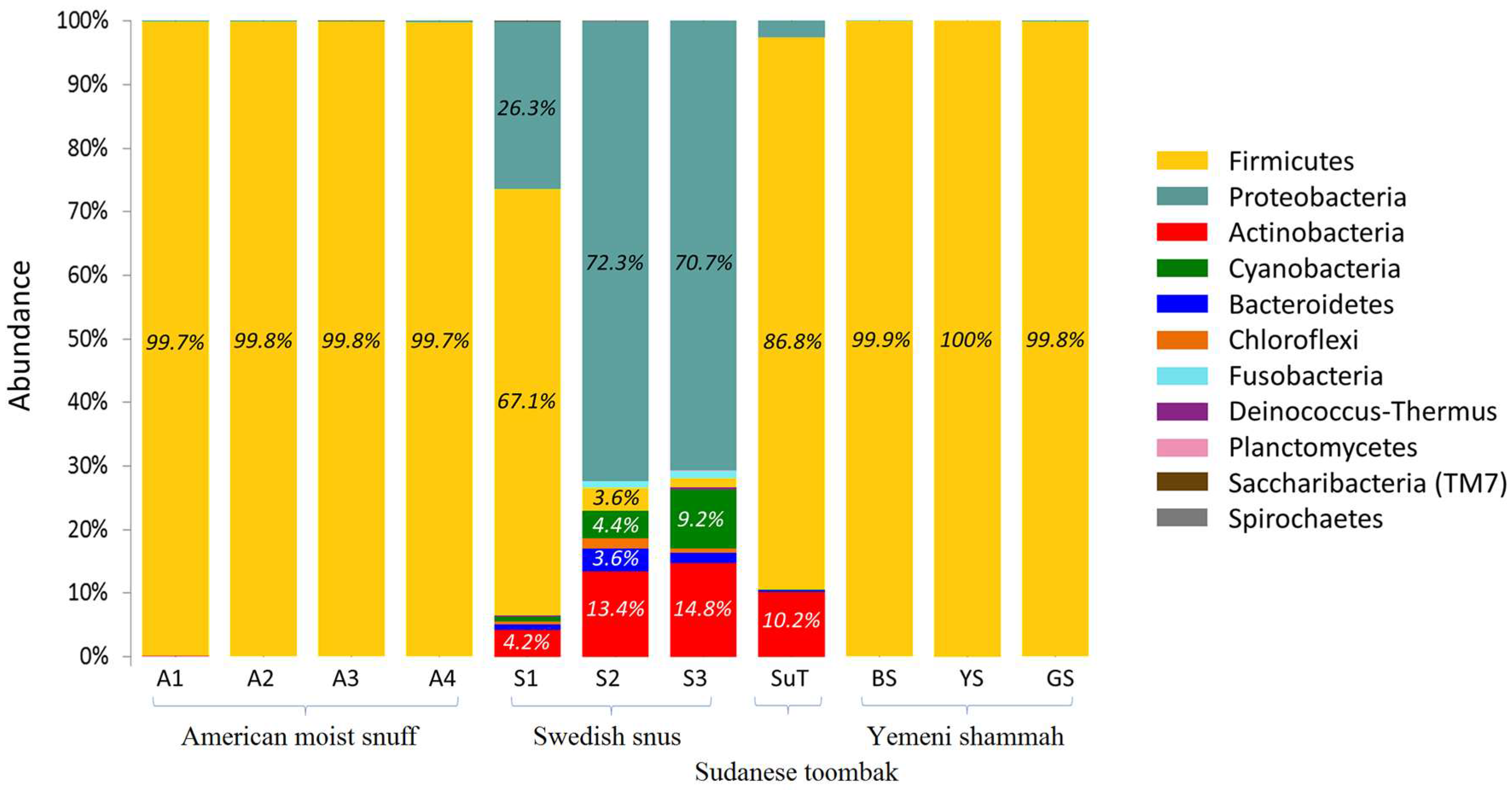
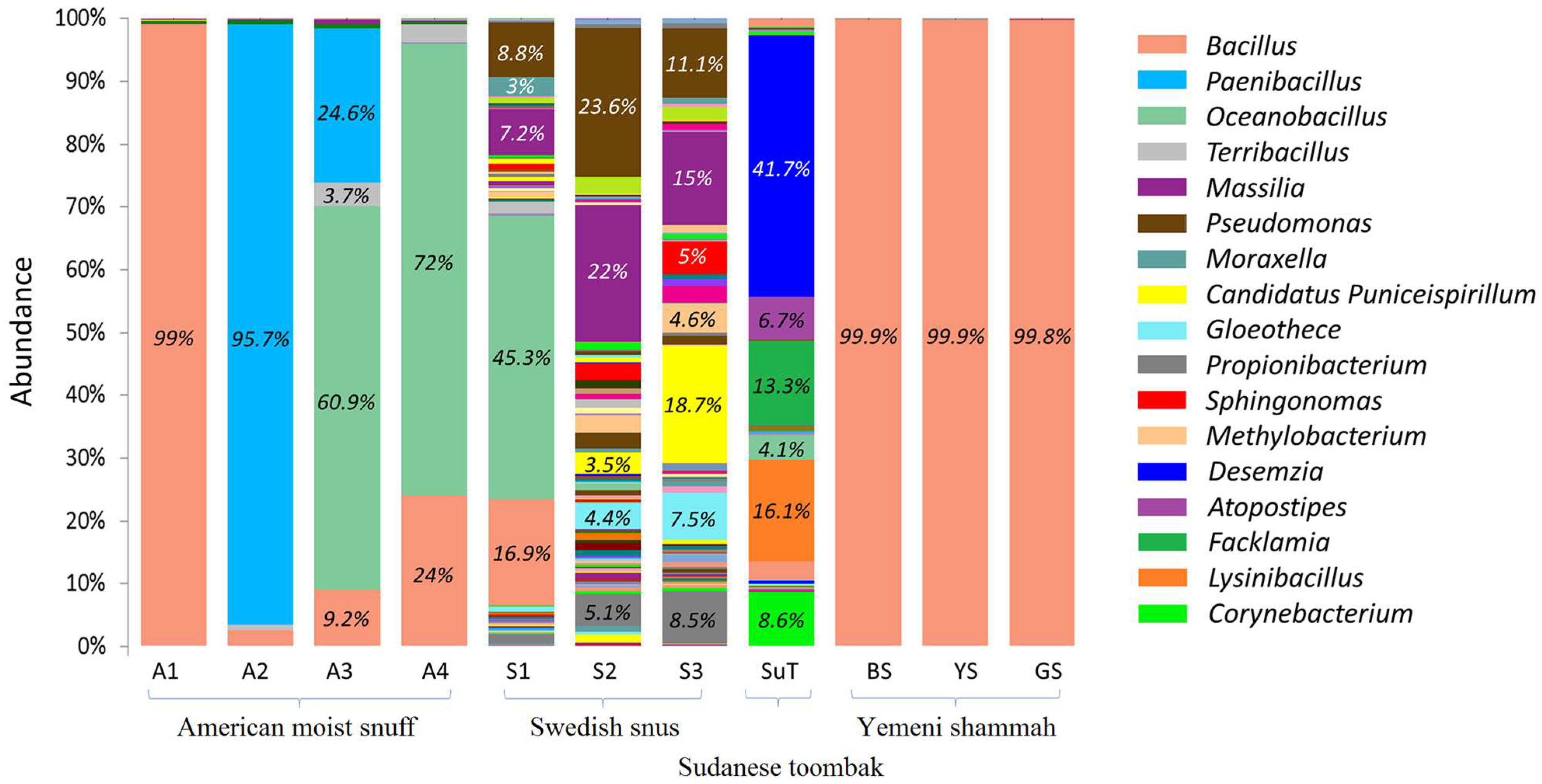
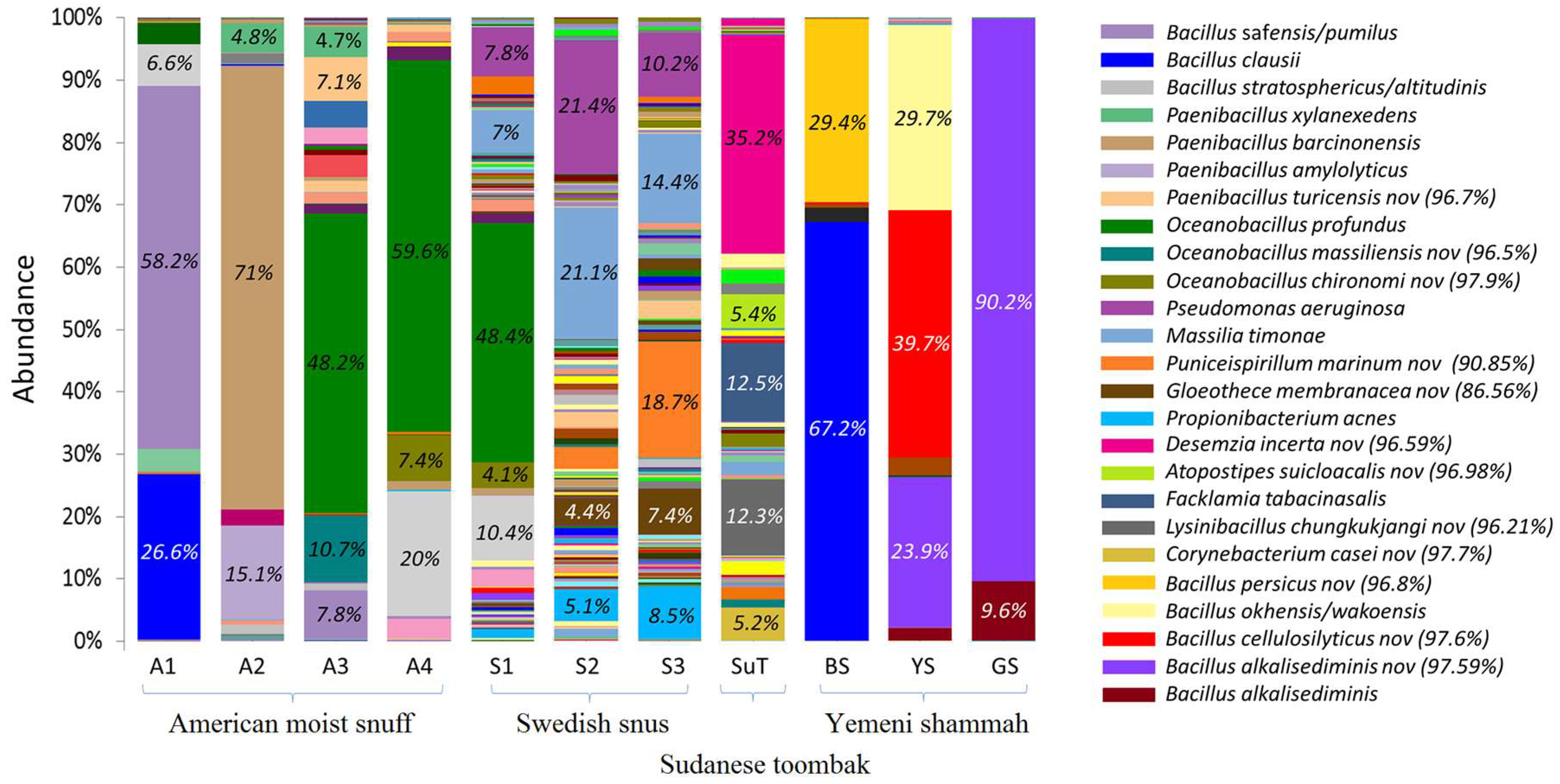
3.4. Bacteriome Identified in the ST Products
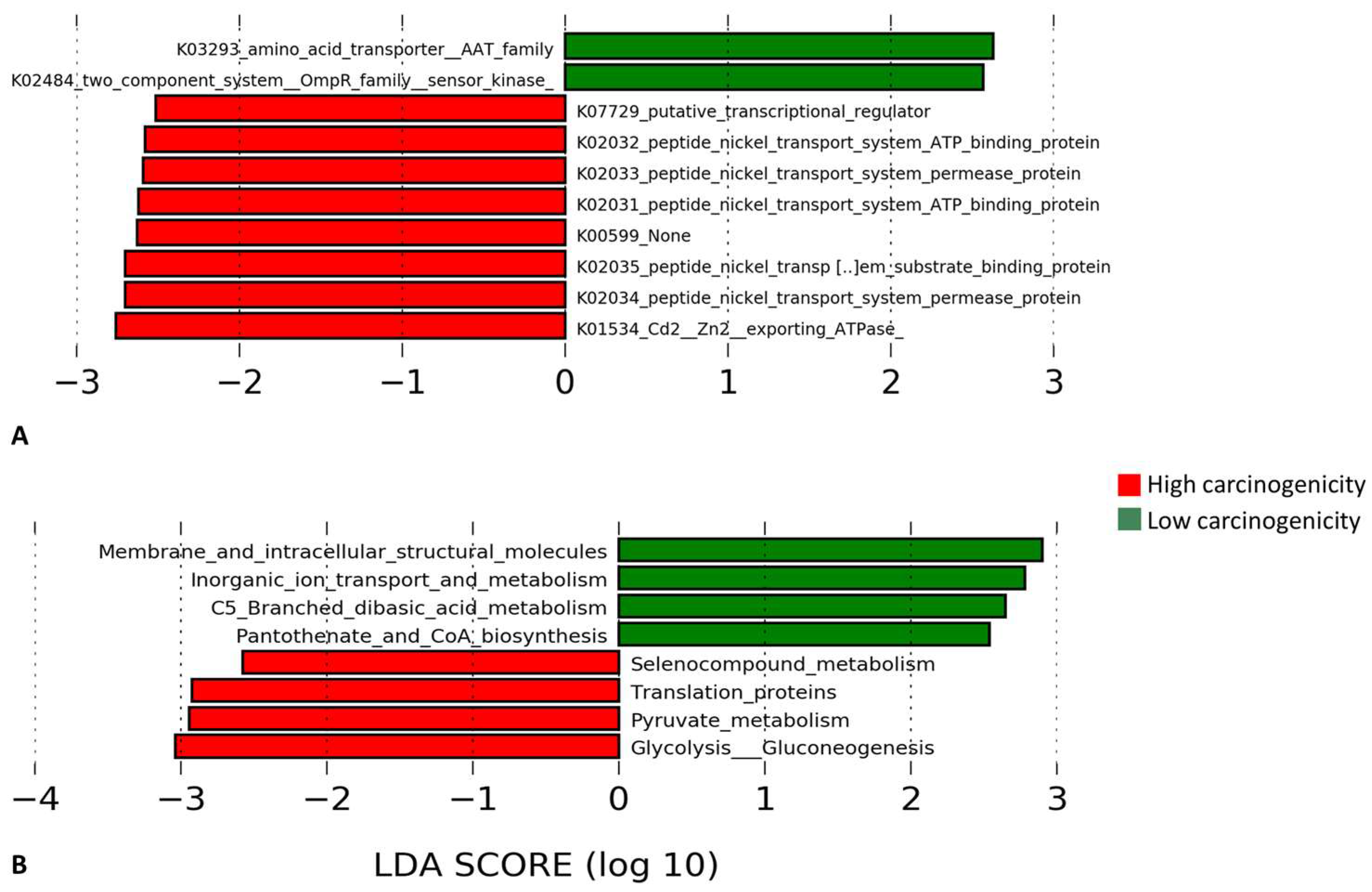
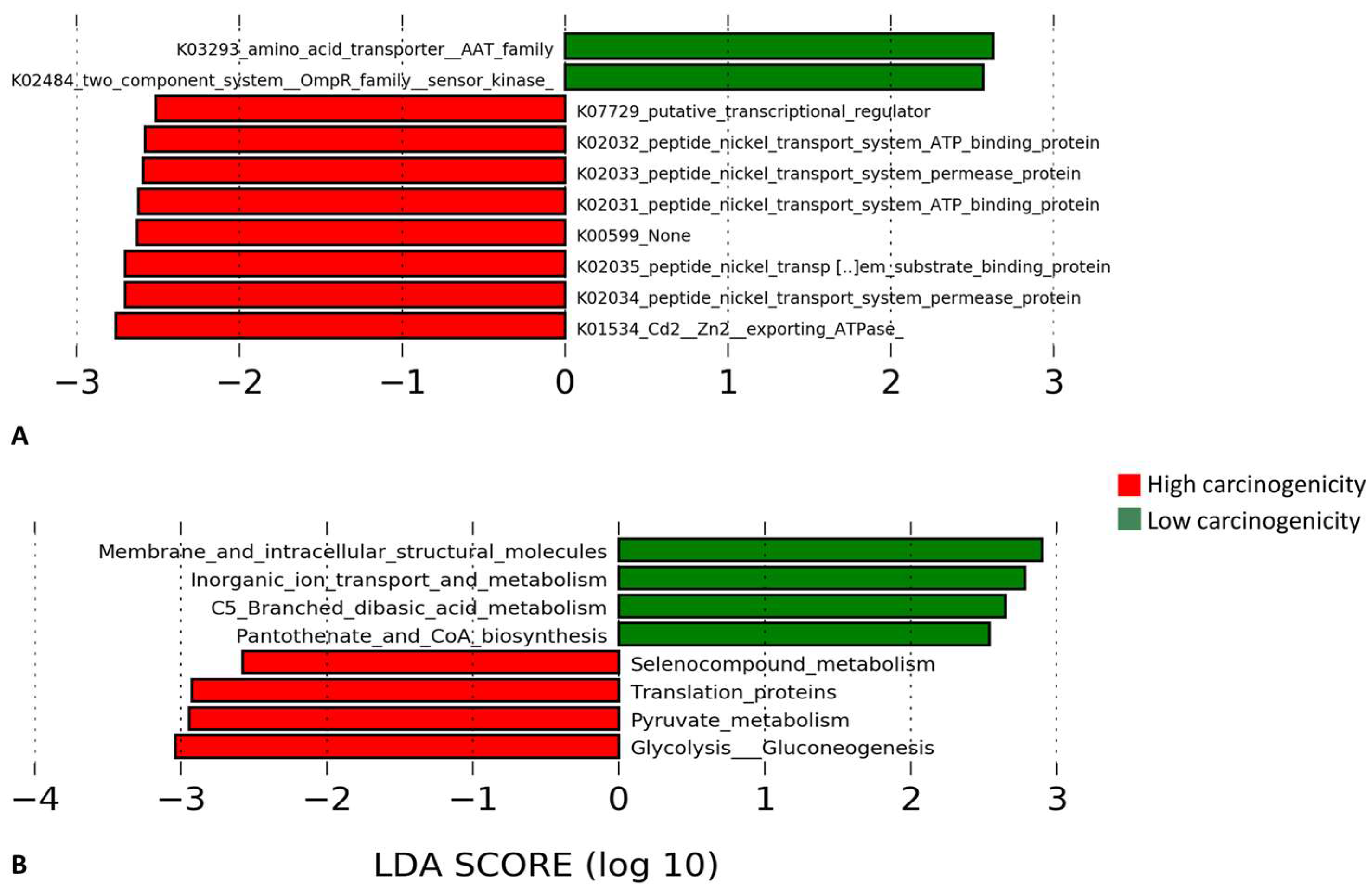
3.5. Differentially Enriched Microbial Genes and Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer. Smokeless tobacco and some tobacco-specific N-nitrosamines. In A Review of Human Carcinogens: Personal Habits and Indoor Combustions, Volume 100 E; World Health Organization; distributed by WHO Press: Lyon, France; Geneva, Switzerland, 2012; pp. 267–321. [Google Scholar]
- Lee, P.N.; Hamling, J. Systematic review of the relation between smokeless tobacco and cancer in Europe and North America. BMC Med. 2009, 7, 36. [Google Scholar] [CrossRef] [PubMed]
- Rodu, B.; Jansson, C. Smokeless tobacco and oral cancer: A review of the risks and determinants. Crit Rev. Oral Biol. Med. 2004, 15, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Weitkunat, R.; Sanders, E.; Lee, P.N. Meta-analysis of the relation between European and American smokeless tobacco and oral cancer. BMC Public Health 2007, 7, 334. [Google Scholar] [CrossRef] [PubMed]
- Elbeshir, E.I.; Abeen, H.A.; Idris, A.M.; Abbas, K. Snuff dipping and oral cancer in Sudan: A retrospective study. Br. J. Oral Maxillofac. Surg. 1989, 27, 243–248. [Google Scholar] [PubMed]
- Idris, A.M.; Ahmed, H.M.; Malik, M.O. Toombak dipping and cancer of the oral cavity in the Sudan: A case-control study. Int J. Cancer 1995, 63, 477–480. [Google Scholar] [CrossRef] [PubMed]
- Idris, A.M.; Ahmed, H.M.; Mukhtar, B.I.; Gadir, A.F.; el-Beshir, E.I. Descriptive epidemiology of oral neoplasms in Sudan 1970-1985 and the role of toombak. Int J. Cancer 1995, 61, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Allard, W.F.; DeVol, E.B.; Te, O.B. Smokeless tobacco (shamma) and oral cancer in Saudi Arabia. Community Dent. Oral Epidemiol. 1999, 27, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Scheifele, C.; Nassar, A.; Reichart, P.A. Prevalence of oral cancer and potentially malignant lesions among shammah users in Yemen. Oral Oncol. 2007, 43, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Nasher, A.T.; Al-Hebshi, N.N.; Al-Moayad, E.E.; Suleiman, A.M. Viral infection and oral habits as risk factors for oral squamous cell carcinoma in Yemen: A case-control study. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2014, 118, 566.e1–572.e1. [Google Scholar] [PubMed]
- Muwonge, R.; Ramadas, K.; Sankila, R.; Thara, S.; Thomas, G.; Vinoda, J.; Sankaranarayanan, R. Role of tobacco smoking, chewing and alcohol drinking in the risk of oral cancer in Trivandrum, India: A nested case-control design using incident cancer cases. Oral Oncol. 2008, 44, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Znaor, A.; Brennan, P.; Gajalakshmi, V.; Mathew, A.; Shanta, V.; Varghese, C.; Boffetta, P. Independent and combined effects of tobacco smoking, chewing and alcohol drinking on the risk of oral, pharyngeal and esophageal cancers in Indian men. Int J. Cancer 2003, 105, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Deng, X.; Cai, D.; Ji, Z.; Wang, C.; Yu, J.; Li, J.; Chen, S. Decreased tobacco-specific nitrosamines by microbial treatment with Bacillus amyloliquefaciens DA9 during the air-curing process of burley tobacco. J. Agric. Food Chem. 2014, 62, 12701–12706. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, I.; Pedersen, G.W. Bacillus species are present in chewing tobacco sold in the United States and evoke plasma exudation from the oral mucosa. Clin. Diagn. Lab. Immunol. 2002, 9, 1057–1060. [Google Scholar] [PubMed]
- Pauly, J.L.; Paszkiewicz, G. Cigarette smoke, bacteria, mold, microbial toxins, and chronic lung inflammation. J. Oncol. 2011, 2011, 819129. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Yang, J.; Duan, Y.; Gu, W.; Gong, X.; Zhe, W.; Su, C.; Zhang, K.Q. Bacterial diversities on unaged and aging flue-cured tobacco leaves estimated by 16S rRNA sequence analysis. Appl. Microbiol. Biotechnol 2010, 88, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Wang, B.; Li, F.; Qiu, L.; Li, F.; Wang, S.; Cui, J. Analysis of bacterial communities on aging flue-cured tobacco leaves by 16S rDNA PCR-DGGE technology. Appl. Microbiol. Biotechnol. 2007, 73, 1435–1440. [Google Scholar] [CrossRef] [PubMed]
- Di Giacomo, M.; Paolino, M.; Silvestro, D.; Vigliotta, G.; Imperi, F.; Visca, P.; Alifano, P.; Parente, D. Microbial community structure and dynamics of dark fire-cured tobacco fermentation. Appl. Environ. Microbiol. 2007, 73, 825–837. [Google Scholar] [PubMed]
- Su, C.; Gu, W.; Zhe, W.; Zhang, K.Q.; Duan, Y.; Yang, J. Diversity and phylogeny of bacteria on Zimbabwe tobacco leaves estimated by 16S rRNA sequence analysis. Appl. Microbiol. Biotechnol. 2011, 92, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Law, A.D.; Fisher, C.; Jack, A.; Moe, L.A. Tobacco, Microbes, and Carcinogens: Correlation Between Tobacco Cure Conditions, Tobacco-Specific Nitrosamine Content, and Cured Leaf Microbial Community. Microb. Ecol. 2016, 72, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Tyx, R.E.; Stanfill, S.B.; Keong, L.M.; Rivera, A.J.; Satten, G.A.; Watson, C.H. Characterization of Bacterial Communities in Selected Smokeless Tobacco Products Using 16S rDNA Analysis. PLoS ONE 2016, 11, e0146939. [Google Scholar] [CrossRef] [PubMed]
- Mizrahi-Man, O.; Davenport, E.R.; Gilad, Y. Taxonomic classification of bacterial 16S rRNA genes using short sequencing reads: Evaluation of effective study designs. PLoS ONE 2013, 8, e53608. [Google Scholar] [CrossRef] [PubMed]
- Vanwonterghem, I.; Jensen, P.D.; Dennis, P.G.; Hugenholtz, P.; Rabaey, K.; Tyson, G.W. Deterministic processes guide long-term synchronised population dynamics in replicate anaerobic digesters. ISME J. 2014, 8, 2015–2028. [Google Scholar] [CrossRef] [PubMed]
- Illumina’s Manual 15044223 Rev. B. Available online: http://support.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/16s/16s-metagenomic-library-prep-guide-15044223-b.pdf (accessed on 17 June 2016).
- Frank, J.A.; Reich, C.I.; Sharma, S.; Weisbaum, J.S.; Wilson, B.A.; Olsen, G.J. Critical evaluation of two primers commonly used for amplification of bacterial 16S rRNA genes. Appl. Environ. Microbiol. 2008, 74, 2461–2470. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J.; Pace, B.; Olsen, G.J.; Stahl, D.A.; Sogin, M.L.; Pace, N.R. Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc. Natl. Acad. Sci. USA 1985, 82, 6955–6959. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2014, 30, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glockner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Gevers, D.; Westcott, S.L. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS ONE 2011, 6, e27310. [Google Scholar] [CrossRef] [PubMed]
- Al-Hebshi, N.N.; Nasher, A.T.; Idris, A.M.; Chen, T. Robust species taxonomy assignment algorithm for 16S rRNA NGS reads: Application to oral carcinoma samples. J. Oral Microbiol. 2015, 7, 28934. [Google Scholar] [CrossRef] [PubMed]
- Human Oral Microbiome Database (HOMD). Available online: http://homd.org/index.php?name=seqDownload&file&type=R (accessed on 29 March 2016).
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. Waste not, want not: Why rarefying microbiome data is inadmissible. PLoS Comput. Biol. 2014, 10, e1003531. [Google Scholar]
- Stepanov, I.; Jensen, J.; Hatsukami, D.; Hecht, S.S. New and traditional smokeless tobacco: Comparison of toxicant and carcinogen levels. Nicotine Tob. Res. 2008, 10, 1773–1782. [Google Scholar] [PubMed]
- Hatsukami, D.K.; Ebbert, J.O.; Feuer, R.M.; Stepanov, I.; Hecht, S.S. Changing smokeless tobacco products new tobacco-delivery systems. Am. J. Prev. Med. 2007, 33, S368–S378. [Google Scholar] [CrossRef] [PubMed]
- Idris, A.M.; Nair, J.; Ohshima, H.; Friesen, M.; Brouet, I.; Faustman, E.M.; Bartsch, H. Unusually high levels of carcinogenic tobacco-specific nitrosamines in Sudan snuff (toombak). Carcinogenesis 1991, 12, 1115–1118. [Google Scholar] [PubMed]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer. Cadmium and cadmium compounds. In A Review of Human Carcinogens: Arsenic, Metals, Fibres and Dusts, Volume 100 C; World Health Organization; distributed by WHO Press: Lyon, France; Geneva, Switzerland, 2012; pp. 121–141. [Google Scholar]
- Janbaz, K.H.; Qadir, M.I.; Basser, H.T.; Bokhari, T.H.; Ahmad, B. Risk for oral cancer from smokeless tobacco. Contemp. Oncol. (Pozn.) 2014, 18, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Grimsrud, T.K.; Peto, J. Persisting risk of nickel related lung cancer and nasal cancer among Clydach refiners. Occup. Environ. Med. 2006, 63, 365–366. [Google Scholar] [CrossRef] [PubMed]
- Su, C.C.; Lin, Y.Y.; Chang, T.K.; Chiang, C.T.; Chung, J.A.; Hsu, Y.Y.; Lian Ie, B. Incidence of oral cancer in relation to nickel and arsenic concentrations in farm soils of patients’ residential areas in Taiwan. BMC Public Health 2010, 10, 67. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.H.; Lian Ie, B.; Tsai, K.Y.; Chang, T.K.; Chiang, C.T.; Su, C.C.; Hwang, Y.H. Possible association between nickel and chromium and oral cancer: A case-control study in central Taiwan. Sci Total Environ. 2011, 409, 1046–1052. [Google Scholar] [CrossRef] [PubMed]
- Arain, S.S.; Kazi, T.G.; Afridi, H.I.; Talpur, F.N.; Kazi, A.G.; Brahman, K.D.; Naeemullah; Arain, M.S.; Sahito, O.M. Estimation of Nickel in Different Smokeless Tobacco Products and Their Impact on Human Health of Oral Cancer Patients. Nutr. Cancer 2015, 67, 1063–1074. [Google Scholar] [CrossRef] [PubMed]
- Andersen, R.A.; Burton, H.R.; Fleming, P.D.; Hamilton-Kemp, T.R. Effect of storage conditions on nitrosated, acylated, and oxidized pyridine alkaloid derivatives in smokeless tobacco products. Cancer Res. 1989, 49, 5895–5900. [Google Scholar] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-hebshi, N.N.; Alharbi, F.A.; Mahri, M.; Chen, T. Differences in the Bacteriome of Smokeless Tobacco Products with Different Oral Carcinogenicity: Compositional and Predicted Functional Analysis. Genes 2017, 8, 106. https://doi.org/10.3390/genes8040106
Al-hebshi NN, Alharbi FA, Mahri M, Chen T. Differences in the Bacteriome of Smokeless Tobacco Products with Different Oral Carcinogenicity: Compositional and Predicted Functional Analysis. Genes. 2017; 8(4):106. https://doi.org/10.3390/genes8040106
Chicago/Turabian StyleAl-hebshi, Nezar Noor, Fahd Ali Alharbi, Mohammed Mahri, and Tsute Chen. 2017. "Differences in the Bacteriome of Smokeless Tobacco Products with Different Oral Carcinogenicity: Compositional and Predicted Functional Analysis" Genes 8, no. 4: 106. https://doi.org/10.3390/genes8040106
APA StyleAl-hebshi, N. N., Alharbi, F. A., Mahri, M., & Chen, T. (2017). Differences in the Bacteriome of Smokeless Tobacco Products with Different Oral Carcinogenicity: Compositional and Predicted Functional Analysis. Genes, 8(4), 106. https://doi.org/10.3390/genes8040106