
Regulation of Replication Fork Advance and Stability by Nucleosome Assembly

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
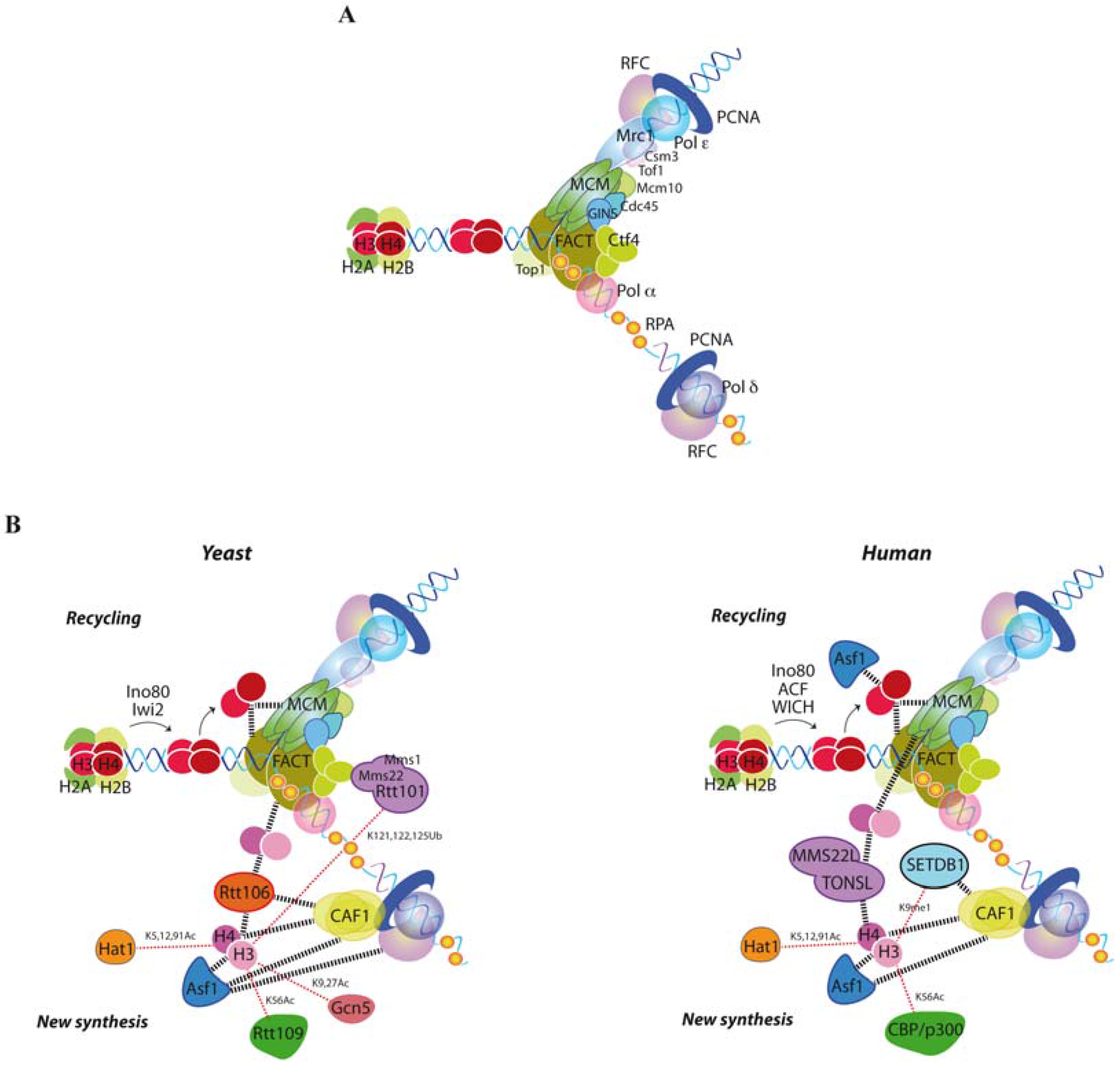
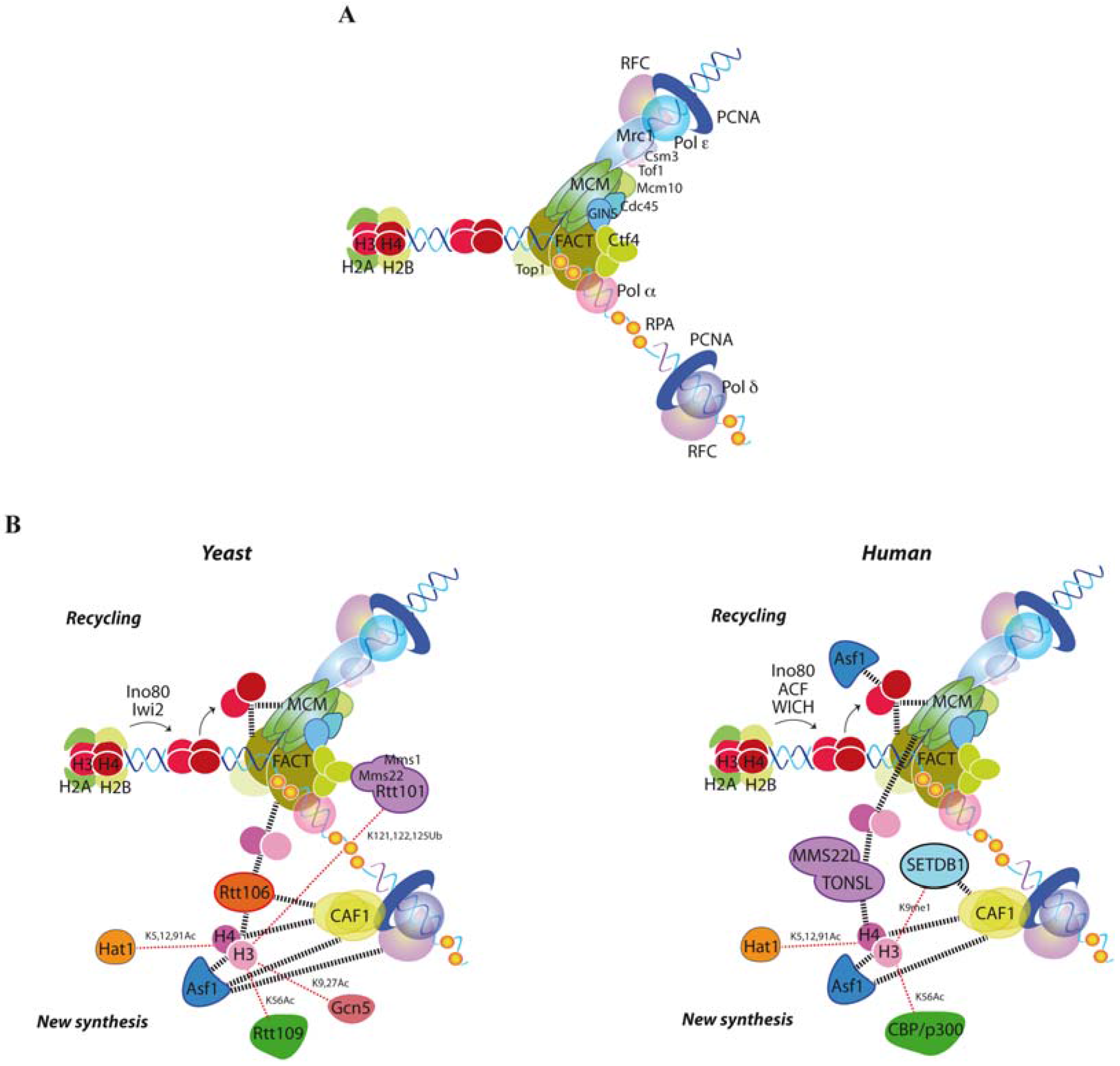
2. Building a Functional Replisome
3. Replication Fork Advance through Chromatin
4. Replication Fork Control by Histone Recycling
5. Mechanisms of New Histone Assembly
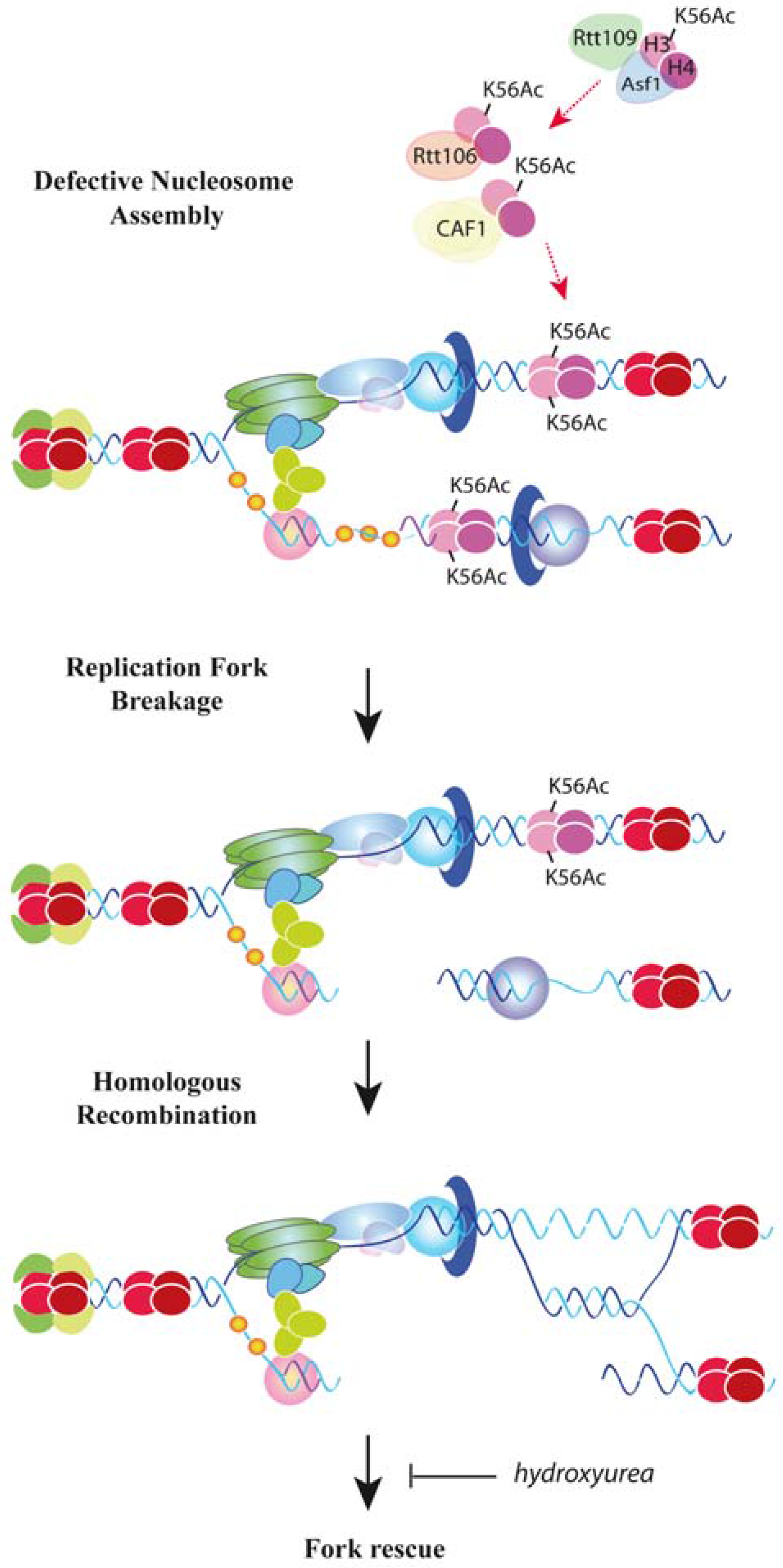
6. Replication Fork Progression and Stability by New Histone Assembly
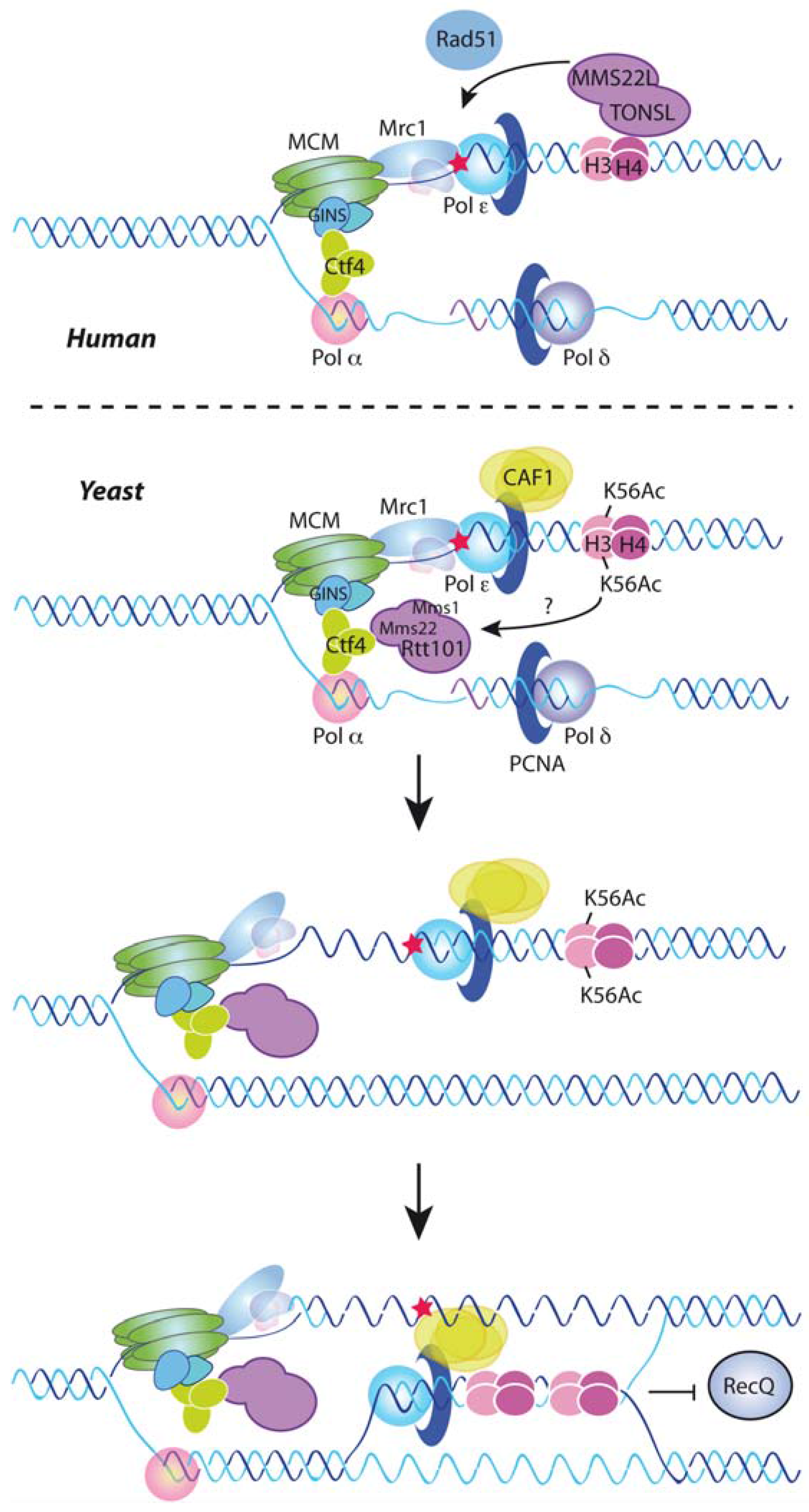
7. DNA Damage Tolerance Control by Nucleosome Assembly
8. Replication-Coupled Chromatin Assembly and Disease
9. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Berti, M.; Vindigni, A. Replication stress: Getting back on track. Nat. Struct. Mol. Biol. 2016, 23, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An Oncogene-Induced DNA Damage Model for Cancer Development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Jasencakova, Z.; Groth, A. Replication stress, a source of epigenetic aberrations in cancer? Bioessays 2010, 32, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Jasencakova, Z.; Scharf, A.N.D.; Ask, K.; Corpet, A.; Imhof, A.; Almouzni, G.; Groth, A. Replication stress interferes with histone recycling and predeposition marking of new histones. Mol. Cell 2010, 37, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Sogo, J.M.; Stahl, H.; Koller, T.; Knippers, R. Structure of replicating simian virus 40 minichromosomes. The replication fork, core histone segregation and terminal structures. J. Mol. Biol. 1986, 189, 189–204. [Google Scholar] [CrossRef]
- Henikoff, S.; Smith, M.M. Histone variants and epigenetics. Cold Spring Harb. Perspect. Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Burgess, R.J.; Zhang, Z. Histone chaperones in nucleosome assembly and human disease. Nat. Struct. Mol. Biol. 2013, 20, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Almouzni, G.; Cedar, H. Maintenance of Epigenetic Information. Cold Spring Harb. Perspect. Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Alabert, C.; Groth, A. Chromatin replication and epigenome maintenance. Nat. Rev. Mol. Cell Biol. 2012, 13, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Deegan, T.D.; Diffley, J.F.X. MCM: One ring to rule them all. Curr. Opin. Struct. Biol. 2016, 37, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Labib, K. How do Cdc7 and cyclin-dependent kinases trigger the initiation of chromosome replication in eukaryotic cells? Genes Dev. 2010, 24, 1208–1219. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.P.; Labib, K. Chromosome Duplication in Saccharomyces cerevisiae. Genetics 2016, 203, 1027–1067. [Google Scholar] [CrossRef] [PubMed]
- Stillman, B. DNA Polymerases at the Replication Fork in Eukaryotes. Mol. Cell 2008, 30, 259–260. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, G.-L.; Pfander, B.; Jentsch, S. PCNA, the Maestro of the Replication Fork. Cell 2007, 129, 665–679. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.I.; Altmeyer, M.; Rask, M.-B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef] [PubMed]
- Simon, A.C.; Zhou, J.C.; Perera, R.L.; van Deursen, F.; Evrin, C.; Ivanova, M.E.; Kilkenny, M.L.; Renault, L.; Kjaer, S.; Matak-Vinkovic, D.; et al. A Ctf4 trimer couples the CMG helicase to DNA polymerase α in the eukaryotic replisome. Nature 2015, 510, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Lou, H.; Komata, M.; Katou, Y.; Guan, Z.; Reis, C.C.; Budd, M.; Shirahige, K.; Campbell, J.L. Mrc1 and DNA Polymerase ɛ Function Together in Linking DNA Replication and the S Phase Checkpoint. Mol. Cell 2008, 32, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Tourriere, H.; Versini, G.; Cordon-Preciado, V.; Alabert, C.; Pasero, P. Mrc1 and Tof1 Promote Replication Fork Progression and Recovery Independently of Rad53. Mol. Cell 2005, 19, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Gambus, A.; van Deursen, F.; Polychronopoulos, D.; Foltman, M.; Jones, R.C.; Edmondson, R.D.; Calzada, A.; Labib, K. A key role for Ctf4 in coupling the MCM2-7 helicase to DNA polymerase alpha within the eukaryotic replisome. EMBO J. 2009, 28, 2992–3004. [Google Scholar] [CrossRef] [PubMed]
- Szyjka, S.J.; Viggiani, C.J.; Aparicio, O.M. Mrc1 Is required for normal progression of replication forks throughout chromatin in S. cerevisiae. Mol. Cell 2005, 19, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Gambus, A.; Jones, R.C.; Sanchez-Diaz, A.; Kanemaki, M.; van Deursen, F.; Edmondson, R.D.; Labib, K. GINS maintains association of Cdc45 with MCM in replisome progression complexes at eukaryotic DNA replication forks. Nat. Cell Biol. 2006, 8, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Sirbu, B.M.; Couch, F.B.; Feigerle, J.T.; Bhaskara, S.; Hiebert, S.W.; Cortez, D. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 2011, 25, 1320–1327. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Contreras, A.J.; Ruppen, I.; Nieto-Soler, M.; Murga, M.; Rodriguez-Acebes, S.; Remeseiro, S.; Rodrigo-Perez, S.; Rojas, A.M.; Mendez, J.; Munoz, J.; et al. A Proteomic Characterization of Factors Enriched at Nascent DNA Molecules. Cell Rep. 2013, 3, 1105–1116. [Google Scholar] [CrossRef] [PubMed]
- Alabert, C.; Bukowski-Wills, J.-C.; Lee, S.-B.; Kustatscher, G.; Nakamura, K.; de Lima Alves, F.; Menard, P.; Mejlvang, J.; Rappsilber, J.; Groth, A. Nascent chromatin capture proteomics determines chromatin dynamics during DNA replication and identifies unknown fork components. Nat. Cell Biol. 2014, 16, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Gomez, M.; Antequera, F. Overreplication of short DNA regions during S phase in human cells. Genes Dev. 2008, 22, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Jimeno-Gonzalez, S.; Payan-Bravo, L.; Munoz-Cabello, A.M.; Guijo, M.; Gutierrez, G.; Prado, F.; Reyes, J.C. Defective histone supply causes changes in RNA polymerase II elongation rate and cotranscriptional pre-mRNA splicing. Proc. Natl. Acad. Sci. USA 2015, 112, 14840–14845. [Google Scholar] [CrossRef] [PubMed]
- Almeida, R.; SantaMaría, C.; Gomez, M.; Centro de Biología Molecular “Severo Ochoa” (CBMSO/CSIC), Madrid, Spain. Personal communication, 2017.
- Annunziato, A. The Fork in the Road: Histone Partitioning During DNA Replication. Genes 2015, 6, 353–371. [Google Scholar] [CrossRef] [PubMed]
- Gasser, R.; Koller, T.; Sogo, J.M. The stability of nucleosomes at the replication fork. J. Mol. Biol. 1996, 258, 224–239. [Google Scholar] [CrossRef] [PubMed]
- Corless, S.; Gilbert, N. Effects of DNA supercoiling on chromatin architecture. Biophys. Rev. 2016, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.A.; Kwong, T.J.; Tsukiyama, T. ATP-dependent chromatin remodeling shapes the DNA replication landscape. Nat. Struct. Mol. Biol. 2008, 15, 477–484. [Google Scholar] [CrossRef]
- Papamichos-Chronakis, M.; Peterson, C.L. The Ino80 chromatin-remodeling enzyme regulates replisome function and stability. Nat. Struct. Mol. Biol. 2008, 15, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Oma, Y.; Schleker, T.; Kugou, K.; Ohta, K.; Harata, M.; Gasser, S.M. Ino80 Chromatin Remodeling Complex Promotes Recovery of Stalled Replication Forks. Curr. Biol. 2008, 18, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-S.; Lee, S.-A.; Hur, S.-K.; Seo, J.-W.; Kwon, J. Stabilization and targeting of INO80 to replication forks by BAP1 during normal DNA synthesis. Nat. Commun. 2014. [Google Scholar] [CrossRef] [PubMed]
- Vassileva, I.; Yanakieva, I.; Peycheva, M.; Gospodinov, A.; Anachkova, B. The mammalian INO80 chromatin remodeling complex is required for replication stress recovery. Nucleic Acids Res. 2014, 42, 9074–9086. [Google Scholar] [CrossRef] [PubMed]
- Poot, R.A.; Bozhenok, L.; van den Berg, D.L.C.; Steffensen, S.; Ferreira, F.; Grimaldi, M.; Gilbert, N.; Ferreira, J.; Varga-Weisz, P.D. The Williams syndrome transcription factor interacts with PCNA to target chromatin remodelling by ISWI to replication foci. Nat. Cell Biol. 2004, 6, 1236–1244. [Google Scholar] [CrossRef] [PubMed]
- Collins, N.; Poot, R.A.; Kukimoto, I.; Garcia-Jimenez, C.; Dellaire, G.; Varga-Weisz, P.D. An ACF1–ISWI chromatin-remodeling complex is required for DNA replication through heterochromatin. Nat. Genet. 2002, 32, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Ohya, T.; Maki, S.; Kawasaki, Y.; Sugino, A. Structure and function of the fourth subunit (Dpb4p) of DNA polymerase epsilon in Saccharomyces cerevisiae. Nucleic Acids Res. 2000, 28, 3846–3852. [Google Scholar] [CrossRef] [PubMed]
- Iida, T.; Araki, H. Noncompetitive counteractions of DNA polymerase epsilon and ISW2/yCHRAC for epigenetic inheritance of telomere position effect in Saccharomyces cerevisiae. Mol. Cell. Biol. 2004, 24, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Long, C.; Chen, X.; Huang, C.; Chen, S.; Zhu, B. Partitioning of Histone H3-H4 Tetramers During DNA Replication-Dependent Chromatin Assembly. Science 2010, 328, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Katan-Khaykovich, Y.; Struhl, K. Splitting of H3-H4 tetramers at transcriptionally active genes undergoing dynamic histone exchange. Proc. Natl. Acad. Sci. USA 2011, 108, 1296–1301. [Google Scholar] [CrossRef] [PubMed]
- Ishimi, Y.; Komamura, Y.; You, Z.; Kimura, H. Biochemical function of mouse minichromosomemaintenance 2 protein. J. Biol. Chem. 1998, 273, 8369–8375. [Google Scholar] [CrossRef] [PubMed]
- Foltman, M.; Evrin, C.; De Piccoli, G.; Jones, R.C.; Edmondson, R.D.; Katou, Y.; Nakato, R.; Shirahige, K.; Labib, K. Eukaryotic Replisome Components Cooperate to Process Histones During Chromosome Replication. Cell Rep. 2013, 3, 892–904. [Google Scholar] [CrossRef] [PubMed]
- Richet, N.; Liu, D.; Legrand, P.; Velours, C.; Corpet, A.; Gaubert, A.; Bakail, M.; Moal-Raisin, G.; Guerois, R.; Compper, C.; et al. Structural insight into how the human helicase subunit MCM2 may act as a histone chaperone together with ASF1 at the replication fork. Nucleic Acids Res. 2015, 43, 1905–1917. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Stromme, C.B.; Saredi, G.; Hodl, M.; Strandsby, A.; Gonzalez-Aguilera, C.; Chen, S.; Groth, A.; Patel, D.J. A unique binding mode enables MCM2 to chaperone histones H3-H4 at replication forks. Nat. Struct. Mol. Biol. 2015, 22, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Formosa, T. The role of FACT in making and breaking nucleosomes. Biochim. Biophys. Acta 2012, 1819, 247–255. [Google Scholar] [CrossRef] [PubMed]
- VanDemark, A.P.; Blanksma, M.; Ferris, E.; Heroux, A.; Hill, C.P.; Formosa, T. The Structure of the yFACT Pob3-M Domain, Its Interaction with the DNA Replication Factor RPA, and a Potential Role in Nucleosome Deposition. Mol. Cell 2006, 22, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Wittmeyer, J.; Formosa, T. The Saccharomyces cerevisiae DNA polymerase alpha catalytic subunit interacts with Cdc68/Spt16 and with Pob3, a protein similar to an HMG1-like protein. Mol. Cell. Biol. 1997, 17, 4178–4190. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.C.-M.; Chien, C.-T.; Hirose, S.; Lee, S.-C. Functional cooperation between FACT and MCM helicase facilitates initiation of chromatin DNA replication. EMBO J. 2006, 25, 3975–3985. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Li, Q.; McCullough, L.; Kettelkamp, C.; Formosa, T.; Zhang, Z. Ubiquitylation of FACT by the Cullin-E3 ligase Rtt101 connects FACT to DNA replication. Genes Dev. 2010, 24, 1485–1490. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, M.B.; Formosa, T. POB3 is required for both transcription and replication in the yeast Saccharomyces cerevisiae. Genetics 2000, 155, 1593–1606. [Google Scholar] [PubMed]
- Le, S.; Davis, C.; Konopka, J.B.; Sternglanz, R. Two new S-phase-specific genes from Saccharomyces cerevisiae. Yeast 1997, 13, 1029–1042. [Google Scholar] [CrossRef]
- Tyler, J.K.; Adams, C.R.; Chen, S.R.; Kobayashi, R.; Kamakaka, R.T.; Kadonaga, J.T. The RCAF complex mediates chromatin assembly during DNA replication and repair. Nature 1999, 402, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Mousson, F.; Ochsenbein, F.; Mann, C. The histone chaperone Asf1 at the crossroads of chromatin and DNA checkpoint pathways. Chromosoma 2006, 116, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Schulz, L.L.; Tyler, J.K. The histone chaperone ASF1 localizes to active DNA replication forks to mediate efficient DNA replication. FASEB J. 2006, 20, 488–490. [Google Scholar] [CrossRef] [PubMed]
- Franco, A.A.; Lam, W.M.; Burgers, P.M.; Kaufman, P.D. Histone deposition protein Asf1 maintains DNA replisome integrity and interacts with replication factor C. Genes Dev. 2005, 19, 1365–1375. [Google Scholar] [CrossRef] [PubMed]
- Groth, A.; Corpet, A.; Cook, A.J.L.; Roche, D.; Bartek, J.; Lukas, J.; Almouzni, G. Regulation of Replication Fork Progression Through Histone Supply and Demand. Science 2007, 318, 1928–1931. [Google Scholar] [CrossRef] [PubMed]
- Natsume, R.; Eitoku, M.; Akai, Y.; Sano, N.; Horikoshi, M.; Senda, T. Structure and function of the histone chaperone CIA/ASF1 complexed with histones H3 and H4. Nature 2007, 446, 338–341. [Google Scholar] [CrossRef] [PubMed]
- English, C.M.; Adkins, M.W.; Carson, J.J.; Churchill, M.E.A.; Tyler, J.K. Structural Basis for the Histone Chaperone Activity of Asf1. Cell 2006, 127, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Clement, C.; Almouzni, G. MCM2 binding to histones H3-H4 and ASF1 supports a tetramer-to-dimer model for histone inheritance at the replication fork. Nat. Struct. Mol. Biol. 2015, 22, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Sanematsu, F.; Takami, Y.; Barman, H.K.; Fukagawa, T.; Ono, T.; Shibahara, K.-I.; Nakayama, T. Asf1 is required for viability and chromatin assembly during DNA replication in vertebrate cells. J. Biol. Chem. 2006, 281, 13817–13827. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Ohsumi, T.; Tada, S.; Natsume, R.; Kundu, L.R.; Nozaki, N.; Senda, T.; Enomoto, T.; Horikoshi, M.; Seki, M. Roles of histone chaperone CIA/Asf1 in nascent DNA elongation during nucleosome replication. Genes Cells 2011, 16, 1050–1062. [Google Scholar] [CrossRef] [PubMed]
- Osley, M.A. The regulation of histone synthesis in the cell cycle. Annu. Rev. Biochem. 1991, 60, 827–861. [Google Scholar] [CrossRef] [PubMed]
- Marzluff, W.F.; Duronio, R.J. Histone mRNA expression: Multiple levels of cell cycle regulation and important developmental consequences. Curr. Opin. Cell Biol. 2002, 14, 692–699. [Google Scholar] [CrossRef]
- Maya, D.; Morillo-Huesca, M.; Delgado, L.; Chavez, S.; Munoz-Centeno, M.-C. A Histone Cycle. In The Mechanisms of DNA Replication; Stuart, D., Ed.; InTech: Rijeka, Croatia, 2013. [Google Scholar]
- Singh, R.K.; Liang, D.; Gajjalaiahvari, U.R.; Kabbaj, M.-H.M.; Paik, J.; Gunjan, A. Excess histone levels mediate cytotoxicity via multiple mechanisms. Cell Cycle 2010, 9, 4236–4244. [Google Scholar] [CrossRef] [PubMed]
- Ling, X.; Harkness, T.A.; Schultz, M.C.; Fisher-Adams, G.; Grunstein, M. Yeast histone H3 and H4 amino termini are important for nucleosome assembly in vivo and in vitro: Redundant and position-independent functions in assembly but not in gene regulation. Genes Dev. 1996, 10, 686–699. [Google Scholar] [CrossRef] [PubMed]
- Ai, X.; Parthun, M.R. The nuclear Hat1p/Hat2p complex: A molecular link between type B histone acetyltransferases and chromatin assembly. Mol. Cell 2004, 14, 195–205. [Google Scholar] [CrossRef]
- Sobel, R.E.; Cook, R.G.; Perry, C.A.; Annunziato, A.T.; Allis, C.D. Conservation of deposition-related acetylation sites in newly synthesized histones H3 and H4. Proc. Natl. Acad. Sci. USA 1995, 92, 1237–1241. [Google Scholar] [CrossRef] [PubMed]
- Fillingham, J.; Recht, J.; Silva, A.C.; Suter, B.; Emili, A.; Stagljar, I.; Krogan, N.J.; Allis, C.D.; Keogh, M.C.; Greenblatt, J.F. Chaperone Control of the Activity and Specificity of the Histone H3 Acetyltransferase Rtt109. Mol. Cell. Biol. 2008, 28, 4342–4353. [Google Scholar] [CrossRef] [PubMed]
- Burgess, R.J.; Zhou, H.; Han, J.; Zhang, Z. A Role for Gcn5 in Replication-Coupled Nucleosome Assembly. Mol. Cell 2010, 37, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Ai, X.; Eugeni, E.E.; Zhang, L.; Carpenter, L.R.; Jelinek, M.A.; Freitas, M.A.; Parthun, M.R. Histone H4 Lysine 91 Acetylation. Mol. Cell 2005, 18, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Zhou, H.; Horazdovsky, B.; Zhang, K.; Xu, R.M.; Zhang, Z. Rtt109 Acetylates Histone H3 Lysine 56 and Functions in DNA Replication. Science 2007, 315, 653–655. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, R.; Hudson, A.; Jackson, S.P. Yeast Rtt109 promotes genome stability by acetylating histone H3 on lysine 56. Science 2007, 315, 649–652. [Google Scholar] [CrossRef] [PubMed]
- Masumoto, H.; Hawke, D.; Kobayashi, R.; Verreault, A. A role for cell-cycle-regulated histone H3 lysine 56 acetylation in the DNA damage response. Nature 2005, 436, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Tsubota, T.; Berndsen, C.E.; Erkmann, J.A.; Smith, C.L.; Yang, L.; Freitas, M.A.; Denu, J.M.; Kaufman, P.D. Histone H3-K56 Acetylation Is Catalyzed by Histone Chaperone-Dependent Complexes. Mol. Cell 2007, 25, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhou, H.; Wurtele, H.; Davies, B.; Horazdovsky, B.; Verreault, A.; Zhang, Z. Acetylation of Histone H3 Lysine 56 Regulates Replication-Coupled Nucleosome Assembly. Cell 2008, 134, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Recht, J.; Tsubota, T.; Tanny, J.C.; Diaz, R.L.; Berger, J.M.; Zhang, X.; Garcia, B.A.; Shabanowitz, J.; Burlingame, A.L.; Hunt, D.F.; et al. Histone chaperone Asf1 is required for histone H3 lysine 56 acetylation, a modification associated with S phase in mitosis and meiosis. Proc. Natl. Acad. Sci. USA 2006, 103, 6988–6993. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Zhou, H.; Li, Z.; Xu, R.-M.; Zhang, Z. Acetylation of lysine 56 of histone H3 catalyzed by RTT109 and regulated by ASF1 is required for replisome integrity. J. Biol. Chem. 2007, 282, 28587–28596. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Zhang, H.; Zhang, H.; Wang, Z.; Zhou, H.; Zhang, Z. A Cul4 E3 ubiquitin ligase regulates histone hand-off during nucleosome assembly. Cell 2013, 155, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Buser, R.; Kellner, V.; Melnik, A.; Wilson-Zbinden, C.; Schellhaas, R.; Kastner, L.; Piwko, W.; Dees, M.; Picotti, P.; Maric, M.; et al. The Replisome-Coupled E3 Ubiquitin Ligase Rtt101Mms22 Counteracts Mrc1 Function to Tolerate Genotoxic Stress. PLoS Genet. 2016, 12, e1005843. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, P.D.; Kobayashi, R.; Stillman, B. Ultraviolet radiation sensitivity and reduction of telomeric silencing in Saccharomyces cerevisiae cells lacking chromatin assembly factor-I. Genes Dev. 1997, 11, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.; Stillman, B. Purification and characterization of CAF-I, a human cell factor required for chromatin assembly during DNA replication in vitro. Cell 1989, 58, 15–25. [Google Scholar] [CrossRef]
- Smith, S.; Stillman, B. Stepwise assembly of chromatin during DNA replication in vitro. EMBO J. 1991, 10, 971–980. [Google Scholar] [PubMed]
- Shibahara, K.; Verreault, A.; Stillman, B. The N-terminal domains of histones H3 and H4 are not necessary for chromatin assembly factor-1-mediated nucleosome assembly onto replicated DNA in vitro. Proc. Natl. Acad. Sci. USA 2000, 97, 7766–7771. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, P.D.; Kobayashi, R.; Kessler, N.; Stillman, B. The p150 and p60 subunits of chromatin assembly factor I: A molecular link between newly synthesized histones and DNA replication. Cell 1995, 81, 1105–1114. [Google Scholar] [CrossRef]
- Shibahara, K.; Stillman, B. Replication-dependent marking of DNA by PCNA facilitates CAF-1-coupled inheritance of chromatin. Cell 1999, 96, 575–585. [Google Scholar] [CrossRef]
- Moggs, J.G.; Grandi, P.; Quivy, J.P.; Jónsson, Z.O.; Hübscher, U.; Becker, P.B.; Almouzni, G. A CAF-1-PCNA-mediated chromatin assembly pathway triggered by sensing DNA damage. Mol. Cell. Biol. 2000, 20, 1206–1218. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Shibahara, K.; Stillman, B. PCNA connects DNA replication to epigenetic inheritance in yeast. Nature 2000, 408, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Tagami, H.; Ray-Gallet, D.; Almouzni, G.; Nakatani, Y. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell 2004, 116, 51–61. [Google Scholar] [CrossRef]
- Tyler, J.K.; Collins, K.A.; Prasad-Sinha, J.; Amiott, E.; Bulger, M.; Harte, P.J.; Kobayashi, R.; Kadonaga, J.T. Interaction between the Drosophila CAF-1 and ASF1 chromatin assembly factors. Mol. Cell. Biol. 2001, 21, 6574–6584. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.H.; Roemer, S.C.; Port, A.M.; Churchill, M.E.A. CAF-1-induced oligomerization of histones H3/H4 and mutually exclusive interactions with Asf1 guide H3/H4 transitions among histone chaperones and DNA. Nucleic Acids Res. 2012, 40, 11229–11239. [Google Scholar] [CrossRef] [PubMed]
- Quivy, J.P.; Grandi, P.; Almouzni, G. Dimerization of the largest subunit of chromatin assembly factor 1: Importance in vitro and during Xenopus early development. EMBO J. 2001, 20, 2015–2027. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.H.; Roemer, S.C.; Zhou, Y.; Shen, Z.-J.; Dennehey, B.K.; Balsbaugh, J.L.; Liddle, J.C.; Nemkov, T.; Ahn, N.G.; Hansen, K.C.; et al. The Cac1 subunit of histone chaperone CAF-1 organizes CAF-1-H3/H4 architecture and tetramerizes histones. eLife 2016, 5, e18023. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Zhou, H.; Katzmann, D.; Hochstrasser, M.; Atanasova, E.; Zhang, Z. Rtt106p is a histone chaperone involved in heterochromatin-mediated silencing. Proc. Natl. Acad. Sci. USA 2005, 102, 13410–13415. [Google Scholar] [CrossRef] [PubMed]
- Su, D.; Hu, Q.; Li, Q.; Thompson, J.R.; Cui, G.; Fazly, A.; Davies, B.A.; Botuyan, M.V.; Zhang, Z.; Mer, G. Structural basis for recognition of H3K56-acetylated histone H3-H4 by the chaperone Rtt106. Nature 2012, 482, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Fazly, A.; Li, Q.; Hu, Q.; Mer, G.; Horazdovsky, B.; Zhang, Z. Histone chaperone Rtt106 promotes nucleosome formation using (H3-H4)2 tetramers. J. Biol. Chem. 2012, 287, 10753–10760. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, X.; Feng, J.; Leng, H.; Li, S.; Xiao, J.; Liu, S.; Xu, Z.; Xu, J.; Di, L.; et al. The Histone Chaperone FACT Contributes to DNA Replication-Coupled Nucleosome Assembly. Cell Rep. 2016, 14, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, K.M.; Osley, M.A. A Role for H2B Ubiquitylation in DNA Replication. Mol. Cell 2012, 48, 734–746. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, L.; Panier, S.; Wildenhain, J.; Tkach, J.M.; Al-Hakim, A.; Landry, M.-C.; Escribano-Diaz, C.; Szilard, R.K.; Young, J.T.F.; Munro, M.; et al. The MMS22L-TONSL Complex Mediates Recovery from Replication Stress and Homologous Recombination. Mol. Cell 2010, 40, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Duro, E.; Lundin, C.; Ask, K.; Sanchez-Pulido, L.; MacArtney, T.J.; Toth, R.; Ponting, C.P.; Groth, A.; Helleday, T.; Rouse, J. Identification of the MMS22L-TONSL Complex that Promotes Homologous Recombination. Mol. Cell 2010, 40, 632–644. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, B.C.; Adamson, B.; Lydeard, J.R.; Sowa, M.E.; Ciccia, A.; Bredemeyer, A.L.; Schlabach, M.; Gygi, S.P.; Elledge, S.J.; Harper, J.W. A Genome-wide Camptothecin Sensitivity Screen Identifies a Mammalian MMS22L-NFKBIL2 Complex Required for Genomic Stability. Mol. Cell 2010, 40, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Piwko, W.; Olma, M.H.; Held, M.; Bianco, J.N.; Pedrioli, P.G.A.; Hofmann, K.; Pasero, P.; Gerlich, D.W.; Peter, M. RNAi-based screening identifies the Mms22L-Nfkbil2 complex as a novel regulator of DNA replication in human cells. EMBO J. 2010, 29, 4210–4222. [Google Scholar] [CrossRef] [PubMed]
- Saredi, G.; Huang, H.; Hammond, C.M.; Alabert, C.; Bekker-Jensen, S.; Forne, I.; Reverón-Gómez, N.; Foster, B.M.; Mlejnkova, L.; Bartke, T.; et al. H4K20me0 marks post-replicative chromatin and recruits the TONSL–MMS22L DNA repair complex. Nature 2016, 534, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Ray-Gallet, D.; Woolfe, A.; Vassias, I.; Pellentz, C.; Lacoste, N.; Puri, A.; Schultz, D.C.; Pchelintsev, N.A.; Adams, P.D.; Jansen, L.E.T.; et al. Dynamics of histone H3 deposition in vivo reveal a nucleosome gap-filling mechanism for H3.3 to maintain chromatin integrity. Mol. Cell 2011, 44, 928–941. [Google Scholar] [CrossRef] [PubMed]
- Hoek, M.; Stillman, B. Chromatin assembly factor 1 is essential and couples chromatin assembly to DNA replication in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 12183–12188. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Franco, A.A.; Santos, H.; Nelson, D.M.; Kaufman, P.D.; Adams, P.D. Defective S phase chromatin assembly causes DNA damage, activation of the S phase checkpoint, and S phase arrest. Mol. Cell 2003, 11, 341–351. [Google Scholar] [CrossRef]
- Nelson, D.M.; Ye, X.; Hall, C.; Santos, H.; Ma, T.; Kao, G.D.; Yen, T.J.; Harper, J.W.; Adams, P.D. Coupling of DNA synthesis and histone synthesis in S phase independent of cyclin/cdk2 activity. Mol. Cell. Biol. 2002, 22, 7459–7472. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; McKillop-Smith, S.; Müller, B. The human histone gene expression regulator HBP/SLBP is required for histone and DNA synthesis, cell cycle progression and cell proliferation in mitotic cells. J. Cell Sci. 2004, 117, 6043–6051. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.J.; Berkow, A.; Marzluff, W.F. Expression of an RNAi-resistant SLBP restores proper S-phase progression. Biochem. Soc. Trans 2005, 33, 471–473. [Google Scholar] [CrossRef] [PubMed]
- Barcaroli, D.; Bongiorno-Borbone, L.; Terrinoni, A.; Hofmann, T.G.; Rossi, M.; Knight, R.A.; Matera, A.G.; Melino, G.; De Laurenzi, V. FLASH is required for histone transcription and S-phase progression. Proc. Natl. Acad. Sci. USA 2006, 103, 14808–14812. [Google Scholar] [CrossRef] [PubMed]
- Mejlvang, J.; Feng, Y.; Alabert, C.; Neelsen, K.J.; Jasencakova, Z.; Zhao, X.; Lees, M.; Sandelin, A.; Pasero, P.; Lopes, M.; et al. New histone supply regulates replication fork speed and PCNA unloading. J. Cell Biol. 2014, 204, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Ghule, P.N.; Xie, R.L.; Medina, R.; Colby, J.L.; Jones, S.N.; Lian, J.B.; Stein, J.L.; van Wijnen, A.J.; Stein, G.S. Fidelity of Histone Gene Regulation Is Obligatory for Genome Replication and Stability. Mol. Cell. Biol. 2014, 34, 2650–2659. [Google Scholar] [CrossRef] [PubMed]
- Günesdogan, U.; Jäckle, H.; Herzig, A. Histone supply regulates S phase timing and cell cycle progression. eLife 2014, 3, e02443. [Google Scholar] [CrossRef] [PubMed]
- Ask, K.; Jasencakova, Z.; Menard, P.; Feng, Y.; Almouzni, G.E.V.; Groth, A. Codanin-1, mutated in the anaemic disease CDAI, regulates Asf1 function in S-phase histone supply. EMBO J. 2012, 31, 2013–2023. [Google Scholar] [CrossRef] [PubMed]
- Klimovskaia, I.M.; Young, C.; Strømme, C.B.; Menard, P.; Jasencakova, Z.; Mejlvang, J.; Ask, K.; Ploug, M.; Nielsen, M.L.; Jensen, O.N.; et al. Tousled-like kinases phosphorylate Asf1 to promote histone supply during DNA replication. Nat. Comm. 2014, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Clemente-Ruiz, M.; Prado, F. Chromatin assembly controls replication fork stability. EMBO Rep. 2009, 10, 790–796. [Google Scholar] [CrossRef] [PubMed]
- Clemente-Ruiz, M.; González-Prieto, R.; Prado, F. Histone H3K56 acetylation, CAF1, and Rtt106 coordinate nucleosome assembly and stability of advancing replication forks. PLoS Genet. 2011, 7, e1002376. [Google Scholar] [CrossRef] [PubMed]
- Kats, E.S.; Albuquerque, C.P.; Zhou, H.; Kolodner, R.D. Checkpoint functions are required for normal S-phase progression in Saccharomyces cerevisiae RCAF- and CAF-I-defective mutants. Proc. Natl. Acad. Sci. USA 2006, 103, 3710–3715. [Google Scholar] [CrossRef] [PubMed]
- Prado, F.; Aguilera, A. Partial depletion of histone H4 increases homologous recombination-mediated genetic instability. Mol. Cell. Biol. 2005, 25, 1526–1536. [Google Scholar] [CrossRef] [PubMed]
- Prado, F.; Cortés-Ledesma, F.; Aguilera, A. The absence of the yeast chromatin assembly factor Asf1 increases genomic instability and sister chromatid exchange. EMBO Rep. 2004, 5, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Myung, K.; Pennaneach, V.; Kats, E.S.; Kolodner, R.D. Saccharomyces cerevisiae chromatin-assembly factors that act during DNA replication function in the maintenance of genome stability. Proc. Natl. Acad. Sci. USA 2003, 100, 6640–6645. [Google Scholar] [CrossRef] [PubMed]
- Polo, S.E.; Almouzni, G. Chromatin dynamics after DNA damage: The legacy of the access-repair–restore model. DNA Repair 2015, 36, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Dahlin, J.L.; Chen, X.; Walters, M.A.; Zhang, Z. Histone-modifying enzymes, histone modifications and histone chaperones in nucleosome assembly: Lessons learned from Rtt109 histone acetyltransferases. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 31–53. [Google Scholar] [CrossRef] [PubMed]
- Cabello-Lobato, M. J.; Prado, F.; Centro Andaluz de Biología Molecular y Medicina Regenerativa (CABIMER/CSIC), Seville, Spain. Personal communication, 2017.
- Smith, D.J.; Whitehouse, I. Intrinsic coupling of lagging-strand synthesis to chromatin assembly. Nature 2012, 483, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Kolodner, R.D. Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants. Nat. Genet. 1999, 23, 81–85. [Google Scholar] [PubMed]
- Tishkoff, D.X.; Filosi, N.; Gaida, G.M.; Kolodner, R.D. A novel mutation avoidance mechanism dependent on S. cerevisiae RAD27 is distinct from DNA mismatch repair. Cell 1997, 88, 253–263. [Google Scholar] [CrossRef]
- Prado, F.; Clemente-Ruiz, M. Nucleosome assembly and genome integrity. BioArchitecture 2012, 2, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Yadav, T.; Whitehouse, I. Replication-Coupled Nucleosome Assembly and Positioning by ATP-Dependent Chromatin- Remodeling Enzymes. Cell Rep. 2016, 15, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Freudenreich, C.H. The Rtt109 histone acetyltransferase facilitates error-free replication to prevent CAG/CTG repeat contractions. DNA Repair 2010, 9, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Houseley, J.; Tollervey, D. Repeat expansion in the budding yeast ribosomal DNA can occur independently of the canonical homologous recombination machinery. Nucleic Acids Res. 2011, 39, 8778–8791. [Google Scholar] [CrossRef] [PubMed]
- Ramey, C.J.; Howar, S.; Adkins, M.; Linger, J.; Spicer, J.; Tyler, J.K. Activation of the DNA damage checkpoint in yeast lacking the histone chaperone anti-silencing function 1. Mol. Cell. Biol. 2004, 24, 10313–10327. [Google Scholar] [CrossRef] [PubMed]
- Hashash, N.; Johnson, A.L.; Cha, R.S. Topoisomerase II- and Condensin-Dependent Breakage of MEC1ATR-Sensitive Fragile Sites Occurs Independently of Spindle Tension, Anaphase, or Cytokinesis. PLoS Genet. 2012, 8, e1002978. [Google Scholar] [CrossRef] [PubMed]
- El Achkar, E.; Gerbault-Seureau, M.; Muleris, M.; Dutrillaux, B.; Debatisse, M. Premature condensation induces breaks at the interface of early and late replicating chromosome bands bearing common fragile sites. Proc. Natl. Acad. Sci. USA 2005, 102, 18069–18074. [Google Scholar] [CrossRef]
- Ivanov, D.; Schleiffer, A.; Eisenhaber, F.; Mechtler, K.; Haering, C.H.; Nasmyth, K. Eco1 is a novel acetyltransferase that can acetylate proteins involved in cohesion. Curr. Biol. 2002, 12, 323–328. [Google Scholar] [CrossRef]
- Lopez-Serra, L.; Kelly, G.; Patel, H.; Stewart, A.; Uhlmann, F. The Scc2–Scc4 complex acts in sister chromatid cohesion and transcriptional regulation by maintaining nucleosome-free regions. Nat. Genet. 2014, 46, 1147–1151. [Google Scholar] [CrossRef] [PubMed]
- Murillo-Pineda, M.; Cabello-Lobato, M.J.; Clemente-Ruiz, M.; Monje-Casas, F.; Prado, F. Defective histone supply causes condensin-dependent chromatin alterations, SAC activation and chromosome decatenation impairment. Nucleic Acids Res. 2014, 42, 12469–12482. [Google Scholar] [CrossRef] [PubMed]
- Thaminy, S.; Newcomb, B.; Kim, J.; Gatbonton, T.; Foss, E.; Simon, J.; Bedalov, A. Hst3 is regulated by Mec1-dependent proteolysis and controls the S phase checkpoint and sister chromatid cohesion by deacetylating histone H3 at lysine 56. J. Biol. Chem. 2007, 282, 37805–37814. [Google Scholar] [CrossRef] [PubMed]
- Prado, F. Homologous recombination maintenance of genome integrity during DNA damage tolerance. Mol. Cell. Oncol. 2014, 1, e957039. [Google Scholar] [CrossRef] [PubMed]
- Sale, J.E.; Lehmann, A.R.; Woodgate, R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol. 2012, 13, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Minca, E.C.; Kowalski, D. Multiple Rad5 Activities Mediate Sister Chromatid Recombination to Bypass DNA Damage at Stalled Replication Forks. Mol. Cell 2010, 38, 649–661. [Google Scholar] [CrossRef] [PubMed]
- González-Prieto, R.; Muñoz-Cabello, A.M.; Cabello-Lobato, M.J.; Prado, F. Rad51 replication fork recruitment is required for DNA damage tolerance. EMBO J. 2013, 32, 1307–1321. [Google Scholar] [CrossRef] [PubMed]
- Vázquez, M.V.; Rojas, V.; Tercero, J.A. Multiple pathways cooperate to facilitate DNA replication fork progression through alkylated DNA. DNA Repair 2008, 7, 1693–1704. [Google Scholar] [CrossRef] [PubMed]
- Alabert, C.; Bianco, J.N.; Pasero, P. Differential regulation of homologous recombination at DNA breaks and replication forks by the Mrc1 branch of the S-phase checkpoint. EMBO J. 2009, 28, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Henry-Mowatt, J.; Jackson, D.; Masson, J.-Y.; Johnson, P.A.; Clements, P.M.; Benson, F.E.; Thompson, L.H.; Takeda, S.; West, S.C.; Caldecott, K.W. XRCC3 and Rad51 modulate replication fork progression on damaged vertebrate chromosomes. Mol. Cell 2003, 11, 1109–1117. [Google Scholar] [CrossRef]
- Daboussi, F.; Courbet, S.; Benhamou, S.; Kannouche, P.; Zdzienicka, M.Z.; Debatisse, M.; Lopez, B.S. A homologous recombination defect affects replication-fork progression in mammalian cells. J. Cell Sci. 2008, 121, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Maas, N.L.; Miller, K.M.; DeFazio, L.G.; Toczyski, D.P. Cell Cycle and Checkpoint Regulation of Histone H3 K56 Acetylation by Hst3 and Hst4. Mol. Cell 2006, 23, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Celic, I.; Masumoto, H.; Griffith, W.P.; Meluh, P.; Cotter, R.J.; Boeke, J.D.; Verreault, A. The Sirtuins Hst3 and Hst4p Preserve Genome Integrity by Controlling Histone H3 Lysine 56 Deacetylation. Curr. Biol. 2006, 16, 1280–1289. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.R.; Miller, K.M.; Maas, N.L.; Roguev, A.; Fillingham, J.; Chu, C.S.; Schuldiner, M.; Gebbia, M.; Recht, J.; Shales, M.; et al. Functional dissection of protein complexes involved in yeast chromosome biology using a genetic interaction map. Nature 2007, 446, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Wurtele, H.; Kaiser, G.S.; Bacal, J.; St-Hilaire, E.; Lee, E.H.; Tsao, S.; Dorn, J.; Maddox, P.; Lisby, M.; Pasero, P.; et al. Histone H3 Lysine 56 Acetylation and the Response to DNA Replication Fork Damage. Mol. Cell. Biol. 2011, 32, 154–172. [Google Scholar] [CrossRef] [PubMed]
- Luke, B.; Versini, G.; Jaquenoud, M.; Zaidi, I.W.; Kurz, T.; Pintard, L.; Pasero, P.; Peter, M. The Cullin Rtt101p Promotes Replication Fork Progression through Damaged DNA and Natural Pause Sites. Curr. Biol. 2006, 16, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, I.W.; Rabut, G.; Poveda, A.; Scheel, H.; Malmström, J.; Ulrich, H.; Hofmann, K.; Pasero, P.; Peter, M.; Luke, B. Rtt101 and Mms1 in budding yeast form a CUL4DDB1-like ubiquitin ligase that promotes replication through damaged DNA. EMBO Rep. 2008, 9, 1034–1040. [Google Scholar] [CrossRef] [PubMed]
- Luciano, P.; Dehé, P.-M.; Audebert, S.; Géli, V.; Corda, Y. Replisome function during replicative stress is modulated by histone h3 lysine 56 acetylation through Ctf4. Genetics 2015, 199, 1047–1063. [Google Scholar] [CrossRef] [PubMed]
- Ivessa, A.S.; Lenzmeier, B.A.; Bessler, J.B.; Goudsouzian, L.K.; Schnakenberg, S.L.; Zakian, V.A. The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past non histone protein-DNA complexes. Mol. Cell 2003, 12, 1525–1536. [Google Scholar] [CrossRef]
- Erkmann, J.A.; Kaufman, P.D. A negatively charged residue in place of histone H3K56 supports chromatin assembly factor association but not genotoxic stress resistance. DNA Repair 2009, 8, 1371–1379. [Google Scholar] [CrossRef] [PubMed]
- Duro, E.; Vaisica, J.A.; Brown, G.W.; Rouse, J. Budding yeast Mms22 and Mms1 regulate homologous recombination induced by replisome blockage. DNA Repair 2008, 7, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhai, L.; Xu, J.; Joo, H.-Y.; Jackson, S.; Erdjument-Bromage, H.; Tempst, P.; Xiong, Y.; Zhang, Y. Histone H3 and H4 Ubiquitylation by the CUL4-DDB-ROC1 Ubiquitin Ligase Facilitates Cellular Response to DNA Damage. Mol. Cell 2006, 22, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Tjeertes, J.V.; Miller, K.M.; Jackson, S.P. Screen for DNA-damage-responsive histone modifications identifies H3K9Ac and H3K56Ac in human cells. EMBO J. 2009, 28, 1878–1889. [Google Scholar] [CrossRef] [PubMed]
- Pietrobon, V.; Fréon, K.; Hardy, J.; Costes, A.; Iraqui, I.; Ochsenbein, F.; Lambert, S.A.E. The Chromatin Assembly Factor 1 Promotes Rad51-Dependent Template Switches at Replication Forks by Counteracting D-Loop Disassembly by the RecQ-Type Helicase Rqh1. PLoS Biol. 2014, 12, e1001968. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Foiani, M. RecQ helicases queuing with Srs2 to disrupt Rad51 filaments and suppress recombination. Genes Dev. 2007, 21, 3019–3026. [Google Scholar] [CrossRef] [PubMed]
- Jiao, R.; Bachrati, C.Z.; Pedrazzi, G.; Kuster, P.; Petkovic, M.; Li, J.L.; Egli, D.; Hickson, I.D.; Stagljar, I. Physical and Functional Interaction between the Bloom’s Syndrome Gene Product and the Largest Subunit of Chromatin Assembly Factor 1. Mol. Cell. Biol. 2004, 24, 4710–4719. [Google Scholar] [CrossRef] [PubMed]
- Ronan, J.L.; Wu, W.; Crabtree, G.R. From neural development to cognition: Unexpected roles for chromatin. Nat. Rev. Genet. 2013, 14, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.A.; Shilatifard, A. Chromatin signatures of cancer. Genes Dev. 2015, 29, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Prado, F.; Jimeno-Gonzalez, S.; Reyes, J.C. Histone availability as a strategy to control gene expression. RNA Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, R.J.; Karlseder, J. The great unravelling: Chromatin as a modulator of the aging process. Trends Biochem. Sci. 2012, 37, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Gasparian, A.V.; Burkhart, C.A.; Purmal, A.A.; Brodsky, L.; Pal, M.; Saranadasa, M.; Bosykh, D.A.; Commane, M.; Guryanova, O.A.; Pal, S.; et al. Curaxins: Anticancer compounds that simultaneously suppress NF-κB and activate p53 by targeting FACT. Sci. Transl. Med. 2011. [Google Scholar] [CrossRef] [PubMed]
- Polo, S.E.; Theocharis, S.E.; Grandin, L.; Gambotti, L.; Antoni, G.; Savignoni, A.; Asselain, B.; Patsouris, E.; Almouzni, G. Clinical significance and prognostic value of chromatin assembly factor-1 overexpression in human solid tumours. Histopathology 2010, 57, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Corpet, A.; De Koning, L.; Toedling, J.; Savignoni, A.; Berger, F.; Lemaître, C.; O’Sullivan, R.J.; Karlseder, J.; Barillot, E.; Asselain, B.; et al. Asf1b, the necessary Asf1 isoform for proliferation, is predictive of outcome in breast cancer. EMBO J. 2010, 30, 480–493. [Google Scholar] [CrossRef] [PubMed]
- Kerzendorfer, C.; Hannes, F.; Colnaghi, R.; Abramowicz, I.; Carpenter, G.; Vermeesch, J.R.; O’Driscoll, M. Characterizing the functional consequences of haploinsufficiency of NELF-A (WHSC2) and SLBP identifies novel cellular phenotypes in Wolf-Hirschhorn syndrome. Hum. Mol. Genet. 2012, 21, 2181–2193. [Google Scholar] [CrossRef] [PubMed]
- Marzluff, W.F.; Wagner, E.J.; Duronio, R.J. Metabolism and regulation of canonical histone mRNAs: Life without a poly(A) tail. Nat. Rev. Genet. 2008, 9, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Martinez, V.D.; Vucic, E.A.; Becker-Santos, D.D.; Gil, L.; Lam, W.L. Arsenic exposure and the induction of human cancers. J. Toxicol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Brocato, J.; Chen, D.; Liu, J.; Fang, L.; Jin, C.; Costa, M. A Potential New Mechanism of Arsenic Carcinogenesis: Depletion of Stem-Loop Binding Protein and Increase in Polyadenylated Canonical Histone H3.1 mRNA. Biol. Trace Element Res. 2015, 166, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Knipe, D.M.; Lieberman, P.M.; Jung, J.U.; McBride, A.A.; Morris, K.V.; Ott, M.; Margolis, D.; Nieto, A.; Nevels, M.; Parks, R.J.; et al. Snapshots: Chromatin control of viral infection. Virology 2013, 435, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Bogenberger, J.M.; Laybourn, P.J. Human T Lymphotropic Virus Type 1 protein Tax reduces histone levels. Retrovirology 2008. [Google Scholar] [CrossRef] [PubMed]
- Grogg, K.L.; Miller, R.F.; Dogan, A. HIV infection and lymphoma. J. Clin. Pathol. 2007, 60, 1365–1372. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Tucker, L.D.; Asara, J.M.; Cheruiyot, C.K.; Lu, H.; Wu, Z.J.; Newstein, M.C.; Dooner, M.S.; Friedman, J.; Lally, M.A.; et al. Stem-loop binding protein is a multifaceted cellular regulator of HIV-1 replication. J. Clin. Investig. 2016, 126, 3117–3129. [Google Scholar] [CrossRef] [PubMed]
- Dgany, O.; Avidan, N.; Delaunay, J.; Krasnov, T.; Shalmon, L.; Shalev, H.; Eidelitz-Markus, T.; Kapelushnik, J.; Cattan, D.; Pariente, A.; et al. Congenital Dyserythropoietic Anemia Type I Is Caused by Mutations in Codanin-1. Am. J. Hum. Genet. 2002, 71, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Cesare, A.J.; Reddel, R.R. Alternative lengthening of telomeres: Models, mechanisms and implications. Nat. Rev. Genet. 2010, 11, 319–330. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, R.J.; Arnoult, N.; Lackner, D.H.; Oganesian, L.; Haggblom, C.; Corpet, A.; Almouzni, G.; Karlseder, J. Rapid induction of alternative lengthening of telomeres by depletion of the histone chaperone ASF1. Nat. Struct. Mol. Biol. 2014, 21, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Platt, J.M.; Ryvkin, P.; Wanat, J.J.; Donahue, G.; Ricketts, M.D.; Barrett, S.P.; Waters, H.J.; Song, S.; Chavez, A.; Abdallah, K.O.; et al. Rap1 relocalization contributes to the chromatin-mediated gene expression profile and pace of cell senescence. Genes Dev. 2013, 27, 1406–1420. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, R.J.; Kubicek, S.; Schreiber, S.L.; Karlseder, J. Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat. Struct. Mol. Biol. 2010, 17, 1218–1225. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.; Pawlikowski, J.; Manoharan, I.; van Tuyn, J.; Nelson, D.M.; Rai, T.S.; Shah, P.P.; Hewitt, G.; Korolchuk, V.I.; Passos, J.F.; et al. Lysosome-mediated processing of chromatin in senescence. J. Cell Biol. 2013, 202, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Feser, J.; Truong, D.; Das, C.; Carson, J.J.; Kieft, J.; Harkness, T.; Tyler, J.K. Elevated Histone Expression Promotes Life Span Extension. Mol. Cell 2010, 39, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Cheung, T.H.; Charville, G.W.; Hurgo, B.M.C.; Leavitt, T.; Shih, J.; Brunet, A.; Rando, T.A. Chromatin Modifications as Determinants of Muscle Stem Cell Quiescence and Chronological Aging. Cell Rep. 2013, 4, 189–204. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Chen, K.; Xia, Z.; Chavez, M.; Pal, S.; Seol, J.H.; Chen, C.C.; Li, W.; Tyler, J.K. Nucleosome loss leads to global transcriptional up-regulation and genomic instability during yeast aging. Genes Dev. 2014, 28, 396–408. [Google Scholar] [CrossRef] [PubMed]
- Nikolov, I.; Taddei, A. Linking replication stress with heterochromatin formation. Chromosoma 2016, 125, 1–11. [Google Scholar] [CrossRef] [PubMed]
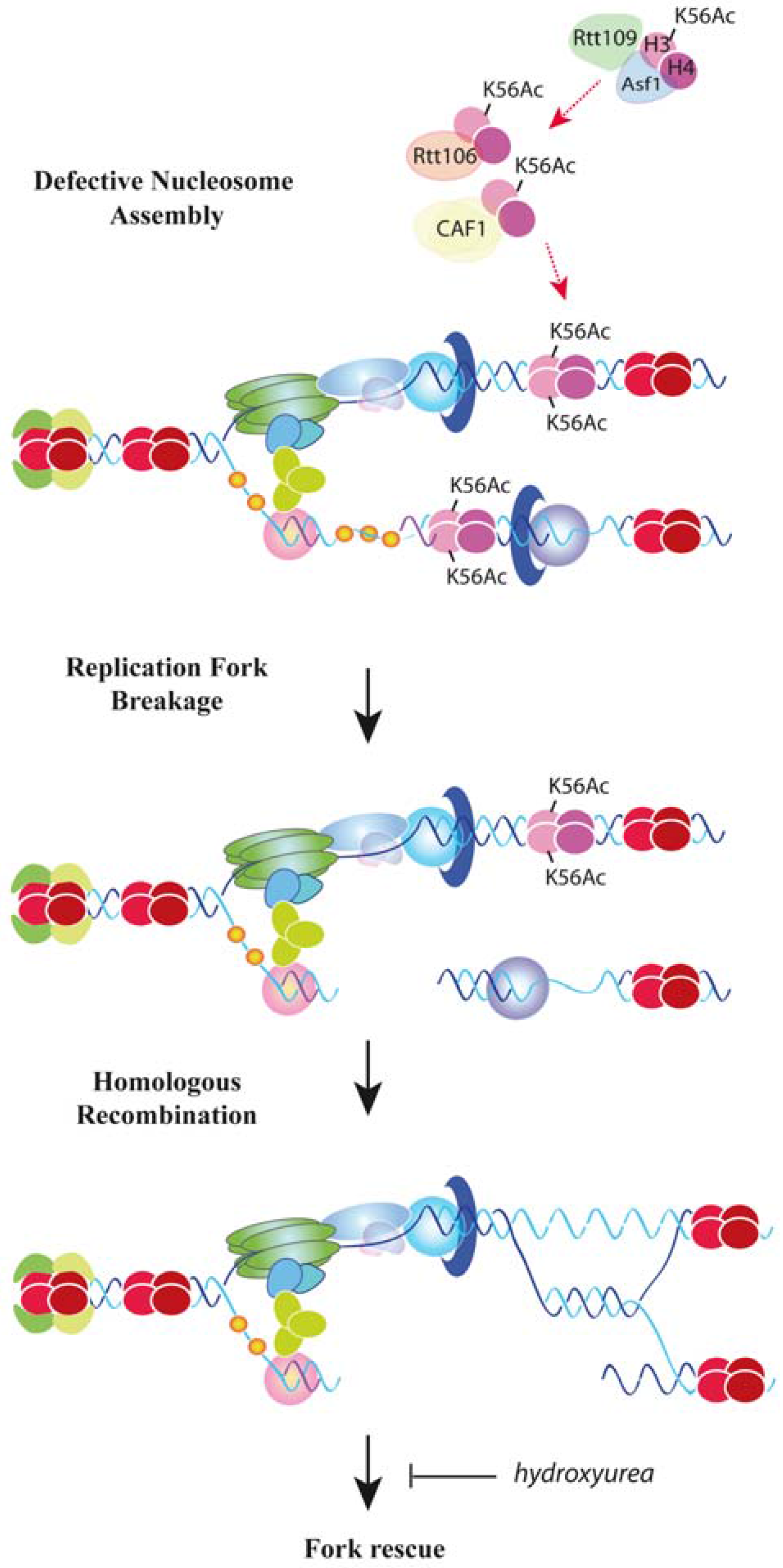
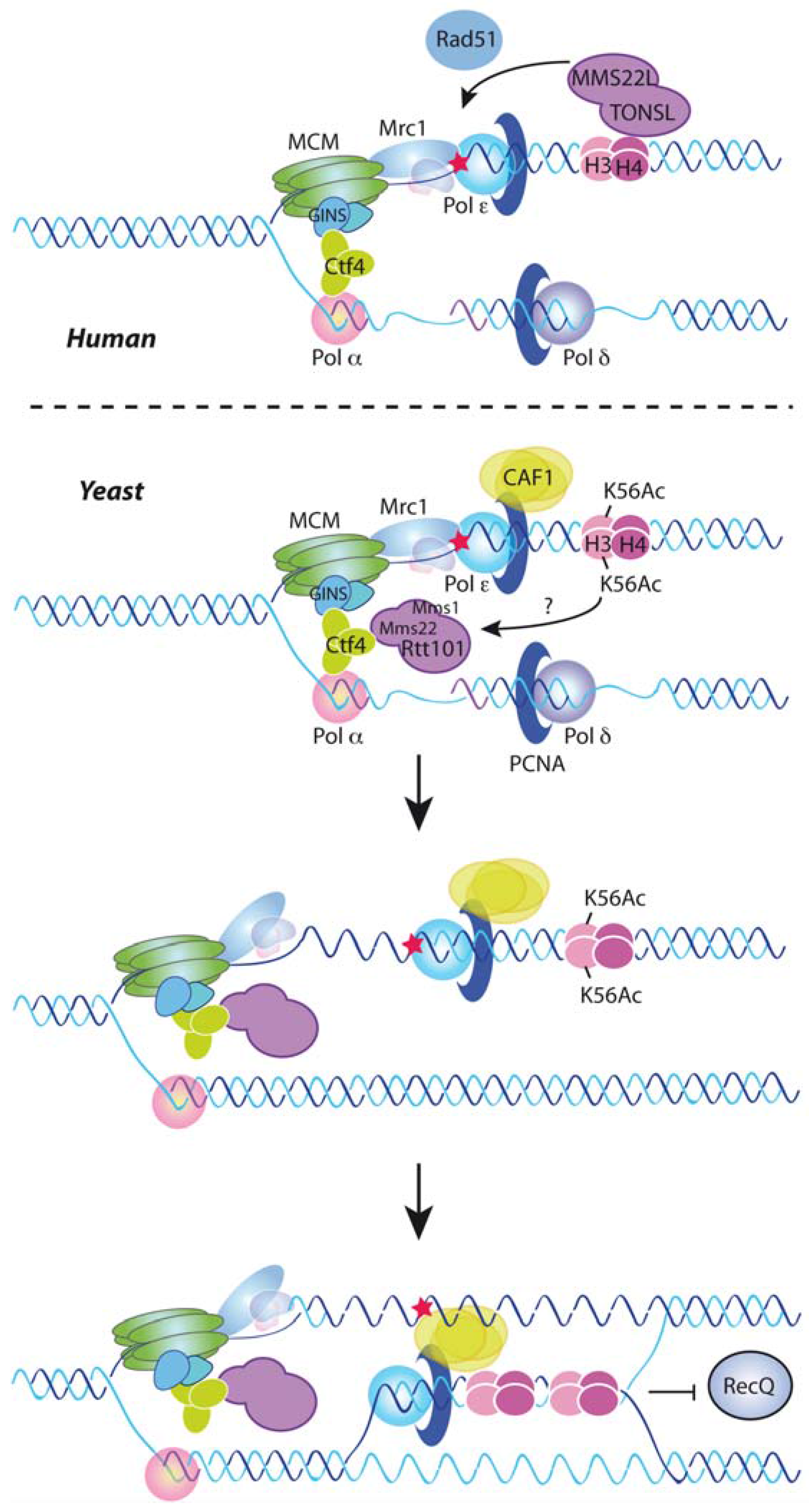
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prado, F.; Maya, D. Regulation of Replication Fork Advance and Stability by Nucleosome Assembly. Genes 2017, 8, 49. https://doi.org/10.3390/genes8020049
Prado F, Maya D. Regulation of Replication Fork Advance and Stability by Nucleosome Assembly. Genes. 2017; 8(2):49. https://doi.org/10.3390/genes8020049
Chicago/Turabian StylePrado, Felix, and Douglas Maya. 2017. "Regulation of Replication Fork Advance and Stability by Nucleosome Assembly" Genes 8, no. 2: 49. https://doi.org/10.3390/genes8020049
APA StylePrado, F., & Maya, D. (2017). Regulation of Replication Fork Advance and Stability by Nucleosome Assembly. Genes, 8(2), 49. https://doi.org/10.3390/genes8020049