
Risks at the DNA Replication Fork: Effects upon Carcinogenesis and Tumor Heterogeneity

Abstract
:1. Introduction
1.1. An Overview of the Eukaryotic DNA Replication Fork
1.2. Mismatch Repair Deficiencies Promote Cancer
1.3. Mutagenic Human Replicative Polymerase Variants Give Rise to Cancer
1.4. Damage to Single-Stranded DNA on the Lagging Strand Template Causes Mutation in Cancer
1.5. Future Directions: Tumor Specific Metabolic Changes as Modulators of Replication Fork-Associated Mutagenesis
2. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Saini, N.; Roberts, S.A.; Klimczak, L.J.; Chan, K.; Grimm, S.A.; Dai, S.; Fargo, D.C.; Boyer, J.C.; Kaufmann, W.K.; Taylor, J.A.; et al. The impact of environmental and endogenous damage on somatic mutation load in human skin fibroblasts. PLoS Genet. 2016, 12, e1006385. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.W.; Charlesworth, B.; Charlesworth, D.; Crow, J.F. Rates of spontaneous mutation. Genetics 1998, 148, 1667–1686. [Google Scholar] [PubMed]
- Herr, A.J.; Ogawa, M.; Lawrence, N.A.; Williams, L.N.; Eggington, J.M.; Singh, M.; Smith, R.A.; Preston, B.D. Mutator suppression and escape from replication error-induced extinction in yeast. PLoS Genet. 2011, 7, e1002282. [Google Scholar] [CrossRef]
- Herr, A.J.; Kennedy, S.R.; Knowels, G.M.; Schultz, E.M.; Preston, B.D. DNA replication error-induced extinction of diploid yeast. Genetics 2014, 196, 677–691. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A. Human cancers express a mutator phenotype: Hypothesis, origin, and consequences. Cancer Res. 2016, 76, 2057–2059. [Google Scholar] [CrossRef] [PubMed]
- Bielas, J.H.; Loeb, K.R.; Rubin, B.P.; True, L.D.; Loeb, L.A. Human cancers express a mutator phenotype. Proc. Natl. Acad. Sci. USA 2006, 103, 18238–18242. [Google Scholar] [CrossRef] [PubMed]
- Theis, J.F.; Newlon, C.S. The ARS309 chromosomal replicator of Saccharomyces cerevisiae depends on an exceptional ars consensus sequence. Proc. Natl. Acad. Sci. USA 1997, 94, 10786–10791. [Google Scholar] [CrossRef] [PubMed]
- Stinchcomb, D.; Struhl, K.; Davis, R. Isolation and characterisation of a yeast chromosomal replicator. Nature 1979, 282, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Nieduszynski, C.A.; Knox, Y.; Donaldson, A.D. Genome-wide identification of replication origins in yeast by comparative genomics. Genes Dev. 2006, 20, 1874–1879. [Google Scholar] [CrossRef] [PubMed]
- Huberman, J.A.; Riggs, A.D. Autoradiography of chromosomal DNA fibers from chinese hamster cells. Proc. Natl. Acad. Sci. USA 1966, 55, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Mechali, M. Eukaryotic DNA replication origins: Many choices for appropriate answers. Nat. Rev. Mol. Cell Biol. 2010, 11, 728–738. [Google Scholar] [CrossRef] [PubMed]
- Takisawa, H.; Mimura, S.; Kubota, Y. Eukaryotic DNA replication: From pre-replication complex to initiation complex. Curr. Opin. Cell Biol. 2000, 12, 690–696. [Google Scholar] [CrossRef]
- Bell, S.P. The origin recognition complex: From simple origins to complex functions. Genes Dev. 2002, 16, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Boos, D.; Frigola, J.; Diffley, J.F. Activation of the replicative DNA helicase: Breaking up is hard to do. Curr. Opin. Cell Biol. 2012, 24, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, L. The pol α-primase complex. In The Eukaryotic Replisome: A Guide to Protein Structure and Function; Springer: New York, NY, USA, 2012; pp. 157–169. [Google Scholar]
- Muzi-Falconi, M.; Giannattasio, M.; Foiani, M.; Plevani, P. The DNA polymerase alpha-primase complex: Multiple functions and interactions. Sci. World J. 2003, 3, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Wold, M.S.; Kelly, T. Purification and characterization of replication protein A, a cellular protein required for in vitro replication of simian virus 40 DNA. Proc. Natl. Acad. Sci. USA 1988, 85, 2523–2527. [Google Scholar] [CrossRef] [PubMed]
- Wobbe, C.R.; Weissbach, L.; Borowiec, J.A.; Dean, F.B.; Murakami, Y.; Bullock, P.; Hurwitz, J. Replication of simian virus 40 origin-containing DNA in vitro with purified proteins. Proc. Natl. Acad. Sci. USA 1987, 84, 1834–1838. [Google Scholar] [CrossRef] [PubMed]
- Fairman, M.P.; Stillman, B. Cellular factors required for multiple stages of SV40 DNA replication in vitro. EMBO J. 1988, 7, 1211–1218. [Google Scholar] [PubMed]
- Brill, S.J.; Stillman, B. Yeast replication factor-A functions in the unwinding of the SV40 origin of DNA replication. Nature 1989, 342, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Fanning, E.; Klimovich, V.; Nager, A.R. A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nuclic Acids Res. 2006, 34, 4126–4137. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.J.; Whitehouse, I. Intrinsic coupling of lagging-strand synthesis to chromatin assembly. Nature 2012, 483, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, G.L.; Pfander, B.; Jentsch, S. PCNA, the maestro of the replication fork. Cell 2007, 129, 665–679. [Google Scholar] [CrossRef] [PubMed]
- Baranovskiy, A.G.; Babayeva, N.D.; Liston, V.G.; Rogozin, I.B.; Koonin, E.V.; Pavlov, Y.I.; Vassylyev, D.G.; Tahirov, T.H. X-ray structure of the complex of regulatory subunits of human DNA polymerase delta. Cell Cycle 2008, 7, 3026–3036. [Google Scholar] [CrossRef] [PubMed]
- Doublie, S.; Zahn, K.E. Structural insights into eukaryotic DNA replication. Front. Microbiol. 2014, 5, 444. [Google Scholar] [CrossRef] [PubMed]
- Hogg, M.; Osterman, P.; Bylund, G.O.; Ganai, R.A.; Lundstrom, E.B.; Sauer-Eriksson, A.E.; Johansson, E. Structural basis for processive DNA synthesis by yeast DNA polymerase varepsilon. Nat. Struct. Mol. Biol. 2014, 21, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Isoz, I.; Persson, U.; Volkov, K.; Johansson, E. The C-terminus of Dpb2 is required for interaction with Pol2 and for cell viability. Nuclic Acids Res. 2012, 40, 11545–11553. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Rajashankar, K.R.; Buku, A.; Johnson, R.E.; Prakash, L.; Prakash, S.; Aggarwal, A.K. Crystal structure of yeast DNA polymerase epsilon catalytic domain. PLoS ONE 2014, 9, e94835. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.H.; Wang, X.; Zhang, S.; Zhang, Z.; Lee, E.Y.; Lee, M.Y. Dynamics of enzymatic interactions during short flap human okazaki fragment processing by two forms of human DNA polymerase delta. DNA Repair 2013, 12, 922–935. [Google Scholar] [CrossRef] [PubMed]
- Swan, M.K.; Johnson, R.E.; Prakash, L.; Prakash, S.; Aggarwal, A.K. Structural basis of high-fidelity DNA synthesis by yeast DNA polymerase delta. Nat. Struct. Mol. Biol. 2009, 16, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Tsurimoto, T.; Stillman, B. Purification of a cellular replication factor, RF-C, that is required for coordinated synthesis of leading and lagging strands during simian virus 40 DNA replication in vitro. Mol. Cell. Biol. 1989, 9, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Shen, B. Okazaki fragment maturation: Nucleases take centre stage. J. Mol. Cell Biol. 2011, 3, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Ellenberger, T.; Tomkinson, A.E. Eukaryotic DNA ligases: Structural and functional insights. Annu. Rev. Biochem. 2008, 77, 313–338. [Google Scholar] [CrossRef] [PubMed]
- Shcherbakova, P.V.; Pavlov, Y.I. 3′→5′ exonucleases of DNA polymerases ϵ and δ correct base analog induced DNA replication errors on opposite DNA strands in Saccharomyces cerevisiae. Genetics 1996, 142, 717–726. [Google Scholar] [PubMed]
- Jin, Y.H.; Obert, R.; Burgers, P.M.; Kunkel, T.A.; Resnick, M.A.; Gordenin, D.A. The 3′→5′ exonuclease of DNA polymerase δ can substitute for the 5′ flap endonuclease Rad27/Fen1 in processing Okazaki fragments and preventing genome instability. Proc. Natl. Acad. Sci. USA 2001, 98, 5122–5127. [Google Scholar] [CrossRef] [PubMed]
- Garg, P.; Stith, C.M.; Sabouri, N.; Johansson, E.; Burgers, P.M. Idling by DNA polymerase delta maintains a ligatable nick during lagging-strand DNA replication. Genes Dev. 2004, 18, 2764–2773. [Google Scholar] [CrossRef] [PubMed]
- Georgescu, R.E.; Langston, L.; Yao, N.Y.; Yurieva, O.; Zhang, D.; Finkelstein, J.; Agarwal, T.; O’Donnell, M.E. Mechanism of asymmetric polymerase assembly at the eukaryotic replication fork. Nat. Struct. Mol. Biol. 2014, 21, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Nick McElhinny, S.A.; Gordenin, D.A.; Stith, C.M.; Burgers, P.M.; Kunkel, T.A. Division of labor at the eukaryotic replication fork. Mol. Cell 2008, 30, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Pursell, Z.F.; Isoz, I.; Lundstrom, E.B.; Johansson, E.; Kunkel, T.A. Yeast DNA polymerase e participates in leading-strand DNA replication. Science 2007, 317, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Lujan, S.A.; Clausen, A.R.; Clark, A.B.; MacAlpine, H.K.; MacAlpine, D.M.; Malc, E.P.; Mieczkowski, P.A.; Burkholder, A.B.; Fargo, D.C.; Gordenin, D.A.; et al. Heterogeneous polymerase fidelity and mismatch repair bias genome variation and composition. Genome Res. 2014, 24, 1751–1764. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, Y.I.; Shcherbakova, P.V. DNA polymerases at the eukaryotic fork-20 years later. Mutat. Res. 2010, 685, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Reijns, M.A.; Kemp, H.; Ding, J.; de Proce, S.M.; Jackson, A.P.; Taylor, M.S. Lagging-strand replication shapes the mutational landscape of the genome. Nature 2015, 518, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Niimi, A.; Limsirichaikul, S.; Yoshida, S.; Iwai, S.; Masutani, C.; Hanaoka, F.; Kool, E.T.; Nishiyama, Y.; Suzuki, M. Palm mutants in DNA polymerases alpha and eta alter DNA replication fidelity and translesion activity. Mol. Cell. Biol. 2004, 24, 2734–2746. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, Y.I.; Frahm, C.; Nick McElhinny, S.A.; Niimi, A.; Suzuki, M.; Kunkel, T.A. Evidence that errors made by DNA polymerase alpha are corrected by DNA polymerase delta. Curr. Biol. 2006, 16, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Shinbrot, E.; Henninger, E.E.; Weinhold, N.; Covington, K.R.; Goksenin, A.Y.; Schultz, N.; Chao, H.; Doddapaneni, H.; Muzny, D.M.; Gibbs, R.A.; et al. Exonuclease mutations in DNA polymerase epsilon reveal replication strand specific mutation patterns and human origins of replication. Genome Res. 2014, 24, 1740–1750. [Google Scholar] [CrossRef] [PubMed]
- Prakash, S.; Johnson, R.E.; Prakash, L. Eukaryotic translesion synthesis DNA polymerases: Specificity of structure and function. Annu. Rev. Biochem. 2005, 74, 317–353. [Google Scholar] [CrossRef] [PubMed]
- Northam, M.R.; Garg, P.; Baitin, D.M.; Burgers, P.M.; Shcherbakova, P.V. A novel function of DNA polymerase ζ regulated by PCNA. EMBO J. 2006, 25, 4316–4325. [Google Scholar] [CrossRef] [PubMed]
- Northam, M.R.; Robinson, H.A.; Kochenova, O.V.; Shcherbakova, P.V. Participation of DNA polymerase zeta in replication of undamaged DNA in Saccharomyces cerevisiae. Genetics 2010, 184, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Neuberger, M.S.; Rada, C. Somatic hypermutation: Activation-induced deaminase for C/G followed by polymerase eta for A/T. J. Exp. Med. 2007, 204, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Despras, E.; Sittewelle, M.; Pouvelle, C.; Delrieu, N.; Cordonnier, A.M.; Kannouche, P.L. Rad18-dependent sumoylation of human specialized DNA polymerase eta is required to prevent under-replicated DNA. Nat. Commun. 2016, 7, 13326. [Google Scholar] [CrossRef] [PubMed]
- Urban, V.; Dobrovolna, J.; Janscak, P. Distinct functions of human recq helicases during DNA replication. Biophys. Chem. 2016. [Google Scholar] [CrossRef] [PubMed]
- Bansbach, C.E.; Betous, R.; Lovejoy, C.A.; Glick, G.G.; Cortez, D. The annealing helicase smarcal1 maintains genome integrity at stalled replication forks. Genes Dev. 2009, 23, 2405–2414. [Google Scholar] [CrossRef] [PubMed]
- Betous, R.; Mason, A.C.; Rambo, R.P.; Bansbach, C.E.; Badu-Nkansah, A.; Sirbu, B.M.; Eichman, B.F.; Cortez, D. Smarcal1 catalyzes fork regression and holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 2012, 26, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Blastyak, A.; Hajdu, I.; Unk, I.; Haracska, L. Role of double-stranded DNA translocase activity of human HLTF in replication of damaged DNA. Mol. Cell. Biol. 2010, 30, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Burkovics, P.; Sebesta, M.; Balogh, D.; Haracska, L.; Krejci, L. Strand invasion by HLTF as a mechanism for template switch in fork rescue. Nuclic Acids Res. 2014, 42, 1711–1720. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Nimonkar, A.V.; Hu, Y.; Hajdu, I.; Achar, Y.J.; Izhar, L.; Petit, S.A.; Adamson, B.; Yoon, J.C.; Kowalczykowski, S.C.; et al. Polyubiquitinated PCNA recruits the ZRANB3 translocase to maintain genomic integrity after replication stress. Mol. Cell 2012, 47, 396–409. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Ghosal, G.; Chen, J. The HARP-like domain-containing protein AH2/ZRANB3 binds to PCNA and participates in cellular response to replication stress. Mol. Cell 2012, 47, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Yusufzai, T.; Kadonaga, J.T. Harp is an atp-driven annealing helicase. Science 2008, 322, 748–750. [Google Scholar] [CrossRef] [PubMed]
- Rassool, F.V.; North, P.S.; Mufti, G.J.; Hickson, I.D. Constitutive DNA damage is linked to DNA replication abnormalities in bloom’s syndrome cells. Oncogene 2003, 22, 8749–8757. [Google Scholar] [CrossRef] [PubMed]
- Urban, V.; Dobrovolna, J.; Huhn, D.; Fryzelkova, J.; Bartek, J.; Janscak, P. Recq5 helicase promotes resolution of conflicts between replication and transcription in human cells. J. Cell Biol. 2016, 214, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Gebhart, E.; Bauer, R.; Raub, U.; Schinzel, M.; Ruprecht, K.W.; Jonas, J.B. Spontaneous and induced chromosomal instability in werner syndrome. Hum. Genet. 1988, 80, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Lauper, J.M.; Krause, A.; Vaughan, T.L.; Monnat, R.J., Jr. Spectrum and risk of neoplasia in werner syndrome: A systematic review. PLoS ONE 2013, 8, e59709. [Google Scholar] [CrossRef] [PubMed]
- Baradaran-Heravi, A.; Raams, A.; Lubieniecka, J.; Cho, K.S.; DeHaai, K.A.; Basiratnia, M.; Mari, P.O.; Xue, Y.; Rauth, M.; Olney, A.H.; et al. Smarcal1 deficiency predisposes to non-hodgkin lymphoma and hypersensitivity to genotoxic agents in vivo. Am. J. Med. Genet. A 2012, 158A, 2204–2213. [Google Scholar] [CrossRef] [PubMed]
- Ellis, N.A.; Groden, J.; Ye, T.Z.; Straughen, J.; Lennon, D.J.; Ciocci, S.; Proytcheva, M.; German, J. The bloom’s syndrome gene product is homologous to recq helicases. Cell 1995, 83, 655–666. [Google Scholar] [CrossRef]
- Cybulski, C.; Carrot-Zhang, J.; Kluzniak, W.; Rivera, B.; Kashyap, A.; Wokolorczyk, D.; Giroux, S.; Nadaf, J.; Hamel, N.; Zhang, S.; et al. Germline RECQL mutations are associated with breast cancer susceptibility. Nat. Genet. 2015, 47, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Futami, K.; Furuichi, Y. RECQL1 and WRN DNA repair helicases: Potential therapeutic targets and proliferative markers against cancers. Front. Genet. 2014, 5, 441. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, M.; Sommers, J.A.; Shoemaker, R.H.; Brosh, R.M., Jr. Inhibition of helicase activity by a small molecule impairs werner syndrome helicase (WRN) function in the cellular response to DNA damage or replication stress. Proc. Natl. Acad. Sci. USA 2011, 108, 1525–1530. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A.; Springgate, C.F.; Battula, N. Errors in DNA replication as a basis of malignant changes. Cancer Res. 1974, 34, 2311–2321. [Google Scholar] [PubMed]
- Kunkel, T.A.; Erie, D.A. DNA mismatch repair. Annu. Rev. Biochem. 2005, 74, 681–710. [Google Scholar] [CrossRef] [PubMed]
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006, 7, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Marsischky, G.T.; Filosi, N.; Kane, M.F.; Kolodner, R. Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair. Genes Dev. 1996, 10, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Harfe, B.D.; Jinks-Robertson, S. DNA mismatch repair and genetic instability. Annu. Rev. Genet. 2000, 34, 359–399. [Google Scholar] [CrossRef] [PubMed]
- Wei, K.; Kucherlapati, R.; Edelmann, W. Mouse models for human DNA mismatch-repair gene defects. Trends Mol. Med. 2002, 8, 346–353. [Google Scholar] [CrossRef]
- Lynch, H.T.; de la Chapelle, A. Hereditary colorectal cancer. N. Engl. J. Med. 2003, 348, 919–932. [Google Scholar] [PubMed]
- Peltomäki, P.; Vasen, H. Mutations associated with HNPCC predisposition—Update of ICG-HNPCC/insight mutation database. Dis. Mark. 2004, 20, 269–276. [Google Scholar] [CrossRef]
- Imai, K.; Yamamoto, H. Carcinogenesis and microsatellite instability: The interrelationship between genetics and epigenetics. Carcinogenesis 2008, 29, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Morrison, A.; Johnson, A.L.; Johnston, L.H.; Sugino, A. Pathway correcting DNA replication errors in Saccharomyces cerevisiae. EMBO J. 1993, 12, 1467–1473. [Google Scholar] [PubMed]
- Williams, L.N.; Herr, A.J.; Preston, B.D. Emergence of DNA polymerase e antimutators that escape error-induced extinction in yeast. Genetics 2013, 193, 751–770. [Google Scholar] [CrossRef] [PubMed]
- Prindle, M.J.; Schmitt, M.W.; Parmeggiani, F.; Loeb, L.A. A substitution in the fingers domain of DNA polymerase d reduces fidelity by altering nucleotide discrimination in the catalytic site. J. Biol. Chem. 2013, 288, 5572–5580. [Google Scholar] [CrossRef] [PubMed]
- Palles, C.; Cazier, J.B.; Howarth, K.M.; Domingo, E.; Jones, A.M.; Broderick, P.; Kemp, Z.; Spain, S.L.; Guarino, E.; Salguero, I.; et al. Germline mutations affecting the proofreading domains of pole and pold1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 2013, 45, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Wimmer, K.; Beilken, A.; Nustede, R.; Ripperger, T.; Lamottke, B.; Ure, B.; Steinmann, D.; Reineke-Plaass, T.; Lehmann, U.; Zschocke, J. A novel germline pole mutation causes an early onset cancer prone syndrome mimicking constitutional mismatch repair deficiency. Fam. Cancer 2016. [Google Scholar] [CrossRef] [PubMed]
- Aoude, L.G.; Heitzer, E.; Johansson, P.; Gartside, M.; Wadt, K.; Pritchard, A.L.; Palmer, J.M.; Symmons, J.; Gerdes, A.M.; Montgomery, G.W.; et al. Pole mutations in families predisposed to cutaneous melanoma. Fam. Cancer 2015, 14, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Bellido, F.; Pineda, M.; Aiza, G.; Valdes-Mas, R.; Navarro, M.; Puente, D.A.; Pons, T.; Gonzalez, S.; Iglesias, S.; Darder, E.; et al. POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: Review of reported cases and recommendations for genetic testing and surveillance. Genet. Med. 2016, 18, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.; Darmawan, H.; Schultz, A.; Fidalgo da Silva, E.; Reha-Krantz, L.J. A method to select for mutator DNA polymerase deltas in Saccharomyces cerevisiae. Genome 2006, 49, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Abdus Sattar, A.K.; Lin, T.C.; Jones, C.; Konigsberg, W.H. Functional consequences and exonuclease kinetic parameters of point mutations in bacteriophage T4 DNA polymerase. Biochemistry 1996, 35, 16621–16629. [Google Scholar] [CrossRef] [PubMed]
- Kane, D.P.; Shcherbakova, P.V. A common cancer-associated DNA polymerase ε mutation causes an exceptionally strong mutator phenotype, indicating fidelity defects distinct from loss of proofreading. Cancer Res. 2014, 74, 1895–1901. [Google Scholar] [CrossRef] [PubMed]
- Rohlin, A.; Zagoras, T.; Nilsson, S.; Lundstam, U.; Wahlstrom, J.; Hulten, L.; Martinsson, T.; Karlsson, G.B.; Nordling, M. A mutation in pole predisposing to a multi-tumour phenotype. Int. J. Oncol. 2014, 45, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Chubb, D.; Broderick, P.; Frampton, M.; Kinnersley, B.; Sherborne, A.; Penegar, S.; Lloyd, A.; Ma, Y.P.; Dobbins, S.E.; Houlston, R.S. Genetic diagnosis of high-penetrance susceptibility for colorectal cancer (CRC) is achievable for a high proportion of familial CRC by exome sequencing. J. Clin. Oncol. 2015, 33, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Spier, I.; Holzapfel, S.; Altmuller, J.; Zhao, B.; Horpaopan, S.; Vogt, S.; Chen, S.; Morak, M.; Raeder, S.; Kayser, K.; et al. Frequency and phenotypic spectrum of germline mutations in pole and seven other polymerase genes in 266 patients with colorectal adenomas and carcinomas. Int. J. Cancer 2015, 137, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.F.; Johansen, J.; Bjornevoll, I.; Sylvander, A.E.; Steinsbekk, K.S.; Saetrom, P.; Sandvik, A.K.; Drablos, F.; Sjursen, W. A novel POLE mutation associated with cancers of colon, pancreas, ovaries and small intestine. Fam. Cancer 2015, 14, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Valle, L.; Hernández-Illán, E.; Bellido, F.; Aiza, G.; Castillejo, A.; Castillejo, M.-I.; Navarro, M.; Seguí, N.; Vargas, G.; Guarinos, C. New insights into pole and pold1 germline mutations in familial colorectal cancer and polyposis. Hum. Mol. Genet. 2014, 23, 3506–3512. [Google Scholar] [CrossRef] [PubMed]
- Rayner, E.; van Gool, I.C.; Palles, C.; Kearsey, S.E.; Bosse, T.; Tomlinson, I.; Church, D.N. A panoply of errors: Polymerase proofreading domain mutations in cancer. Nat. Rev. Cancer 2016, 16, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Church, D.N.; Briggs, S.E.; Palles, C.; Domingo, E.; Kearsey, S.J.; Grimes, J.M.; Gorman, M.; Martin, L.; Howarth, K.M.; Hodgson, S.V.; et al. DNA polymerase ɛ and δ exonuclease domain mutations in endometrial cancer. Hum. Mol. Genet. 2013, 22, 2820–2828. [Google Scholar] [CrossRef] [PubMed]
- Seshagiri, S.; Stawiski, E.W.; Durinck, S.; Modrusan, Z.; Storm, E.E.; Conboy, C.B.; Chaudhuri, S.; Guan, Y.; Janakiraman, V.; Jaiswal, B.S.; et al. Recurrent R-spondin fusions in colon cancer. Nature 2012, 488, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar]
- Briggs, S.; Tomlinson, I. Germline and somatic polymerase ɛ and δ mutations define a new class of hypermutated colorectal and endometrial cancers. J. Pathol. 2013, 230, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Seshagiri, S. The burden of faulty proofreading in colon cancer. Nat. Genet. 2013, 45, 121–122. [Google Scholar] [CrossRef] [PubMed]
- Daee, D.L.; Mertz, T.M.; Shcherbakova, P.V. A cancer-associated DNA polymerase d variant modeled in yeast causes a catastrophic increase in genomic instability. Proc. Natl. Acad. Sci. USA 2010, 107, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cbio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013. [Google Scholar] [CrossRef] [PubMed]
- Shlien, A.; Campbell, B.B.; de Borja, R.; Alexandrov, L.B.; Merico, D.; Wedge, D.; van Loo, P.; Tarpey, P.S.; Coupland, P.; Behjati, S.; et al. Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra-hypermutated cancers. Nat. Genet. 2015, 47, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Mehnert, J.M.; Panda, A.; Zhong, H.; Hirshfield, K.; Damare, S.; Lane, K.; Sokol, L.; Stein, M.N.; Rodriguez-Rodriquez, L.; Kaufman, H.L.; et al. Immune activation and response to pembrolizumab in pole-mutant endometrial cancer. J. Clin. Investig. 2016, 126, 2334–2340. [Google Scholar] [CrossRef] [PubMed]
- Refsland, E.W.; Harris, R.S. The APOBEC3 family of retroelement restriction factors. Curr. Top. Microbiol. Immunol. 2013, 371, 1–27. [Google Scholar] [PubMed]
- Bransteitter, R.; Pham, P.; Scharff, M.D.; Goodman, M.F. Activation-induced cytidine deaminase deaminates deoxycytidine on single-stranded DNA but requires the action of RNase. Proc. Natl. Acad. Sci. USA 2003, 100, 4102–4107. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, S.K.; Market, E.; Besmer, E.; Papavasiliou, F.N. Aid mediates hypermutation by deaminating single stranded DNA. J. Exp. Med. 2003, 197, 1291–1296. [Google Scholar] [CrossRef] [PubMed]
- Suspene, R.; Sommer, P.; Henry, M.; Ferris, S.; Guetard, D.; Pochet, S.; Chester, A.; Navaratnam, N.; Wain-Hobson, S.; Vartanian, J.P. APOBEC3G is a single-stranded DNA cytidine deaminase and functions independently of HIV reverse transcriptase. Nuclic Acids Res. 2004, 32, 2421–2429. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Konig, R.; Pillai, S.; Chiles, K.; Kearney, M.; Palmer, S.; Richman, D.; Coffin, J.M.; Landau, N.R. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat. Struct. Mol. Biol. 2004, 11, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.; Resnick, M.A.; Gordenin, D.A. The choice of nucleotide inserted opposite abasic sites formed within chromosomal DNA reveals the polymerase activities participating in translesion DNA synthesis. DNA Repair 2013, 12, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, J.; Tian, M.; Khuong, C.; Chua, K.; Pinaud, E.; Alt, F.W. Transcription-targeted DNA deamination by the aid antibody diversification enzyme. Nature 2003, 422, 726–730. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Petersen-Mahrt, S.K.; Neuberger, M.S. RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol. Cell 2002, 10, 1247–1253. [Google Scholar] [CrossRef]
- Love, R.P.; Xu, H.; Chelico, L. Biochemical analysis of hypermutation by the deoxycytidine deaminase APOBEC3A. J. Biol. Chem. 2012, 287, 30812–30822. [Google Scholar] [CrossRef] [PubMed]
- Ramiro, A.R.; Stavropoulos, P.; Jankovic, M.; Nussenzweig, M.C. Transcription enhances aid-mediated cytidine deamination by exposing single-stranded DNA on the nontemplate strand. Nat. Immunol. 2003, 4, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.J.M.; Nik-Zainal, S.; Wu, Y.L.; Stebbings, L.A.; Raine, K.; Campbell, P.J.; Rada, C.; Stratton, M.R.; Neuberger, M.S. DNA deaminases induce break-associated mutation showers with implication of APOBEC3B and 3A in breast cancer kataegis. eLife 2013, 2, e00534. [Google Scholar] [CrossRef] [PubMed]
- Hoopes, J.I.; Cortez, L.M.; Mertz, T.M.; Malc, E.P.; Mieczkowski, P.A.; Roberts, S.A. APOBEC3A and APOBEC3B preferentially deaminate the lagging strand template during DNA replication. Cell Rep. 2016, 14, 1273–1282. [Google Scholar] [CrossRef] [PubMed]
- Green, A.M.; Landry, S.; Budagyan, K.; Avgousti, D.C.; Shalhout, S.; Bhagwat, A.S.; Weitzman, M.D. APOBEC3A damages the cellular genome during DNA replication. Cell Cycle 2016, 15, 998–1008. [Google Scholar] [CrossRef] [PubMed]
- Refsland, E.W.; Stenglein, M.D.; Shindo, K.; Albin, J.S.; Brown, W.L.; Harris, R.S. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: Implications for HIV-1 restriction. Nuclic Acids Res. 2010, 38, 4274–4284. [Google Scholar] [CrossRef] [PubMed]
- Bogerd, H.P.; Wiegand, H.L.; Hulme, A.E.; Garcia-Perez, J.L.; O’Shea, K.S.; Moran, J.V.; Cullen, B.R. Cellular inhibitors of long interspersed element 1 and Alu retrotransposition. Proc. Natl. Acad. Sci. USA 2006, 103, 8780–8785. [Google Scholar] [CrossRef] [PubMed]
- Burns, M.B.; Temiz, N.A.; Harris, R.S. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet. 2013, 45, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Lawrence, M.S.; Klimczak, L.J.; Grimm, S.A.; Fargo, D.; Stojanov, P.; Kiezun, A.; Kryukov, G.V.; Carter, S.L.; Saksena, G.; et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013, 45, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Leonard, B.; Hart, S.N.; Burns, M.B.; Carpenter, M.A.; Temiz, N.A.; Rathore, A.; Vogel, R.I.; Nikas, J.B.; Law, E.K.; Brown, W.L.; et al. APOBEC3B upregulation and genomic mutation patterns in serous ovarian carcinoma. Cancer Res. 2013, 73, 7222–7231. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Gordenin, D.A. Clustered and genome-wide transient mutagenesis in human cancers: Hypermutation without permanent mutators or loss of fitness. BioEssays 2014, 36, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.; Chakravarthy, A.; Su, X.P.; Boshoff, C.; Fenton, T.R. Apobec-mediated cytosine deamination links PIK3CA helical domain mutations to human papillomavirus-driven tumor development. Cell Rep. 2014, 7, 1833–1841. [Google Scholar] [CrossRef] [PubMed]
- Okuyama, S.; Marusawa, H.; Matsumoto, T.; Ueda, Y.; Matsumoto, Y.; Endo, Y.; Takai, A.; Chiba, T. Excessive activity of apolipoprotein B mRNA editing enzyme catalytic polypeptide 2 (APOBEC2) contributes to liver and lung tumorigenesis. Int. J. Cancer 2012, 130, 1294–1301. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, S.; Balestra, M.E.; Ferrell, L.D.; Fan, J.; Arnold, K.S.; Taylor, S.; Taylor, J.M.; Innerarity, T.L. Apolipoprotein B mRNA-editing protein induces hepatocellular carcinoma and dysplasia in transgenic animals. Proc. Natl. Acad. Sci. USA 1995, 92, 8483–8487. [Google Scholar] [CrossRef] [PubMed]
- Lackey, L.; Law, E.K.; Brown, W.L.; Harris, R.S. Subcellular localization of the APOBEC3 proteins during mitosis and implications for genomic DNA deamination. Cell Cycle 2013, 12, 762–772. [Google Scholar] [CrossRef] [PubMed]
- Burns, M.B.; Lackey, L.; Carpenter, M.A.; Rathore, A.; Land, A.M.; Leonard, B.; Refsland, E.W.; Kotandeniya, D.; Tretyakova, N.; Nikas, J.B.; et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 2013, 494, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Middlebrooks, C.D.; Banday, A.R.; Matsuda, K.; Udquim, K.I.; Onabajo, O.O.; Paquin, A.; Figueroa, J.D.; Zhu, B.; Koutros, S.; Kubo, M.; et al. Association of germline variants in the APOBEC3 region with cancer risk and enrichment with APOBEC-signature mutations in tumors. Nat. Genet. 2016, 48, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- Caval, V.; Suspene, R.; Shapira, M.; Vartanian, J.P.; Wain-Hobson, S. A prevalent cancer susceptibility APOBEC3A hybrid allele bearing APOBEC3B 3′ UTR enhances chromosomal DNA damage. Nat. Commun. 2014, 5, 5129. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Wedge, D.C.; Alexandrov, L.B.; Petljak, M.; Butler, A.P.; Bolli, N.; Davies, H.R.; Knappskog, S.; Martin, S.; Papaemmanuil, E.; et al. Association of a germline copy number polymorphism of APOBEC3A and APOBEC3B with burden of putative APOBEC-dependent mutations in breast cancer. Nat. Genet. 2014, 46, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Cescon, D.W.; Haibe-Kains, B.; Mak, T.W. Apobec3B expression in breast cancer reflects cellular proliferation, while a deletion polymorphism is associated with immune activation. Proc. Natl. Acad. Sci. USA 2015, 112, 2841–2846. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.X.; Soo, J.S.-S.; Kwan, P.Y.; Hong, E.; Khang, T.F.; Mariapun, S.; Lee, C.S.-M.; Hasan, S.N.; Rajadurai, P.; Yip, C.H. Germline APOBEC3B deletion is associated with breast cancer risk in an Asian multi-ethnic cohort and with immune cell presentation. Breast Cancer Res. 2016, 18, 56. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Cai, J.; Chang, J.; Yu, D.; Wu, C.; Yan, T.; Zhai, K.; Bi, X.; Zhao, H.; Xu, J.; et al. Evidence of associations of APOBEC3B gene deletion with susceptibility to persistent HBV infection and hepatocellular carcinoma. Hum. Mol. Genet. 2013, 22, 1262–1269. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.; Roberts, S.A.; Klimczak, L.J.; Sterling, J.F.; Saini, N.; Malc, E.P.; Kim, J.; Kwiatkowski, D.J.; Fargo, D.C.; Mieczkowski, P.A.; et al. An APOBEC3A hypermutation signature is distinguishable from the signature of background mutagenesis by APOBEC3B in human cancers. Nat. Genet. 2015, 47, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Park, S.R. Activation-induced cytidine deaminase in B cell immunity and cancers. Immune Netw. 2012, 12, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Saraconi, G.; Severi, F.; Sala, C.; Mattiuz, G.; Conticello, S.G. The RNA editing enzyme APOBEC1 induces somatic mutations and a compatible mutational signature is present in esophageal adenocarcinomas. Genome Biol. 2014, 5, 417. [Google Scholar] [CrossRef]
- Starrett, G.J.; Luengas, E.M.; McCann, J.L.; Ebrahimi, D.; Temiz, N.A.; Love, R.P.; Feng, Y.Q.; Adolph, M.B.; Chelico, L.; Law, E.K.; et al. The DNA cytosine deaminase APOBEC3H haplotype I likely contributes to breast and lung cancer mutagenesis. Nat. Commun. 2016, 7, 12918. [Google Scholar] [CrossRef] [PubMed]
- Lada, A.G.; Dhar, A.; Boissy, R.J.; Hirano, M.; Rubel, A.A.; Rogozin, I.B.; Pavlov, Y.I. Aid/APOBEC cytosine deaminase induces genome-wide kataegis. Biol. Direct 2012, 7, 47. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.J.; Wu, Y.L.; Rada, C. Active RNAP pre-initiation sites are highly mutated by cytidine deaminases in yeast, with aid targeting small RNA genes. eLife 2014, 3, e03553. [Google Scholar] [CrossRef] [PubMed]
- Kotsantis, P.; Silva, L.M.; Irmscher, S.; Jones, R.M.; Folkes, L.; Gromak, N.; Petermann, E. Increased global transcription activity as a mechanism of replication stress in cancer. Nat. Commun. 2016, 7, 13087. [Google Scholar] [CrossRef] [PubMed]
- Mimitou, E.P.; Symington, L.S. DNA end resection—Unraveling the tail. DNA Repair 2011, 10, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Caron, P.; Legube, G.; Paull, T.T. Quantitation of DNA double-strand break resection intermediates in human cells. Nuclic Acids Res. 2014, 42, e19. [Google Scholar] [CrossRef] [PubMed]
- Saini, N.; Ramakrishnan, S.; Elango, R.; Ayyar, S.; Zhang, Y.; Deem, A.; Ira, G.; Haber, J.E.; Lobachev, K.S.; Malkova, A. Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature 2013, 502, 389–392. [Google Scholar] [CrossRef] [PubMed]
- Sakofsky, C.J.; Roberts, S.A.; Malc, E.; Mieczkowski, P.A.; Resnick, M.A.; Gordenin, D.A.; Malkova, A. Break-induced replication is a source of mutation clusters underlying kataegis. Cell Rep. 2014, 7, 1640–1648. [Google Scholar] [CrossRef] [PubMed]
- Bhagwat, A.S.; Hao, W.L.; Townes, J.P.; Lee, H.; Tang, H.X.; Foster, P.L. Strand-biased cytosine deamination at the replication fork causes cytosine to thymine mutations in Escherichia coli. Proc. Natl. Acad. Sci. USA 2016, 113, 2176–2181. [Google Scholar] [CrossRef] [PubMed]
- Le, Q.; Maizels, N. Cell cycle regulates nuclear stability of aid and determines the cellular response to aid. PLoS Genet. 2015, 11, e1005411. [Google Scholar] [CrossRef] [PubMed]
- Haradhvala, N.J.; Polak, P.; Stojanov, P.; Covington, K.R.; Shinbrot, E.; Hess, J.M.; Rheinbay, E.; Kim, J.; Maruvka, Y.E.; Braunstein, L.Z.; et al. Mutational strand asymmetries in cancer genomes reveal mechanisms of DNA damage and repair. Cell 2016, 164, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Morganella, S.; Alexandrov, L.B.; Glodzik, D.; Zou, X.; Davies, H.; Staaf, J.; Sieuwerts, A.M.; Brinkman, A.B.; Martin, S.; Ramakrishna, M.; et al. The topography of mutational processes in breast cancer genomes. Nat. Commun. 2016, 7, 11383. [Google Scholar] [CrossRef] [PubMed]
- Seplyarskiy, V.B.; Soldatov, R.A.; Popadin, K.Y.; Antonarakis, S.E.; Bazykin, G.A.; Nikolaev, S.I. Apobec-induced mutations in human cancers are strongly enriched on the lagging DNA strand during replication. Genome Res. 2016, 26, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Dyson, N.; Howley, P.M.; Munger, K.; Harlow, E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 1989, 243, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, A.J.; Nguyen, M.; Liem, A.; Lee, D.; Montagna, C.; Lambert, P.F.; Ried, T.; Difilippantonio, M.J. E6 and E7 oncoproteins induce distinct patterns of chromosomal aneuploidy in skin tumors from transgenic mice. Cancer Res. 2004, 64, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Law, E.K.; Sieuwerts, A.M.; LaPara, K.; Leonard, B.; Starrett, G.J.; Molan, A.M.; Temiz, N.A.; Vogel, R.I.; Meijer-van Gelder, M.E.; Sweep, F.C.; et al. The DNA cytosine deaminase APOBEC3B promotes tamoxifen resistance in ER-positive breast cancer. Sci. Adv. 2016, 2, e1601737. [Google Scholar] [CrossRef] [PubMed]
- Aird, K.M.; Zhang, G.; Li, H.; Tu, Z.; Bitler, B.G.; Garipov, A.; Wu, H.; Wei, Z.; Wagner, S.N.; Herlyn, M.; et al. Suppression of nucleotide metabolism underlies the establishment and maintenance of oncogene-induced senescence. Cell Rep. 2013, 3, 1252–1265. [Google Scholar] [CrossRef] [PubMed]
- Angus, S.P.; Wheeler, L.J.; Ranmal, S.A.; Zhang, X.; Markey, M.P.; Mathews, C.K.; Knudsen, E.S. Retinoblastoma tumor suppressor targets dNTP metabolism to regulate DNA replication. J. Biol. Chem. 2002, 277, 44376–44384. [Google Scholar] [CrossRef] [PubMed]
- Onyenwoke, R.U.; Forsberg, L.J.; Liu, L.; Williams, T.; Alzate, O.; Brenman, J.E. Ampk directly inhibits NDPK through a phosphoserine switch to maintain cellular homeostasis. Mol. Biol. Cell 2012, 23, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Mannava, S.; Grachtchouk, V.; Wheeler, L.J.; Im, M.; Zhuang, D.; Slavina, E.G.; Mathews, C.K.; Shewach, D.S.; Nikiforov, M.A. Direct role of nucleotide metabolism in C-MYC-dependent proliferation of melanoma cells. Cell Cycle 2008, 7, 2392–2400. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Zhou, B.; Liu, X.; Qiu, W.; Jin, Z.; Yen, Y. Wild-type p53 regulates human ribonucleotide reductase by protein-protein interaction with p53R2 as well as hRRM2 subunits. Cancer Res. 2003, 63, 980–986. [Google Scholar] [PubMed]
- Eckert, K.A.; Kunkel, T.A. DNA polymerase fidelity and the polymerase chain reaction. PCR Methods Appl. 1991, 1, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Creighton, S.; Goodman, M.F. Gel kinetic analysis of DNA polymerase fidelity in the presence of proofreading using bacteriophage T4 DNA polymerase. J. Biol. Chem. 1995, 270, 4759–4774. [Google Scholar] [CrossRef] [PubMed]
- Mendelman, L.V.; Petruska, J.; Goodman, M.F. Base mispair extension kinetics. Comparison of DNA polymerase a and reverse transcriptase. J. Biol. Chem. 1990, 265, 2338–2346. [Google Scholar] [PubMed]
- Mertz, T.M.; Sharma, S.; Chabes, A.; Shcherbakova, P.V. Colon cancer-associated mutator DNA polymerase delta variant causes expansion of dNTP pools increasing its own infidelity. Proc. Natl. Acad. Sci. USA 2015, 112, E2467–E2476. [Google Scholar] [CrossRef] [PubMed]
- Gon, S.; Napolitano, R.; Rocha, W.; Coulon, S.; Fuchs, R.P. Increase in dNTP pool size during the DNA damage response plays a key role in spontaneous and induced-mutagenesis in Escherichia coli. Proc. Natl. Acad. Sci. USA 2011, 108, 19311–19316. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, L.J.; Rajagopal, I.; Mathews, C.K. Stimulation of mutagenesis by proportional deoxyribonucleoside triphosphate accumulation in Escherichia coli. DNA Repair 2005, 4, 1450–1456. [Google Scholar] [CrossRef] [PubMed]
- Datta, A.; Schmeits, J.L.; Amin, N.S.; Lau, P.J.; Myung, K.; Kolodner, R.D. Checkpoint-dependent activation of mutagenic repair in Saccharomyces cerevisiae pol3-01 mutants. Mol. Cell 2000, 6, 593–603. [Google Scholar] [CrossRef]
- Hills, S.A.; Diffley, J.F. DNA replication and oncogene-induced replicative stress. Curr. Biol. 2014, 24, R435–R444. [Google Scholar] [CrossRef] [PubMed]
- Kanu, N.; Cerone, M.A.; Goh, G.; Zalmas, L.P.; Bartkova, J.; Dietzen, M.; McGranahan, N.; Rogers, R.; Law, E.K.; Gromova, I.; et al. DNA replication stress mediates APOBEC3 family mutagenesis in breast cancer. Genome Biol. 2016, 17, 185. [Google Scholar] [CrossRef] [PubMed]
- Bianconi, E.; Piovesan, A.; Facchin, F.; Beraudi, A.; Casadei, R.; Frabetti, F.; Vitale, L.; Pelleri, M.C.; Tassani, S.; Piva, F.; et al. An estimation of the number of cells in the human body. Ann. Hum. Biol. 2013, 40, 463–471. [Google Scholar] [CrossRef] [PubMed]
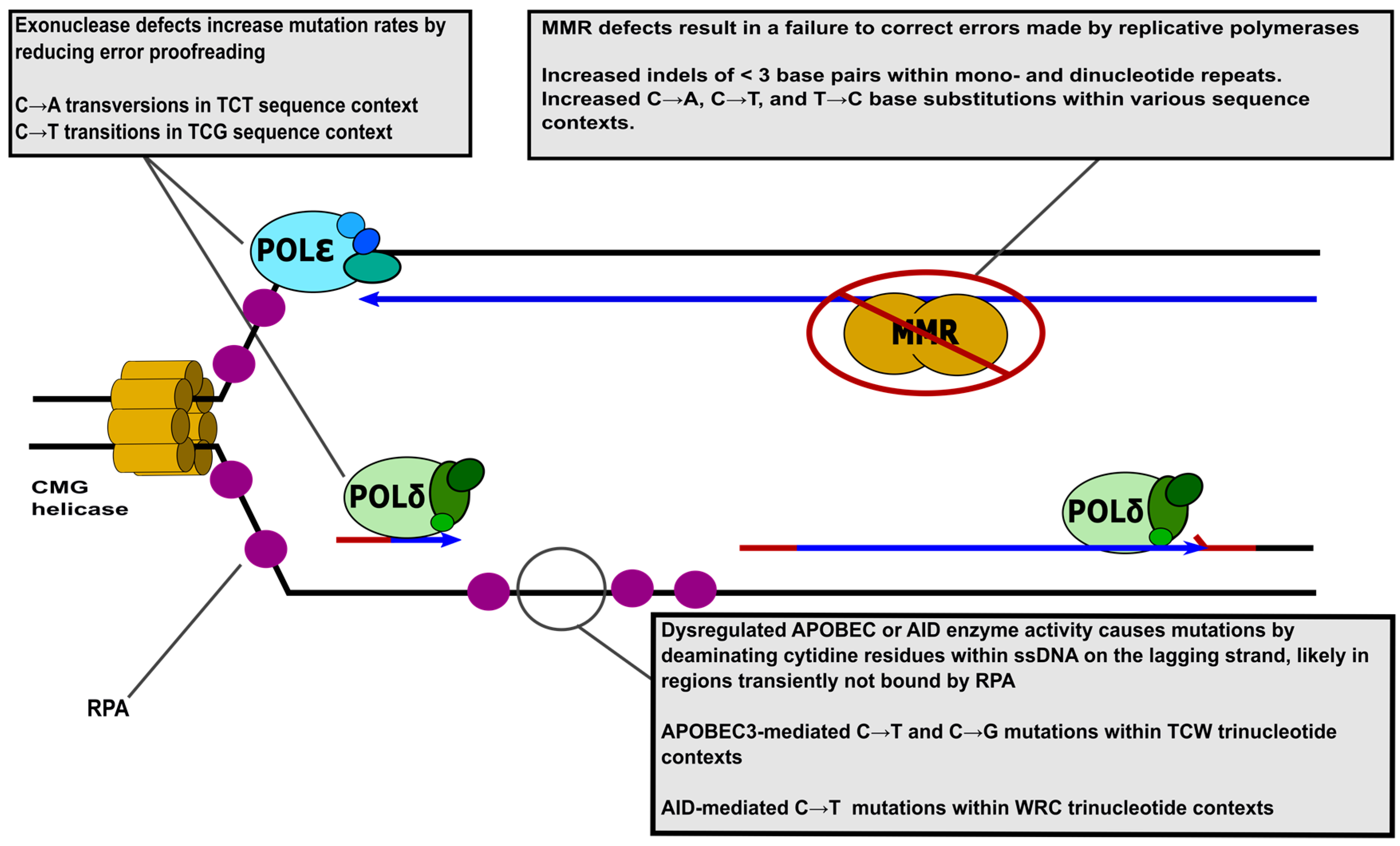

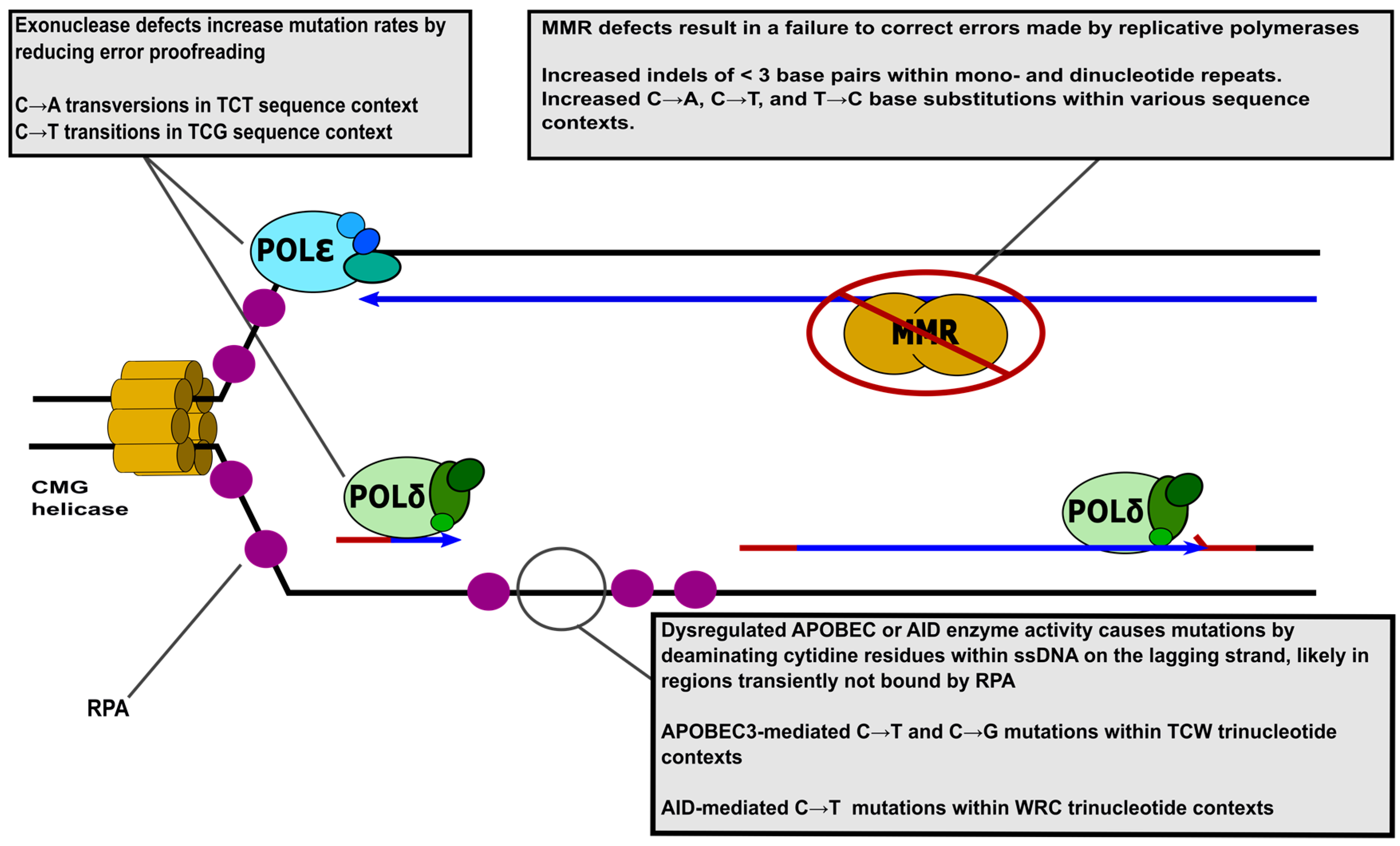{kind=link}
| Amino Acid Change 1 | Somatic/Germline | Cancer Type 2 (n) 3 | Mutator Phenotype in Yeast [References] | Biochemical Support/Enzyme [References] |
|---|---|---|---|---|
| POLD1- | ||||
| D316G | Germline [86] | CRC, EC, and breast | Yes [87] | Yes/T4 polymerase [88] |
| D316H | Germline [86] | CRC and breast | Yes [87] | Yes/T4 polymerase [88] |
| P327L | Germline [83] | None, patient had multiple colonic adenomas | Yes 5 [89] | Yes/human Polε [45] |
| R409W | Germline [86] | CRC | N.d. | N.d. |
| L474P | Germline [86] | CRC and EC | Yes [87] | Yes/human Polε [45] |
| S478N | Germline [83] | CRC and EC | Yes [83] | N.d. |
| POLE- | ||||
| W347C | Germline [85] | Cutaneous melanoma | Yes [85] | N.d. |
| N363K | Germline [90] | CRC and EC | N.d. | N.d. |
| D368V | Germline [91] | CRC | N.d. | Yes/T4 polymerase [88] |
| P436S | Germline [92] | CRC | N.d. | N.d. |
| Y458F | Germline [93] | CRC | N.d. | Yes/T4 polymerase [88] |
| L424V/I | Both [83] | Hereditary CRC, EC (2) 4, breast (1) 4 | Yes 6 [87] | Yes/human Polε [45] |
| P286R/L/H | Somatic | CRC (5), EC (10), breast (1), stomach (1), pancreas (1) | Yes [89] | Yes/human Polε [45] |
| F367S | Somatic | CRC (1) | N.d. | Yes/human Polε [45] |
| V411L | Both [84] | CRC (3), EC (6), stomach (1) | N.d. | Yes/human Polε [45] |
| S459F | Somatic | CRC (4) | N.d. | Yes/human Polε [45] |
| S297F | Somatic | EC (1), cervical (1) | N.d. | N.d. |
| P436R | Somatic | CRC (1) | N.d. | Yes/human Polε [45] |
| M444K | Somatic | EC (1) | N.d. | N.d. |
| A456P | Somatic | EC (1) | N.d. | N.d. |
| APOBEC Family Member | Mutation Motif Preference | Cellular Localization | Expression Correlates with TCW Mutations in Tumors | Evidence for Mutation during Transcription | Evidence for Mutation during Replication | Evidence for Mutation during DSB Repair | References |
|---|---|---|---|---|---|---|---|
| AID | WRC | Cytoplasmic | N/A | Yes | Yes | Yes | [112,113,114,115,116] |
| APOBEC1 | TCW | Pan Cellular | N.d. | N.d. | N.d. | N.d. | ˗ |
| APOBEC2 | N.d. | N.d. | N.d. | N.d. | N.d. | N.d. | ˗ |
| APOBEC3A | TCW | Pan Cellular | Yes | Limited | Yes | Yes | [116,117,118] |
| APOBEC3B | TCW | Nuclear | Yes | Limited | Yes | Yes | [116,117] |
| APOBEC3C | TCW | Pan Cellular | No | N.d. | N.d. | N.d. | ˗ |
| APOBEC3D/E | TCW | Cytoplasmic | No | N.d. | N.d. | N.d. | ˗ |
| APOBEC3F | TCW | Cytoplasmic | No | N.d. | N.d. | N.d. | ˗ |
| APOBEC3G | CC | Cytoplasmic | N/A | Limited | Yes | N.d. | [116] |
| APOBEC3H | TCW | Cytoplasmic | No | N.d. | N.d. | N.d. | ˗ |
| APOBEC4 | N.d. | N.d. | N.d. | N.d. | N.d. | N.d. | ˗ |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mertz, T.M.; Harcy, V.; Roberts, S.A. Risks at the DNA Replication Fork: Effects upon Carcinogenesis and Tumor Heterogeneity. Genes 2017, 8, 46. https://doi.org/10.3390/genes8010046
Mertz TM, Harcy V, Roberts SA. Risks at the DNA Replication Fork: Effects upon Carcinogenesis and Tumor Heterogeneity. Genes. 2017; 8(1):46. https://doi.org/10.3390/genes8010046
Chicago/Turabian StyleMertz, Tony M., Victoria Harcy, and Steven A. Roberts. 2017. "Risks at the DNA Replication Fork: Effects upon Carcinogenesis and Tumor Heterogeneity" Genes 8, no. 1: 46. https://doi.org/10.3390/genes8010046
APA StyleMertz, T. M., Harcy, V., & Roberts, S. A. (2017). Risks at the DNA Replication Fork: Effects upon Carcinogenesis and Tumor Heterogeneity. Genes, 8(1), 46. https://doi.org/10.3390/genes8010046