
Links between DNA Replication, Stem Cells and Cancer

Abstract
:1. Cancer
1.1. What Is Cancer?
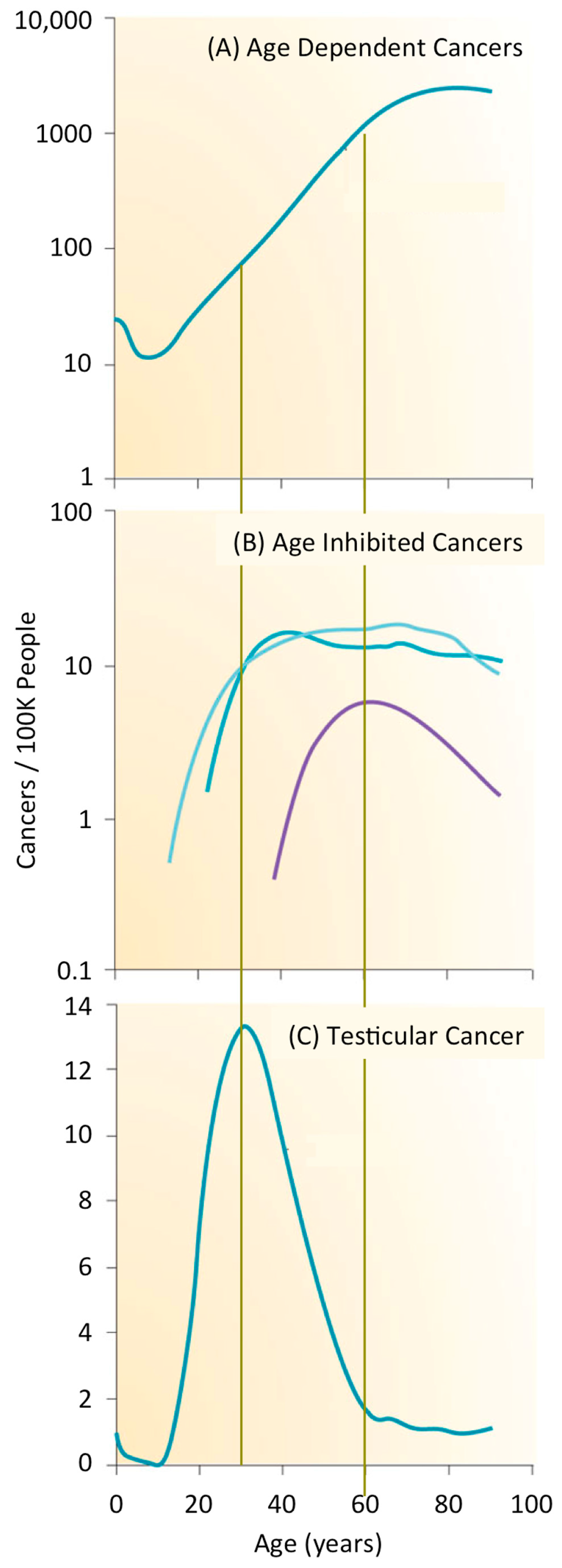
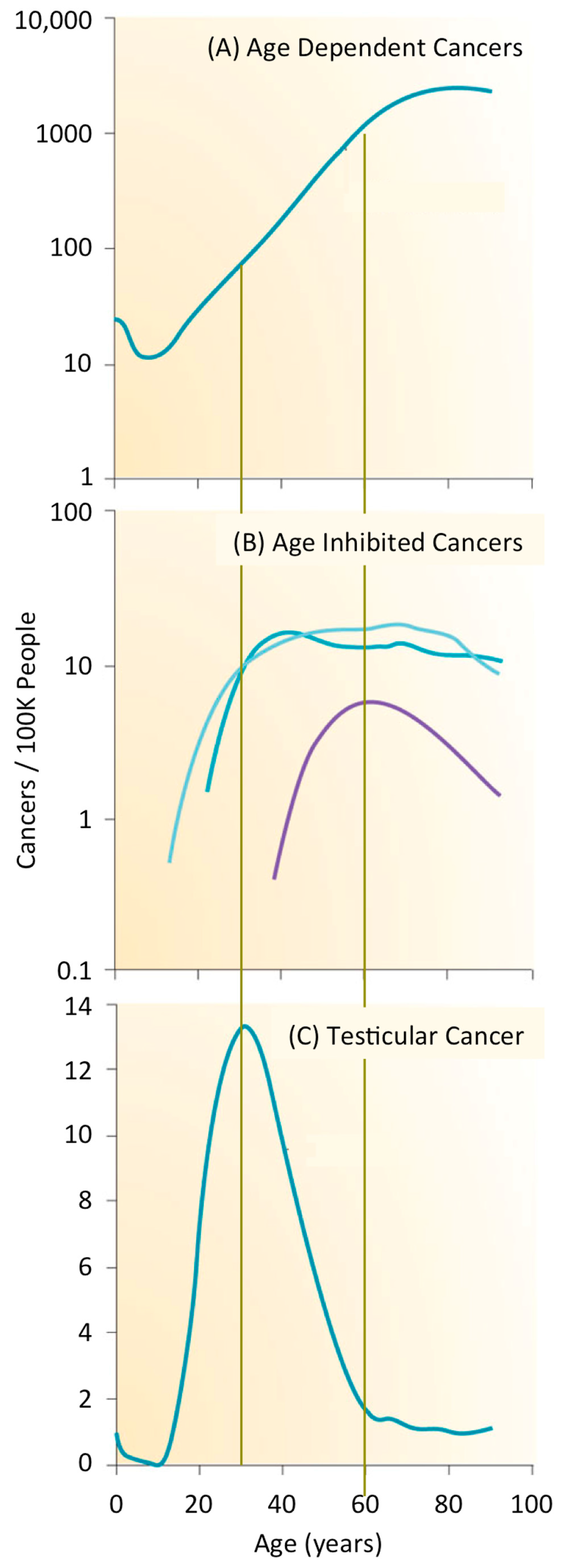
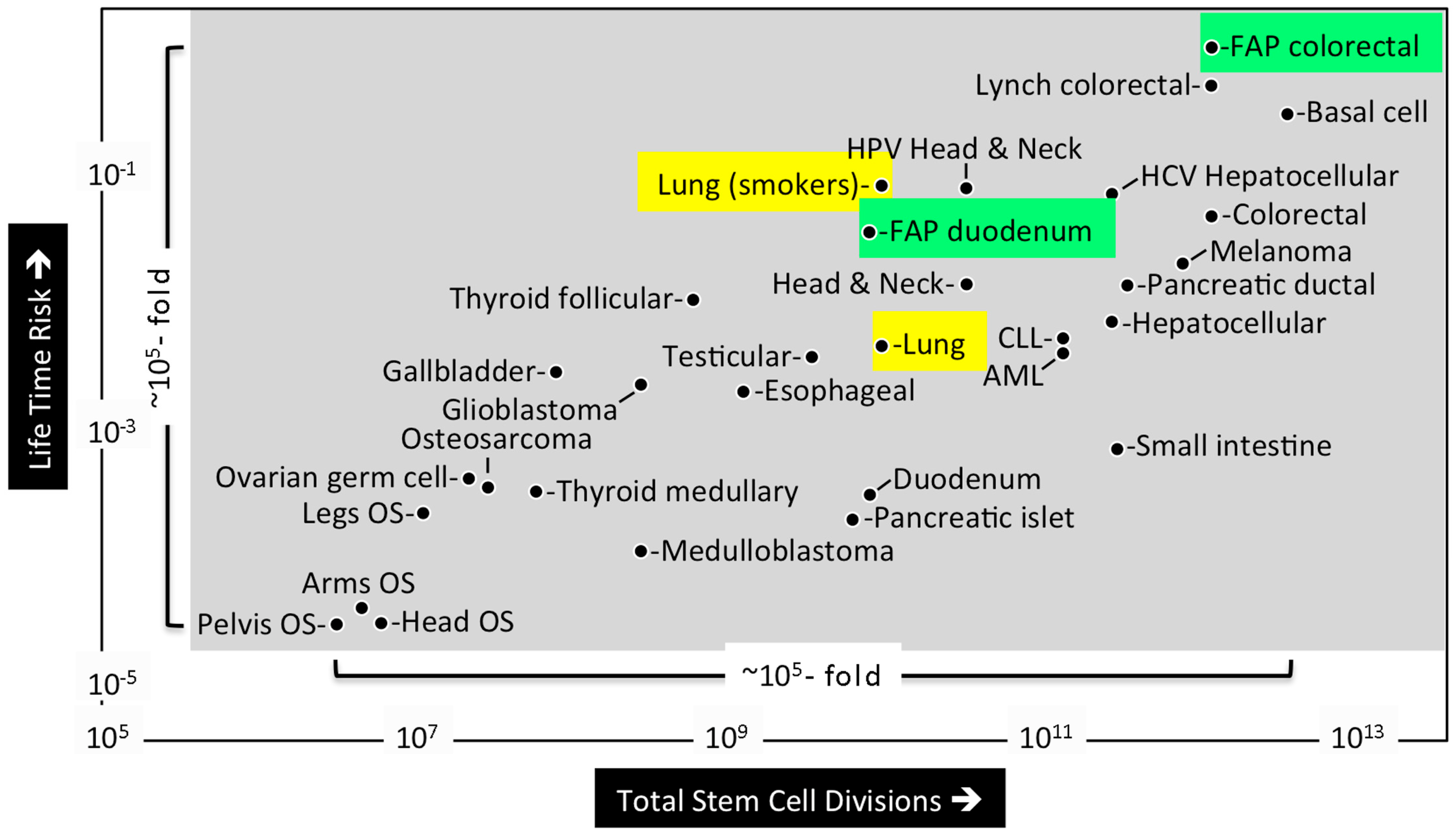
1.2. What Is the Likelihood of Developing a Cancer?
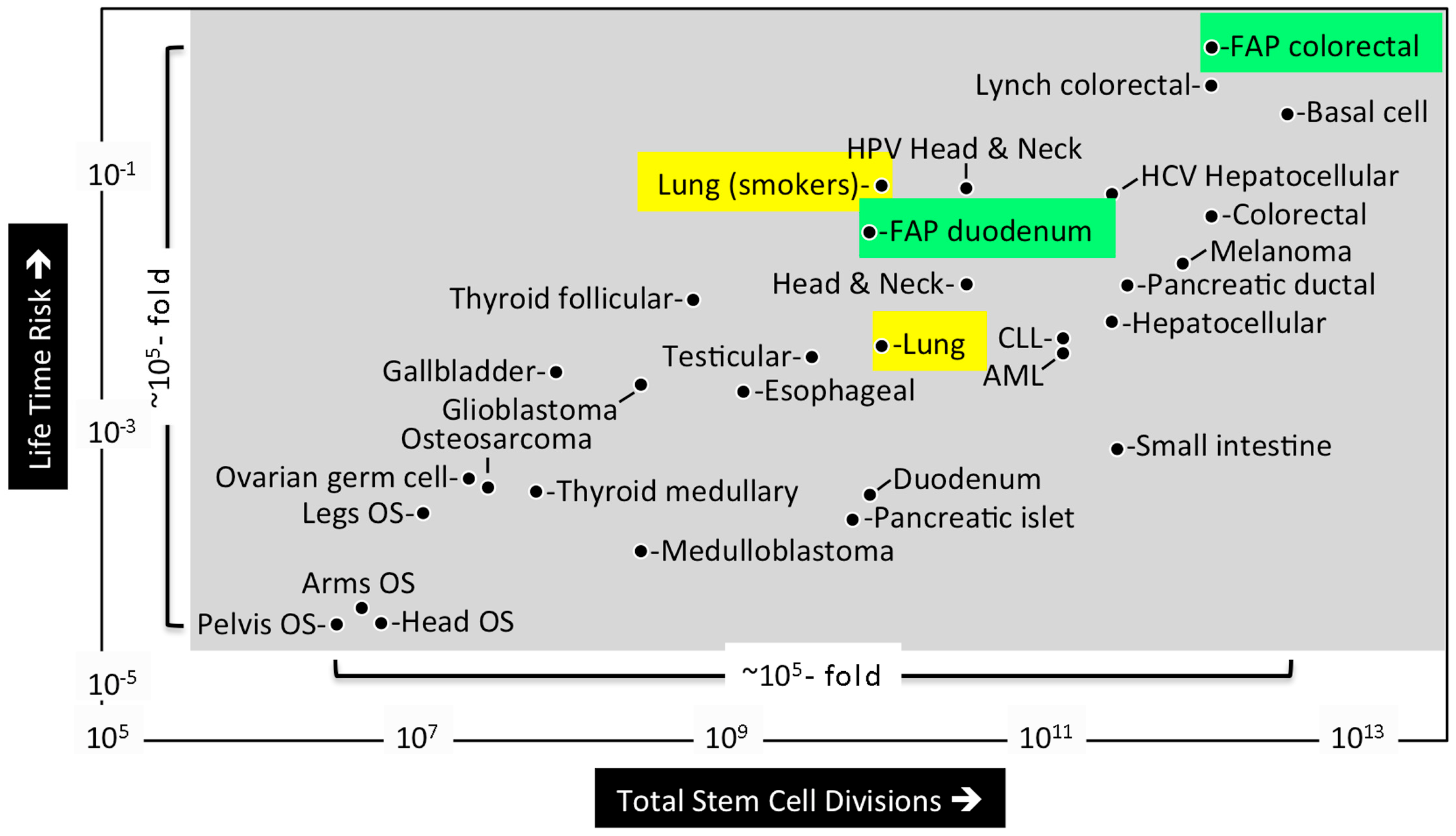
1.3. What Are the Origins of Cancer?
1.4. Intrinsic versus Extrinsic Risk Factors
1.5. Clonal-Evolution of Cancer
1.6. Take-Home Lesson
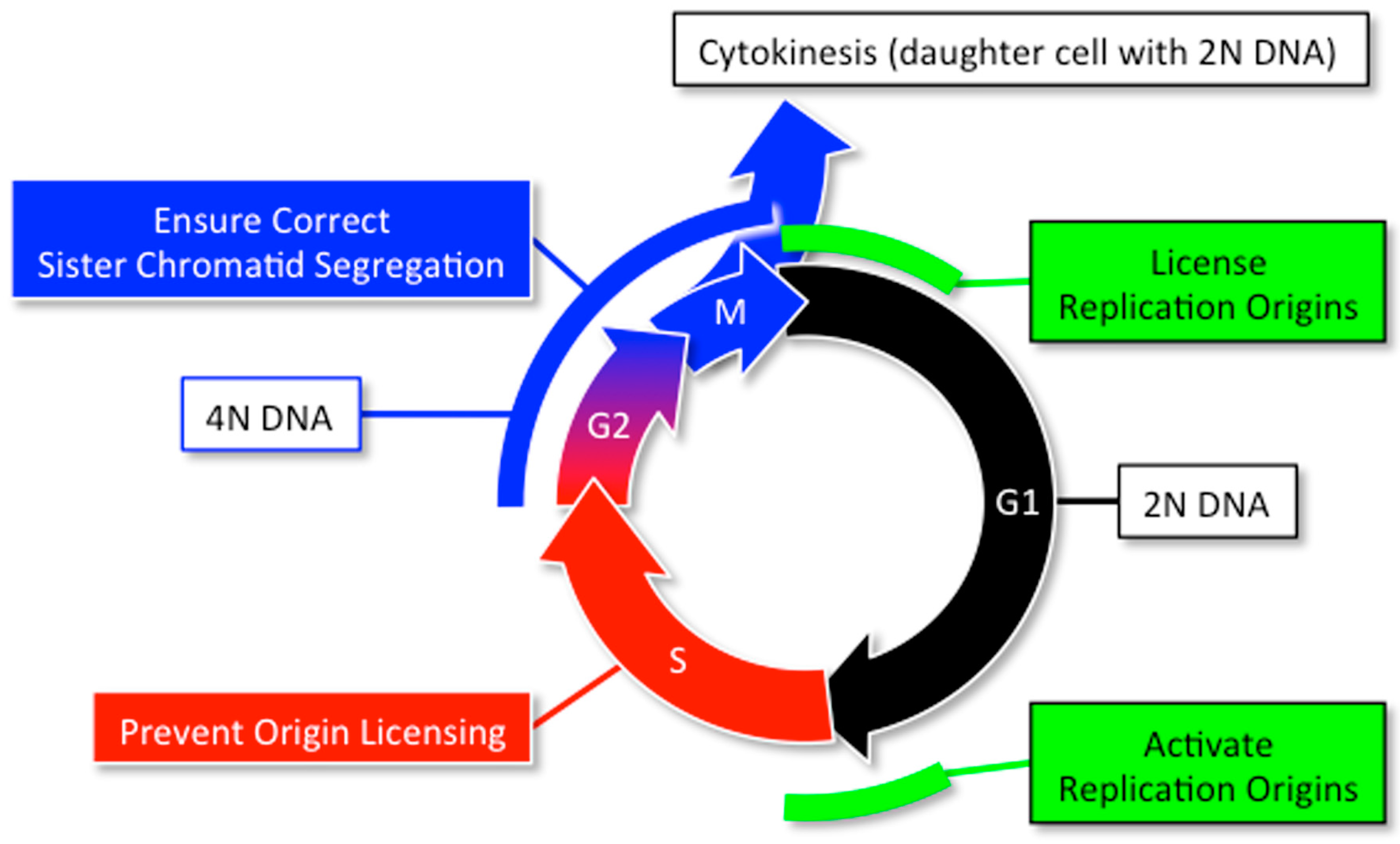
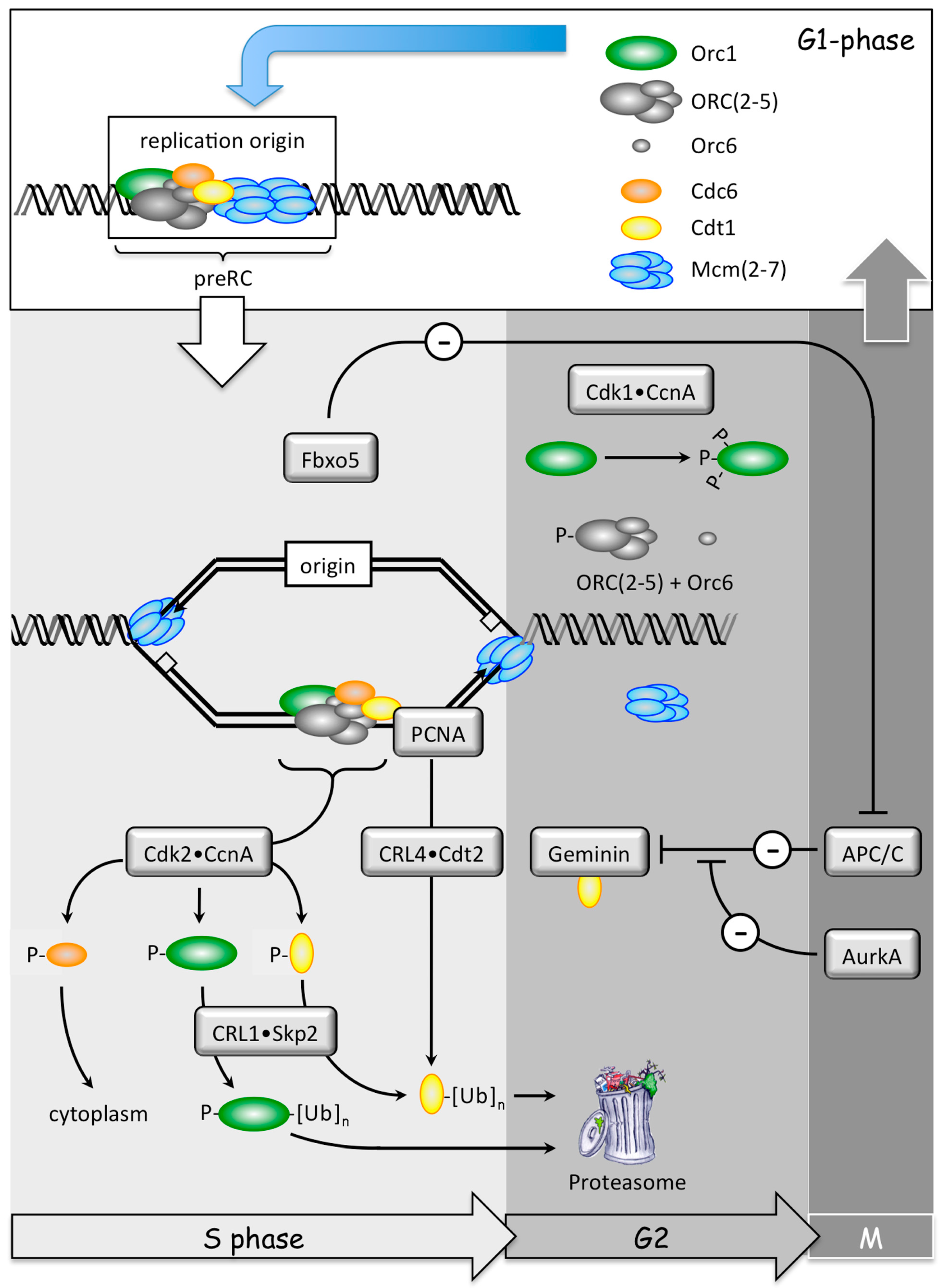
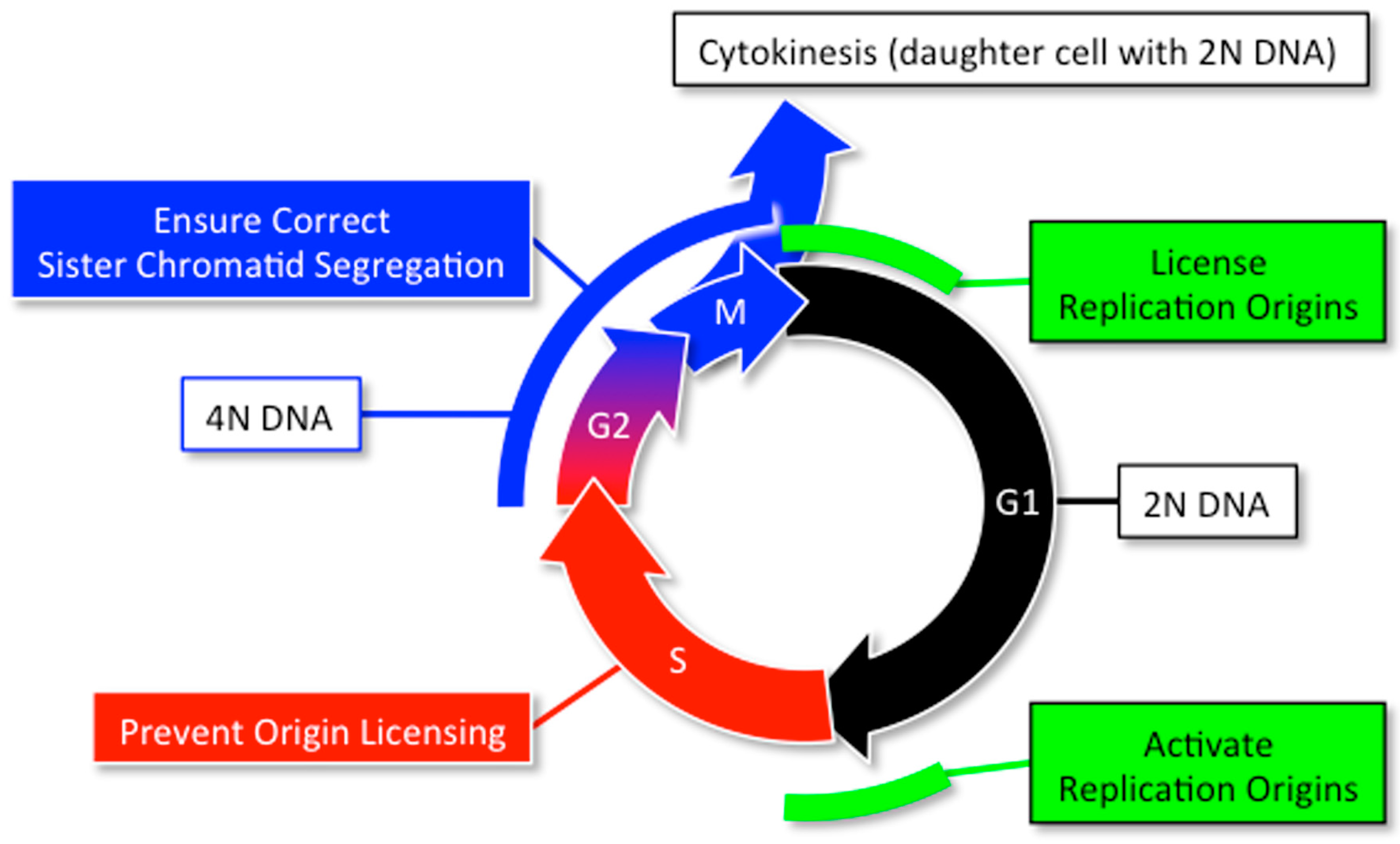
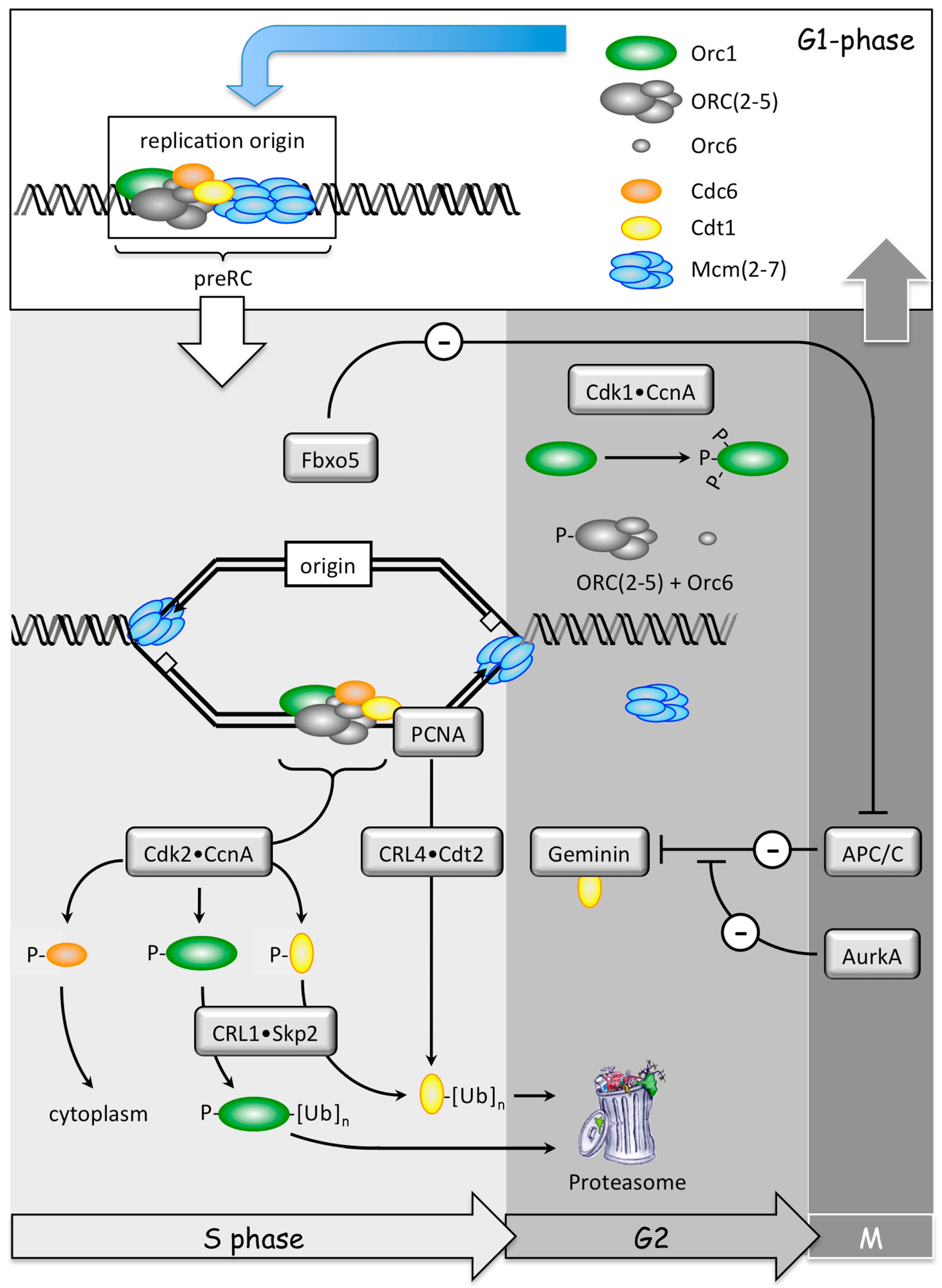
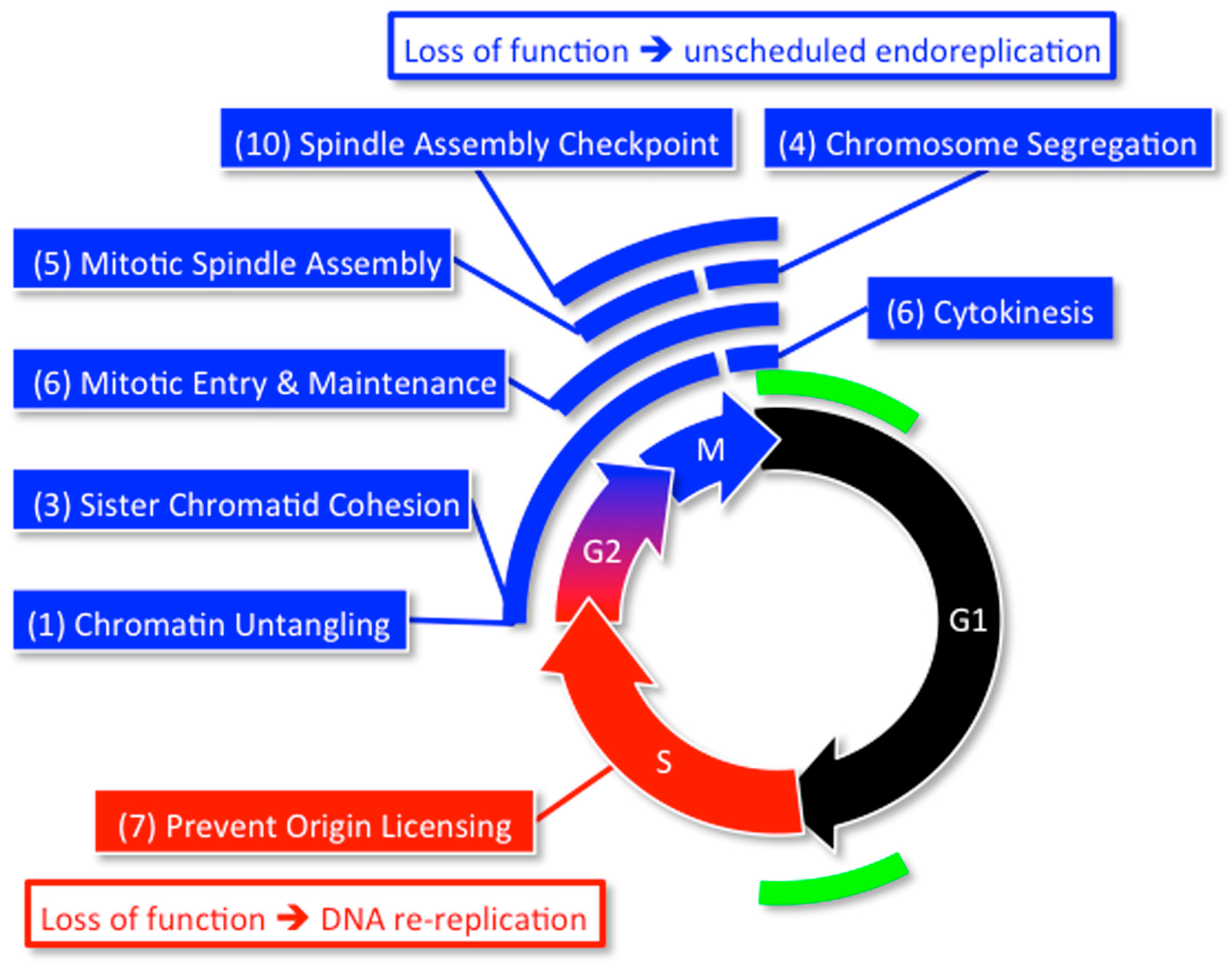
2. DNA Replication and Cancer
BOX 1. Developmentally Regulated Changes in Ploidy
BOX 2. Aberrant Forms Of Genome Duplication
2.1. Normal DNA Replication Produces Genetic Mutations
2.2. Cancer Cells Have Exceptionally High Levels of Genetic Alterations
2.3. Polyploidy Promotes Aneuploidy Which Promotes Cancer
2.4. Preventing Excess Genome Duplication Prevents Aneuploidy and Tumorigenesis
2.5. Excess Genome Duplication (EGD) Promotes Aneuploidy
2.6. Take-Home Lesson
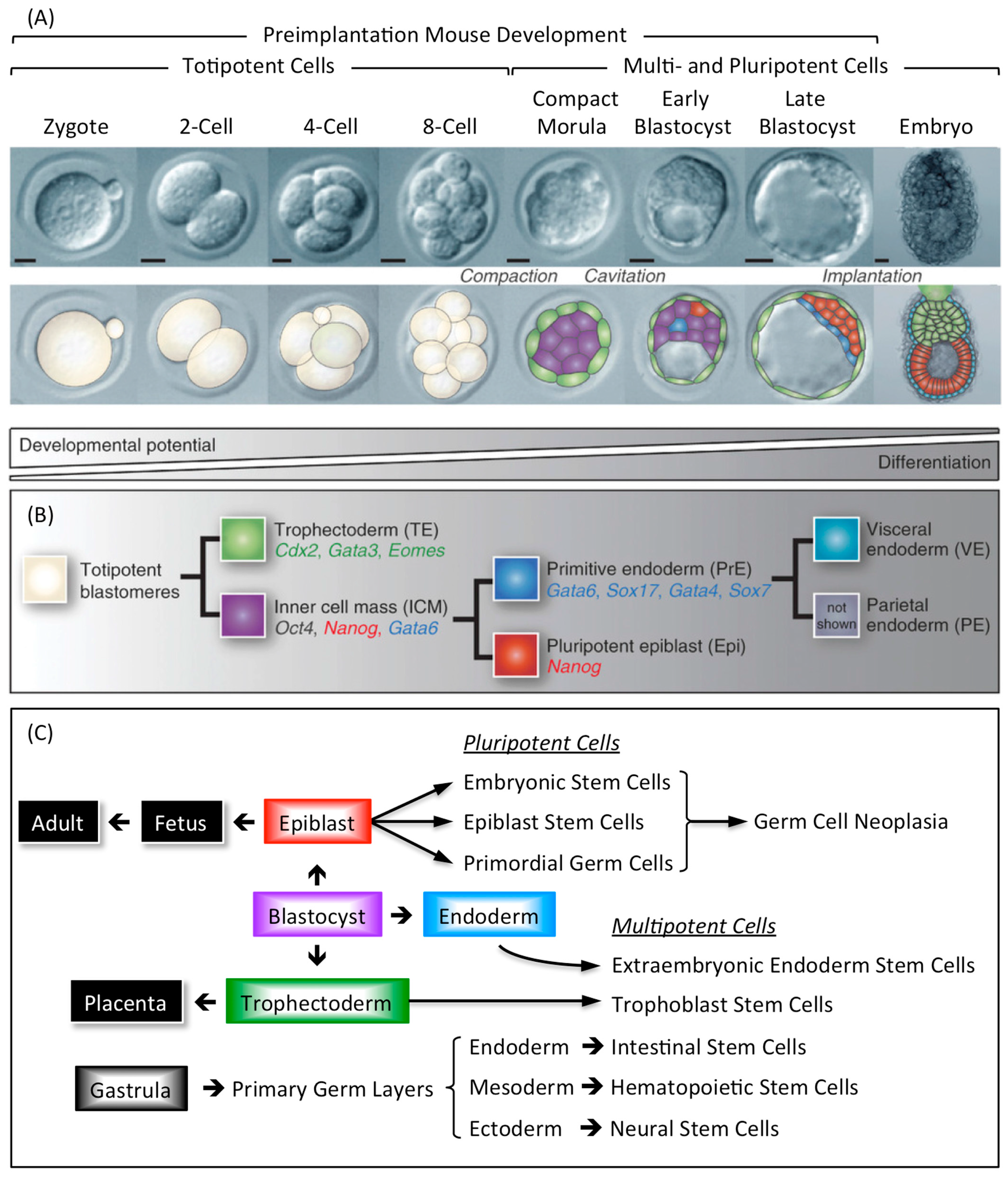
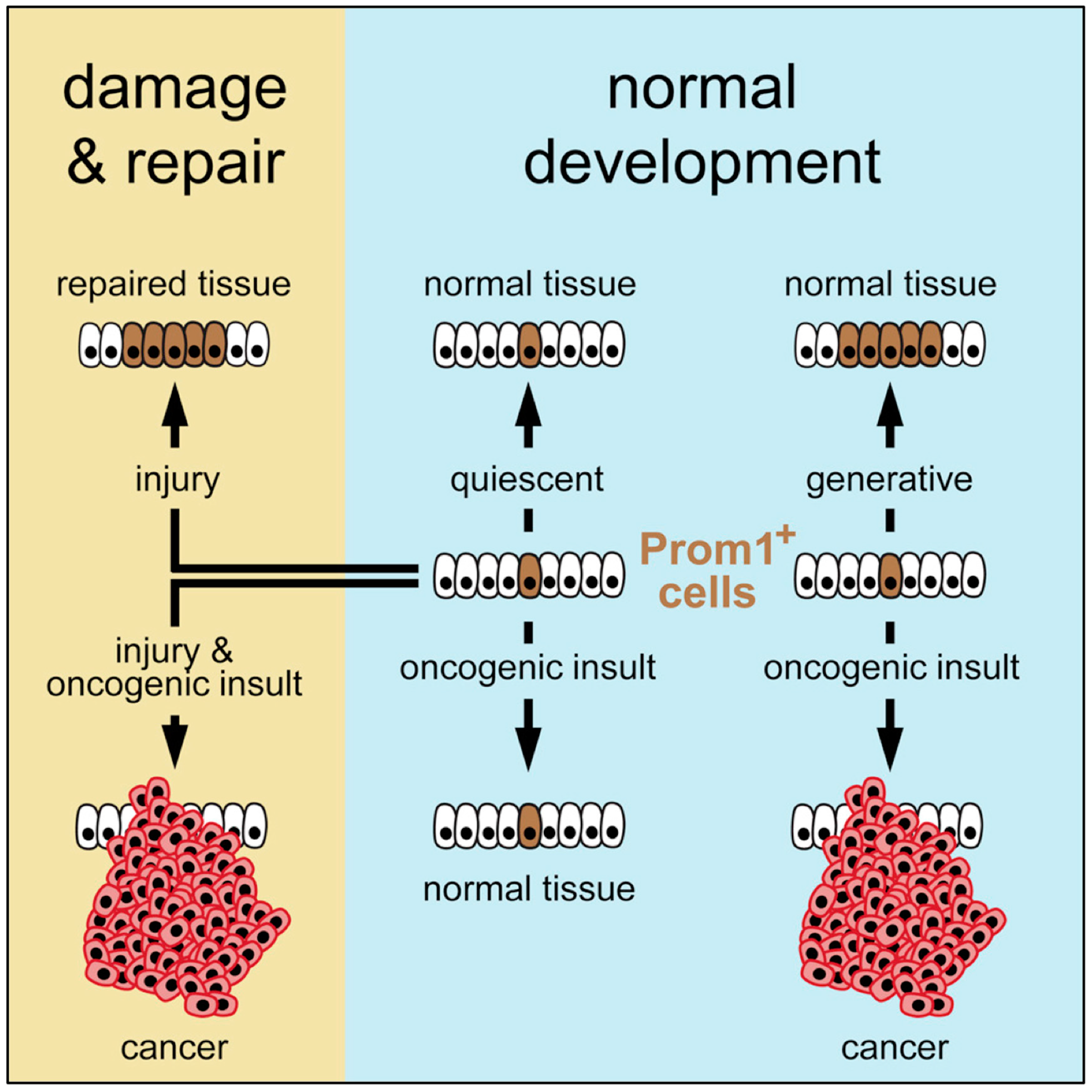
3. Stem Cells and Cancer
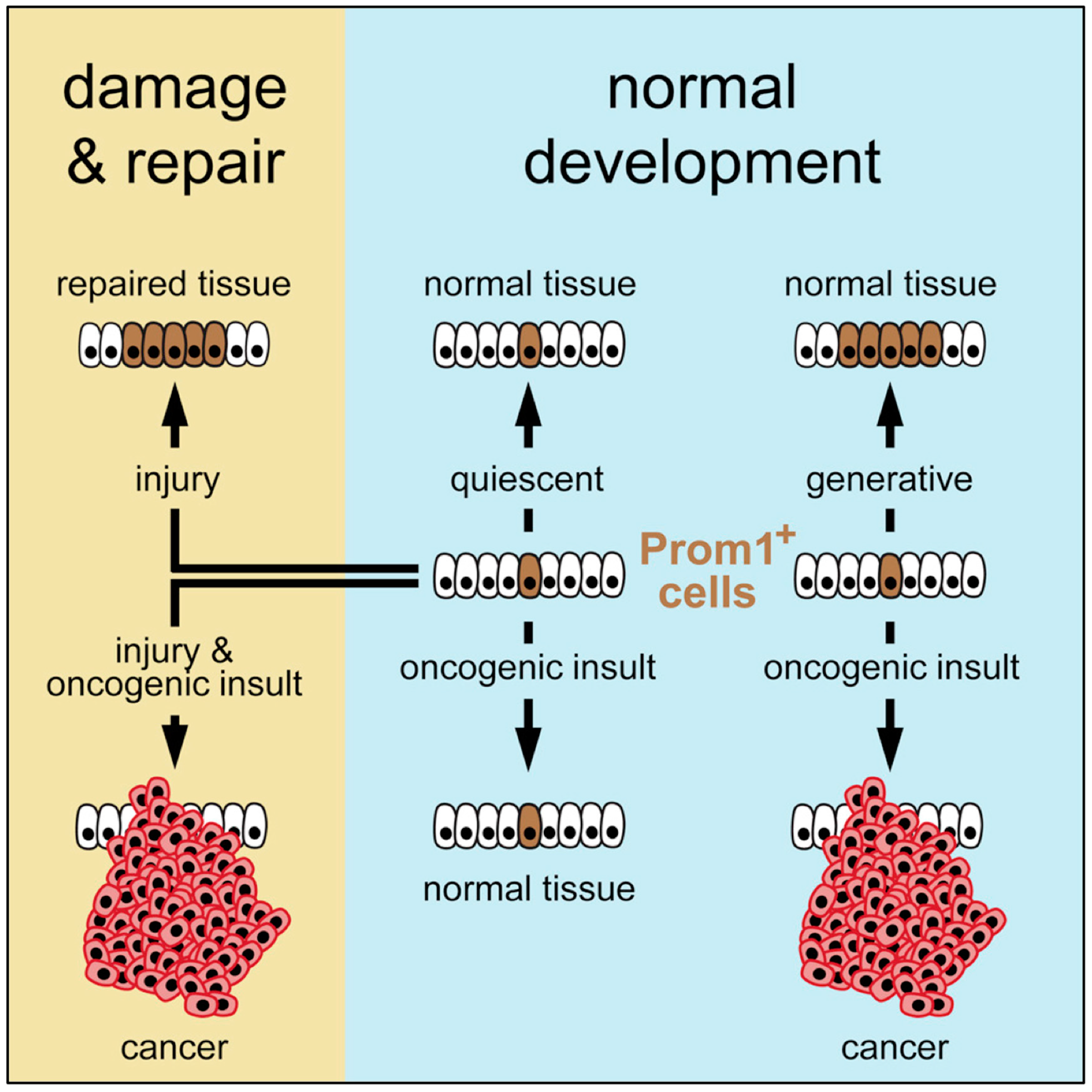
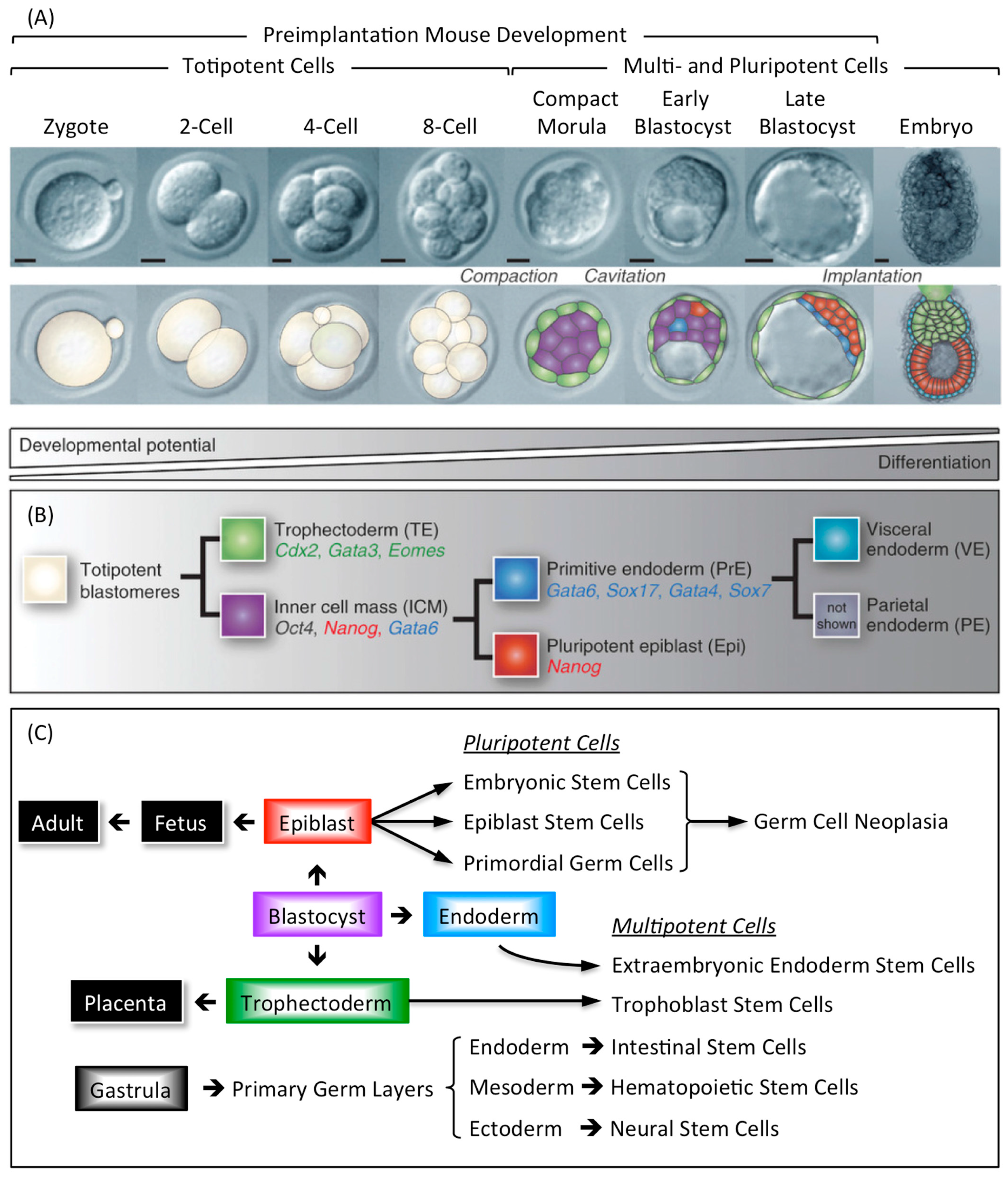
3.1. Tissue Specific Stem Cells
3.2. Embryo Specific Stem Cells
3.3. Pluripotent Stem Cells Are Potential Cancer Stem Cells (CSCs)
3.4. Take-Home Lesson
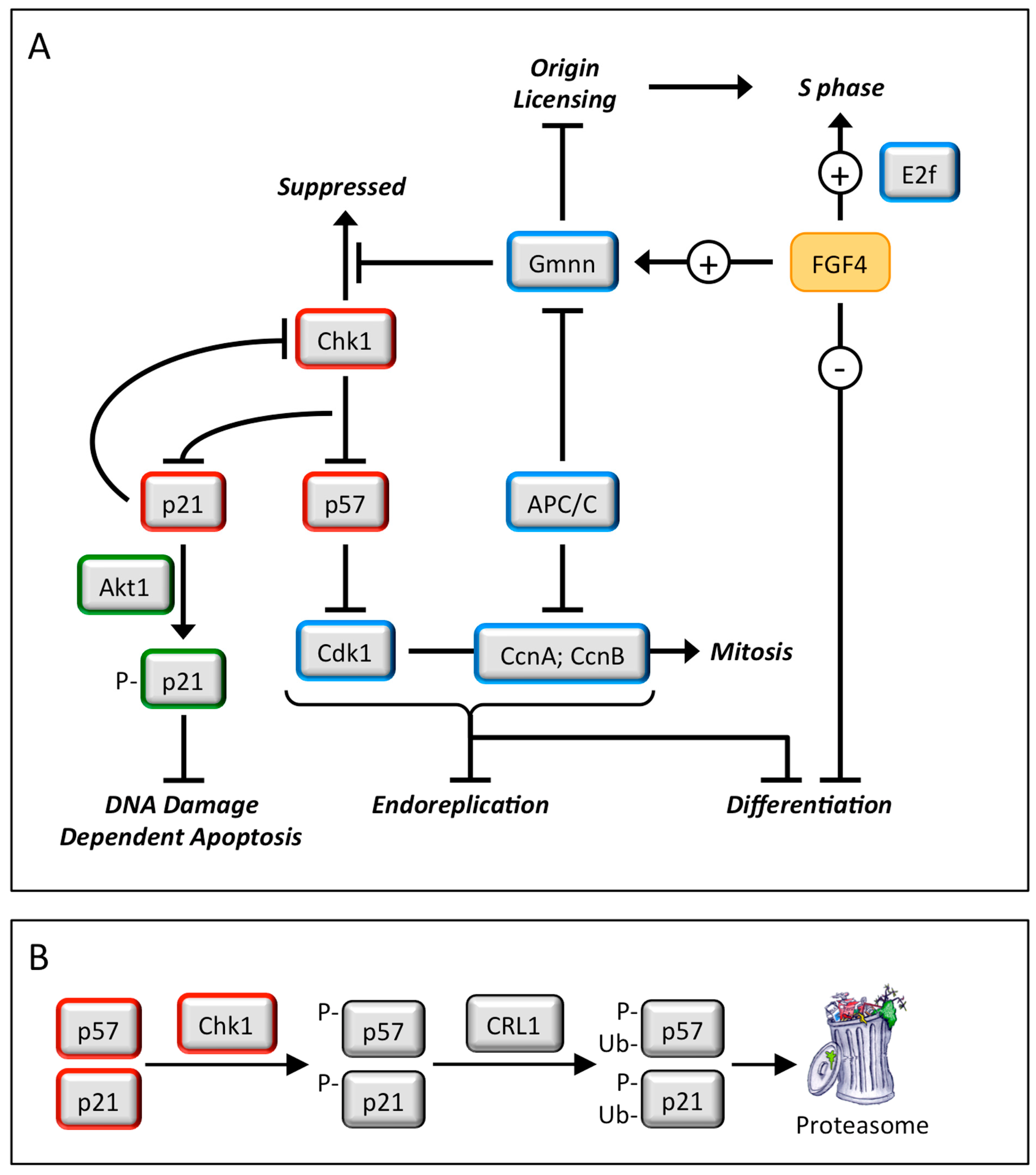
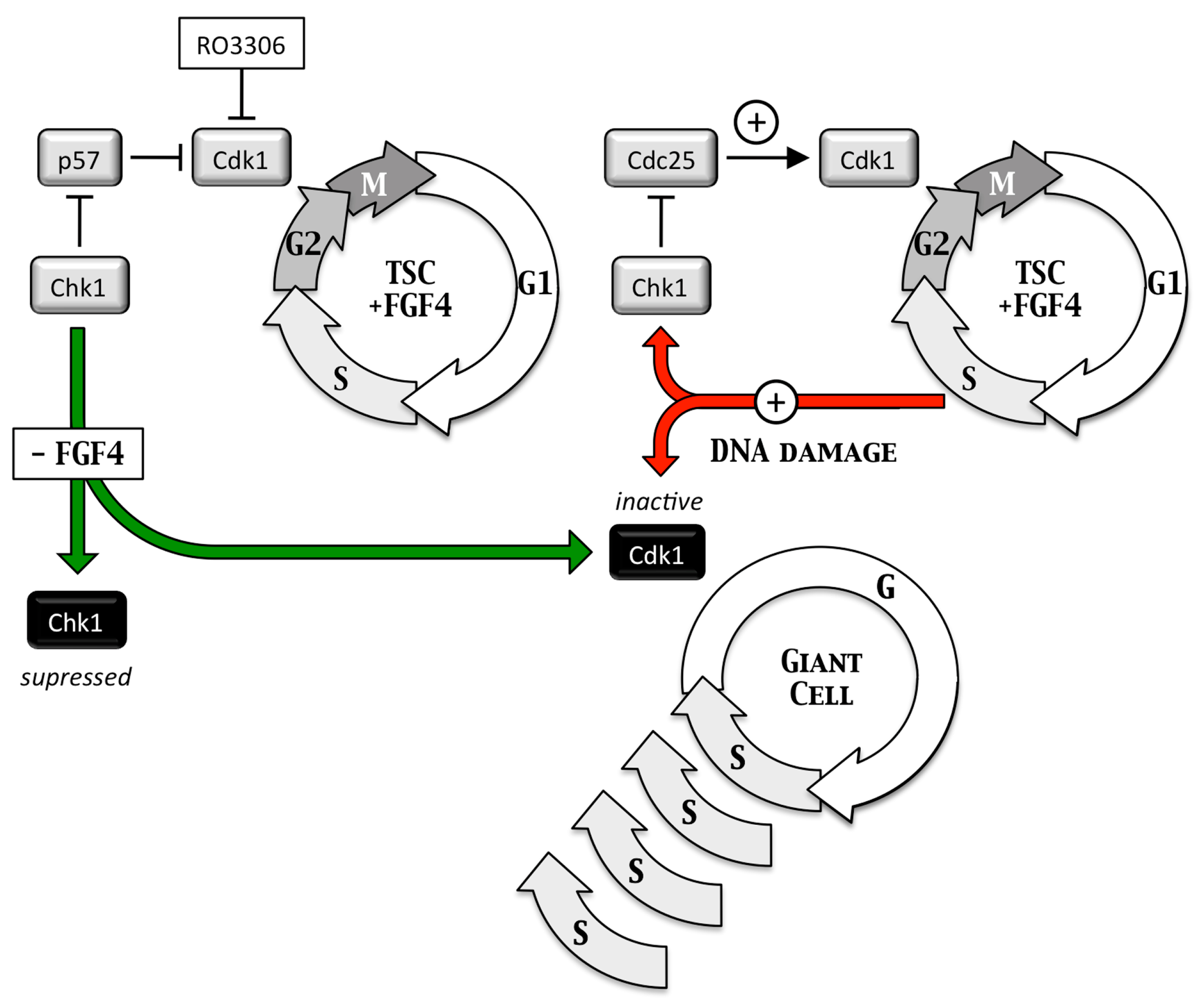
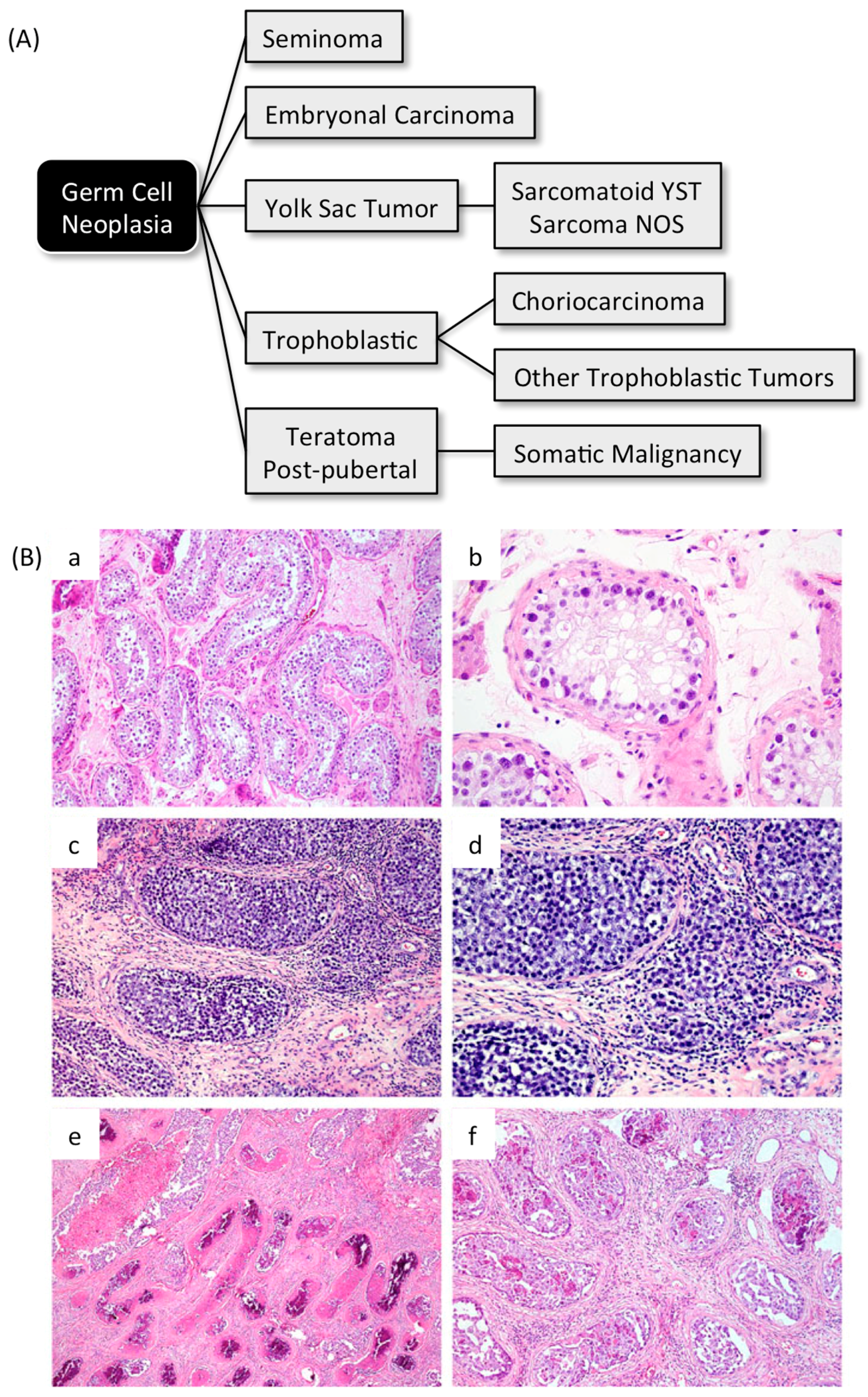
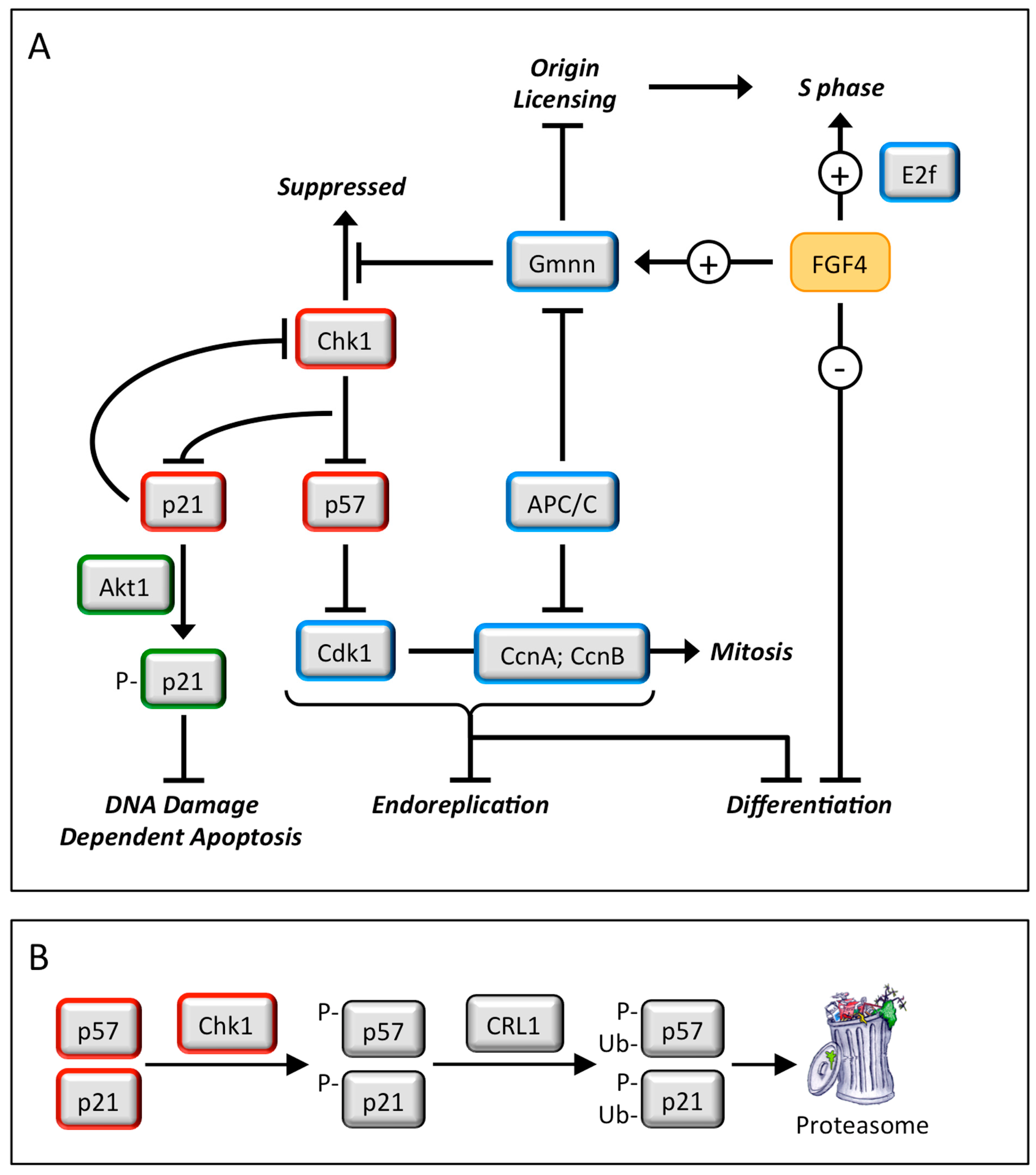
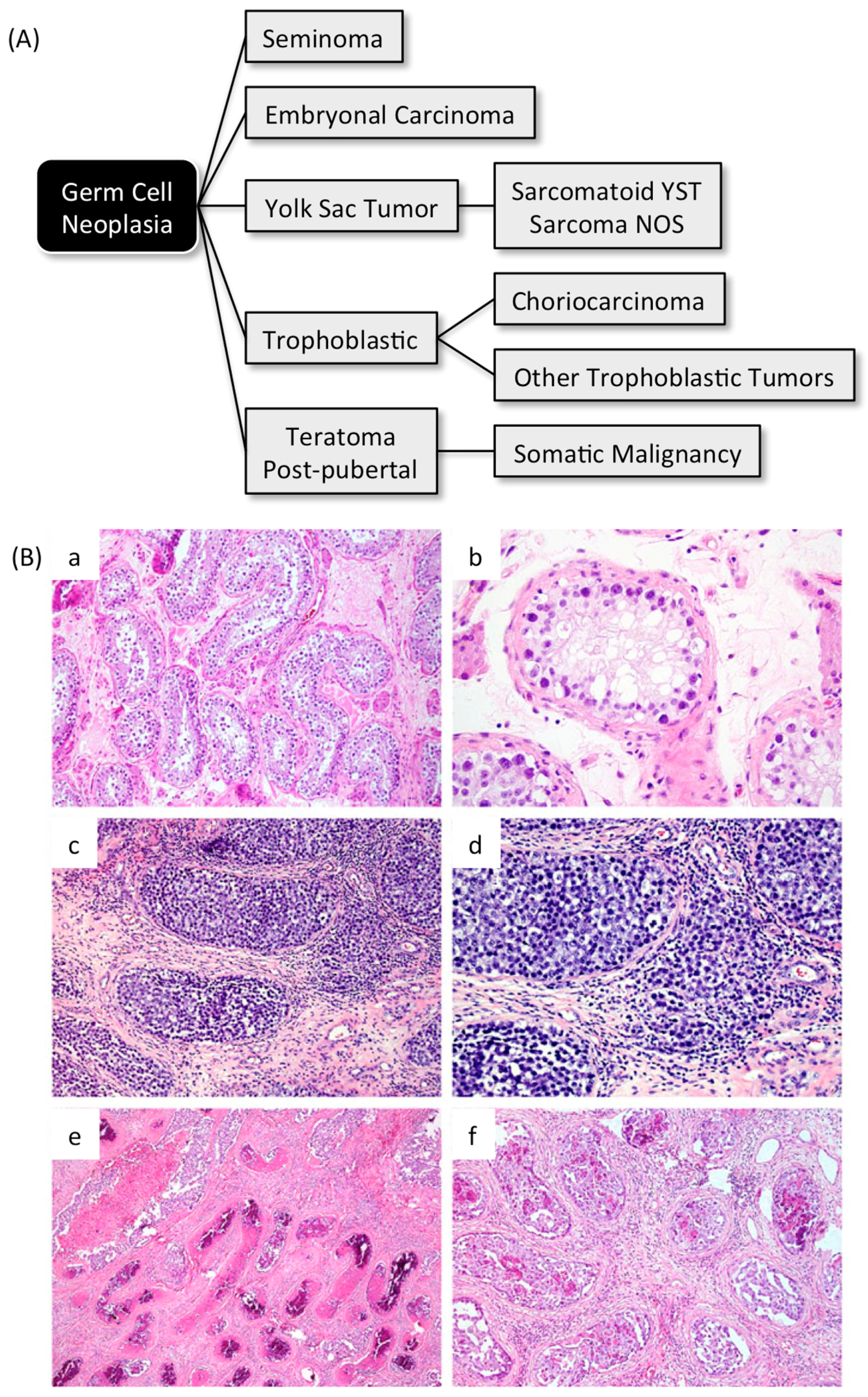
4. Geminin and Germ Cell Neoplasias
4.1. Germ Cell Neoplasias
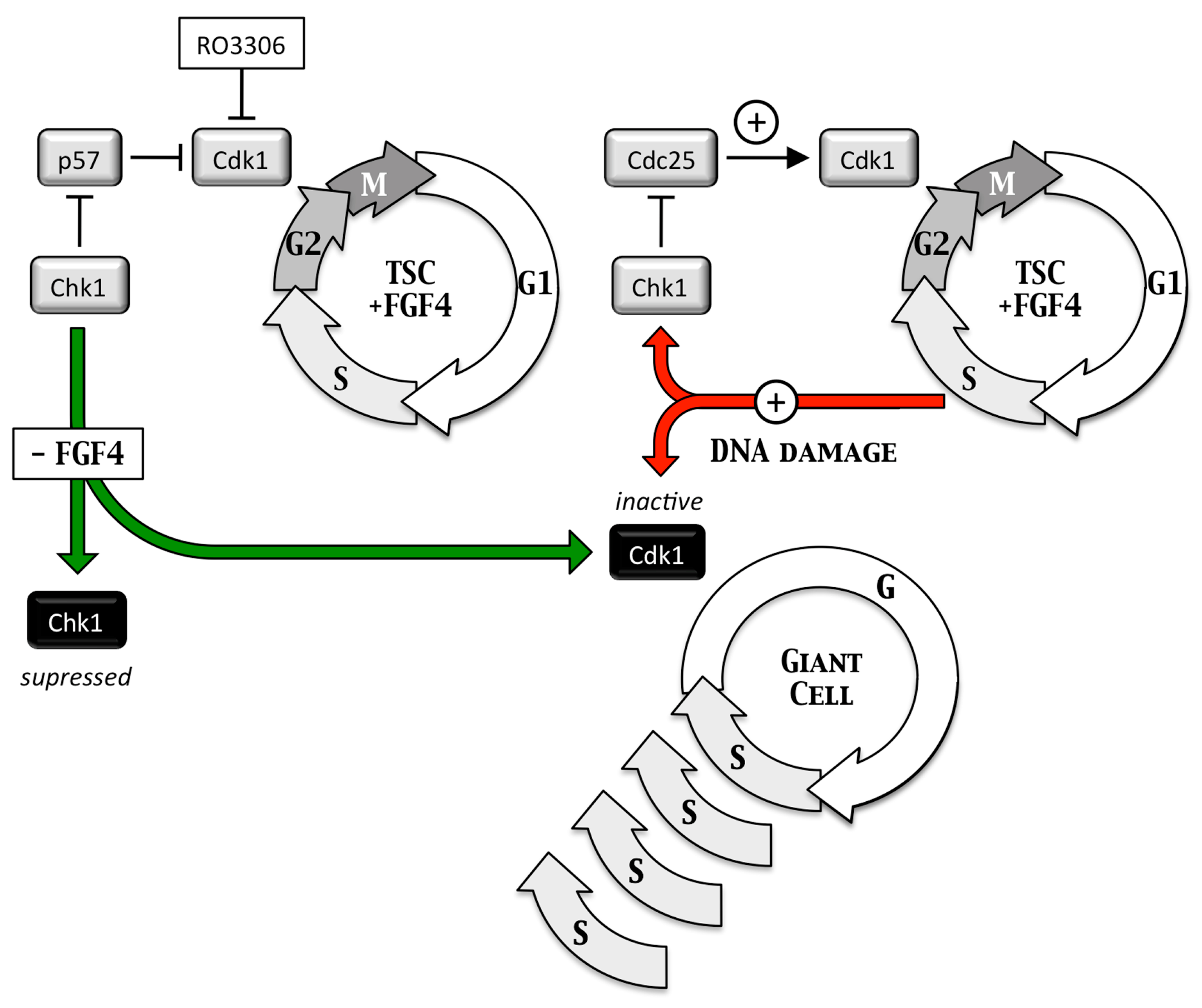
4.2. Geminin Is Essential for Totipotent and Pluripotent Cell Development
4.3. Geminin Prevents DNA Re-Replication Dependent Apoptosis in Pluripotent Cells
4.4. Take-Home Lesson
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- De Magalhaes, J.P. How ageing processes influence cancer. Nat. Rev. Cancer 2013, 13, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Powers, S.; Zhu, W.; Hannun, Y.A. Substantial contribution of extrinsic risk factors to cancer development. Nature 2016, 529, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Vogelstein, B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Rycaj, K.; Tang, D.G. Cell-of-origin of cancer versus cancer stem cells: Assays and interpretations. Cancer Res. 2015, 75, 4003–4011. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into scid mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Liu, X.; Laffin, B.; Chen, X.; Choy, G.; Jeter, C.R.; Calhoun-Davis, T.; Li, H.; Palapattu, G.S.; Pang, S.; et al. The psa(-/lo) prostate cancer cell population harbors self-renewing long-term tumor-propagating cells that resist castration. Cell Stem Cell 2012, 10, 556–569. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Vogelstein, B. Cancer risk: Role of environment-response. Science 2015, 347, 729–731. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Finkelstein, D.; Gao, C.; Shi, L.; Wang, Y.; Lopez-Terrada, D.; Wang, K.; Utley, S.; Pounds, S.; Neale, G.; et al. Multi-organ mapping of cancer risk. Cell 2016, 166, 1132.e7–1146.e7. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Polyak, K. Tumor heterogeneity: Causes and consequences. Biochim. Biophys. Acta 2010, 1805, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Song, L.; Zhu, P.; Zhang, B.; Tao, Y.; Xu, X.; Li, F.; Wu, K.; Liang, J.; Shao, D.; et al. Single-cell exome sequencing and monoclonal evolution of a jak2-negative myeloproliferative neoplasm. Cell 2012, 148, 873–885. [Google Scholar] [CrossRef] [PubMed]
- DePamphilis, M.L.; Bell, S.D. Genome Duplication: Concepts, Mechanisms, Evolution and Disease; Garland Science/Taylor & Francis Group: London, UK, 2011. [Google Scholar]
- Duncan, A.W. Aneuploidy, polyploidy and ploidy reversal in the liver. Semin. Cell Dev. Biol. 2013, 24, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Ullah, Z.; Lee, C.Y.; Depamphilis, M.L. Cip/kip cyclin-dependent protein kinase inhibitors and the road to polyploidy. Cell Div. 2009, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Lordier, L.; Bluteau, D.; Jalil, A.; Legrand, C.; Pan, J.; Rameau, P.; Jouni, D.; Bluteau, O.; Mercher, T.; Leon, C.; et al. Runx1-induced silencing of non-muscle myosin heavy chain IIB contributes to megakaryocyte polyploidization. Nat. Commun. 2012, 3, 717. [Google Scholar] [CrossRef] [PubMed]
- Melendez, J.; Stengel, K.; Zhou, X.; Chauhan, B.K.; Debidda, M.; Andreassen, P.; Lang, R.A.; Zheng, Y. Rhoa gtpase is dispensable for actomyosin regulation but is essential for mitosis in primary mouse embryonic fibroblasts. J. Biol. Chem. 2011, 286, 15132–15137. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Smith, E.; Ker, E.; Campbell, P.; Cheng, E.C.; Zou, S.; Lin, S.; Wang, L.; Halene, S.; Krause, D.S. Role of rhoa-specific guanine exchange factors in regulation of endomitosis in megakaryocytes. Dev. Cell 2012, 22, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Eliades, A.; Papadantonakis, N.; Ravid, K. New roles for cyclin e in megakaryocytic polyploidization. J. Biol. Chem. 2010, 285, 18909–18917. [Google Scholar] [CrossRef] [PubMed]
- Zielke, N.; Edgar, B.A.; DePamphilis, M.L. Endoreplication. Cold Spring Harb. Perspect. Biol. 2013, 5, a012948. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Z.; Ouseph, M.M.; Li, J.; Pecot, T.; Chokshi, V.; Kent, L.; Bae, S.; Byrne, M.; Duran, C.; Comstock, G.; et al. Canonical and atypical e2fs regulate the mammalian endocycle. Nat. Cell Biol. 2012, 14, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Zielke, N.; Kim, K.J.; Tran, V.; Shibutani, S.T.; Bravo, M.J.; Nagarajan, S.; van Straaten, M.; Woods, B.; von Dassow, G.; Rottig, C.; et al. Control of drosophila endocycles by e2f and crl4(cdt2). Nature 2011, 480, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Yang, V.S.; Carter, S.A.; Ng, Y.; Hyland, S.J.; Tachibana-Konwalski, K.; Fisher, R.A.; Sebire, N.J.; Seckl, M.J.; Pedersen, R.A.; Laskey, R.A.; et al. Distinct activities of the anaphase-promoting complex/cyclosome (apc/c) in mouse embryonic cells. Cell Cycle 2012, 11, 846–855. [Google Scholar] [CrossRef] [PubMed]
- De Renty, C.; Kaneko, K.J.; DePamphilis, M.L. The dual roles of geminin during trophoblast proliferation and differentiation. Dev. Biol. 2014, 387, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Ullah, Z.; de Renty, C.; DePamphilis, M.L. Checkpoint kinase 1 prevents cell cycle exit linked to terminal cell differentiation. Mol. Cell. Biol. 2011, 31, 4129–4143. [Google Scholar] [CrossRef] [PubMed]
- Hattori, N.; Davies, T.C.; Anson-Cartwright, L.; Cross, J.C. Periodic expression of the cyclin-dependent kinase inhibitor p57(kip2) in trophoblast giant cells defines a g2-like gap phase of the endocycle. Mol. Biol. Cell 2000, 11, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Ullah, Z.; Kohn, M.J.; Yagi, R.; Vassilev, L.T.; DePamphilis, M.L. Differentiation of trophoblast stem cells into giant cells is triggered by p57/kip2 inhibition of cdk1 activity. Genes Dev. 2008, 22, 3024–3036. [Google Scholar] [CrossRef] [PubMed]
- Ullah, Z.; Lee, C.Y.; Lilly, M.A.; DePamphilis, M.L. Developmentally programmed endoreduplication in animals. Cell Cycle 2009, 8, 1501–1509. [Google Scholar] [CrossRef] [PubMed]
- De Renty, C.; DePamphilis, M.L.; Ullah, Z. Cytoplasmic localization of p21 protects trophoblast giant cells from DNA damage induced apoptosis. PLoS ONE 2014, 9, e97434. [Google Scholar] [CrossRef] [PubMed]
- Gottifredi, V.; Karni-Schmidt, O.; Shieh, S.S.; Prives, C. P53 down-regulates chk1 through p21 and the retinoblastoma protein. Mol. Cell. Biol. 2001, 21, 1066–1076. [Google Scholar] [CrossRef] [PubMed]
- Gascoigne, K.E.; Taylor, S.S. How do anti-mitotic drugs kill cancer cells? J. Cell Sci. 2009, 122, 2579–2585. [Google Scholar] [CrossRef] [PubMed]
- Brito, D.A.; Rieder, C.L. Mitotic checkpoint slippage in humans occurs via cyclin b destruction in the presence of an active checkpoint. Curr. Biol. 2006, 16, 1194–1200. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, J.A.; Margolis, R.L.; Fotedar, R. Substrate degradation by the anaphase promoting complex occurs during mitotic slippage. Cell Cycle 2010, 9, 1792–1801. [Google Scholar] [CrossRef] [PubMed]
- Riffell, J.L.; Zimmerman, C.; Khong, A.; McHardy, L.M.; Roberge, M. Effects of chemical manipulation of mitotic arrest and slippage on cancer cell survival and proliferation. Cell Cycle 2009, 8, 3025–3038. [Google Scholar] [CrossRef] [PubMed]
- Storchova, Z.; Pellman, D. From polyploidy to aneuploidy, genome instability and cancer. Nat. Rev. Mol. Cell Biol. 2004, 5, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Di Leonardo, A.; Khan, S.H.; Linke, S.P.; Greco, V.; Seidita, G.; Wahl, G.M. DNA rereplication in the presence of mitotic spindle inhibitors in human and mouse fibroblasts lacking either p53 or prb function. Cancer Res. 1997, 57, 1013–1019. [Google Scholar] [PubMed]
- Khan, S.H.; Wahl, G.M. P53 and prb prevent rereplication in response to microtubule inhibitors by mediating a reversible g1 arrest. Cancer Res. 1998, 58, 396–401. [Google Scholar] [PubMed]
- Casenghi, M.; Mangiacasale, R.; Tuynder, M.; Caillet-Fauquet, P.; Elhajouji, A.; Lavia, P.; Mousset, S.; Kirsch-Volders, M.; Cundari, E. P53-independent apoptosis and p53-dependent block of DNA rereplication following mitotic spindle inhibition in human cells. Exp. Cell Res. 1999, 250, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Trakala, M.; Rodriguez-Acebes, S.; Maroto, M.; Symonds, C.E.; Santamaria, D.; Ortega, S.; Barbacid, M.; Mendez, J.; Malumbres, M. Functional reprogramming of polyploidization in megakaryocytes. Dev. Cell 2015, 32, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Diril, M.K.; Ratnacaram, C.K.; Padmakumar, V.C.; Du, T.; Wasser, M.; Coppola, V.; Tessarollo, L.; Kaldis, P. Cyclin-dependent kinase 1 (cdk1) is essential for cell division and suppression of DNA re-replication but not for liver regeneration. Proc. Natl. Acad. Sci. USA 2012, 109, 3826–3831. [Google Scholar] [CrossRef] [PubMed]
- Hochegger, H.; Dejsuphong, D.; Sonoda, E.; Saberi, A.; Rajendra, E.; Kirk, J.; Hunt, T.; Takeda, S. An essential role for cdk1 in s phase control is revealed via chemical genetics in vertebrate cells. J. Cell Biol. 2007, 178, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Green, R.A.; Paluch, E.; Oegema, K. Cytokinesis in animal cells. Annu. Rev. Cell Dev. Biol. 2012, 28, 29–58. [Google Scholar] [CrossRef] [PubMed]
- Blow, J.J.; Dutta, A. Preventing re-replication of chromosomal DNA. Nat. Rev. Mol. Cell Biol. 2005, 6, 476–486. [Google Scholar] [CrossRef] [PubMed]
- DePamphilis, M.L.; Blow, J.J.; Ghosh, S.; Saha, T.; Noguchi, K.; Vassilev, A. Regulating the licensing of DNA replication origins in metazoa. Curr. Opin. Cell Biol. 2006, 18, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, K.; On, K.F.; Diffley, J.F. Regulating DNA replication in eukarya. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Sonneville, R.; Querenet, M.; Craig, A.; Gartner, A.; Blow, J.J. The dynamics of replication licensing in live caenorhabditis elegans embryos. J. Cell Biol. 2012, 196, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Abbas, T.; Keaton, M.A.; Dutta, A. Genomic instability in cancer. Cold Spring Harb. Perspect. Biol. 2013, 5, a012914. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, A.; Lee, C.Y.; Vassilev, B.; Zhu, W.; Ormanoglu, P.; Martin, S.E.; DePamphilis, M.L. Identification of genes that are essential to restrict genome duplication to once per cell division. Oncotarget 2016, 7, 34956–34976. [Google Scholar] [CrossRef] [PubMed]
- Andreassen, P.R.; Lohez, O.D.; Lacroix, F.B.; Margolis, R.L. Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in g1. Mol. Biol. Cell 2001, 12, 1315–1328. [Google Scholar] [CrossRef] [PubMed]
- Cross, S.M.; Sanchez, C.A.; Morgan, C.A.; Schimke, M.K.; Ramel, S.; Idzerda, R.L.; Raskind, W.H.; Reid, B.J. A p53-dependent mouse spindle checkpoint. Science 1995, 267, 1353–1356. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.Y.; Grove, L.; Datta, N.S.; Long, M.W.; Prochownik, E.V. C-myc overexpression and p53 loss cooperate to promote genomic instability. Oncogene 1999, 18, 1177–1184. [Google Scholar] [CrossRef] [PubMed]
- Meraldi, P.; Honda, R.; Nigg, E.A. Aurora-a overexpression reveals tetraploidization as a major route to centrosome amplification in p53-/- cells. EMBO J. 2002, 21, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Sphyris, N.; Harrison, D.J. P53 deficiency exacerbates pleiotropic mitotic defects, changes in nuclearity and polyploidy in transdifferentiating pancreatic acinar cells. Oncogene 2005, 24, 2184–2194. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Kienitz, A.; Hofmann, I.; Muller, R.; Bastians, H. Crosstalk of the mitotic spindle assembly checkpoint with p53 to prevent polyploidy. Oncogene 2004, 23, 6845–6853. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.C.; Roberts, J.D.; Sabatino, R.D.; Myers, T.W.; Tan, C.K.; Downey, K.M.; So, A.G.; Bambara, R.A.; Kunkel, T.A. Fidelity of mammalian DNA replication and replicative DNA polymerases. Biochemistry 1991, 30, 11751–11759. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A. A mutator phenotype in cancer. Cancer Res. 2001, 61, 3230–3239. [Google Scholar] [PubMed]
- Prindle, M.J.; Fox, E.J.; Loeb, L.A. The mutator phenotype in cancer: Molecular mechanisms and targeting strategies. Curr. Drug Targets 2010, 11, 1296–1303. [Google Scholar] [CrossRef] [PubMed]
- Milo, R.; Phillips, R. Cell Biology by the Numbers; Garland Science: New York, NY, USA, 2016. [Google Scholar]
- Stoler, D.L.; Chen, N.; Basik, M.; Kahlenberg, M.S.; Rodriguez-Bigas, M.A.; Petrelli, N.J.; Anderson, G.R. The onset and extent of genomic instability in sporadic colorectal tumor progression. Proc. Natl. Acad. Sci. USA 1999, 96, 15121–15126. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Vogelstein, B.; Parmigiani, G. Half or more of the somatic mutations in cancers of self-renewing tissues originate prior to tumor initiation. Proc. Natl. Acad. Sci. USA 2013, 110, 1999–2004. [Google Scholar] [CrossRef] [PubMed]
- Garraway, L.A.; Lander, E.S. Lessons from the cancer genome. Cell 2013, 153, 17–37. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Marchionni, L.; Nowak, M.A.; Parmigiani, G.; Vogelstein, B. Only three driver gene mutations are required for the development of lung and colorectal cancers. Proc. Natl. Acad. Sci. USA 2015, 112, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, A.Y.; Seget, K.; Moeller, G.K.; de Pagter, M.S.; de Roos, J.A.; Durrbaum, M.; Kuffer, C.; Muller, S.; Zaman, G.J.; Kloosterman, W.P.; et al. Chromosomal instability, tolerance of mitotic errors and multidrug resistance are promoted by tetraploidization in human cells. Cell Cycle 2015, 14, 2810–2820. [Google Scholar] [CrossRef] [PubMed]
- Masramon, L.; Vendrell, E.; Tarafa, G.; Capella, G.; Miro, R.; Ribas, M.; Peinado, M.A. Genetic instability and divergence of clonal populations in colon cancer cells in vitro. J. Cell Sci. 2006, 119, 1477–1482. [Google Scholar] [CrossRef] [PubMed]
- Geigl, J.B.; Obenauf, A.C.; Schwarzbraun, T.; Speicher, M.R. Defining ‘chromosomal instability’. Trends Genet. 2008, 24, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Swanton, C. Chromosomal instability, aneuploidy, and cancer. Front. Oncol. 2014, 4, 161. [Google Scholar] [CrossRef] [PubMed]
- Giam, M.; Rancati, G. Aneuploidy and chromosomal instability in cancer: A jackpot to chaos. Cell Div. 2015, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.J.; Resio, B.; Pellman, D. Causes and consequences of aneuploidy in cancer. Nat. Rev. Genet. 2012, 13, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Davoli, T.; de Lange, T. The causes and consequences of polyploidy in normal development and cancer. Annu. Rev. Cell Dev. Biol. 2011, 27, 585–610. [Google Scholar] [CrossRef] [PubMed]
- Gerling, M.; Meyer, K.F.; Fuchs, K.; Igl, B.W.; Fritzsche, B.; Ziegler, A.; Bader, F.; Kujath, P.; Schimmelpenning, H.; Bruch, H.P.; et al. High frequency of aneuploidy defines ulcerative colitis-associated carcinomas: A prognostic comparison to sporadic colorectal carcinomas. Ann. Surg. 2010, 252, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Gemoll, T.; Auer, G.; Ried, T.; Habermann, J.K. Genetic instability and disease prognostication. Recent Results Cancer Res. 2015, 200, 81–94. [Google Scholar] [PubMed]
- Walther, A.; Houlston, R.; Tomlinson, I. Association between chromosomal instability and prognosis in colorectal cancer: A meta-analysis. Gut 2008, 57, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhsng, C.Z.; Wala, J.; Mermel, C.H.; et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Bandi, M.; Nitta, M.; Ivanova, E.V.; Bronson, R.T.; Pellman, D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 2005, 437, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.G.; Makitalo, M.; Yang, D.; Chinnappan, D.; St Hilaire, C.; Ravid, K. Deregulated aurora-b induced tetraploidy promotes tumorigenesis. FASEB J. 2009, 23, 2741–2748. [Google Scholar] [CrossRef] [PubMed]
- Sheltzer, J.M.; Blank, H.M.; Pfau, S.J.; Tange, Y.; George, B.M.; Humpton, T.J.; Brito, I.L.; Hiraoka, Y.; Niwa, O.; Amon, A. Aneuploidy drives genomic instability in yeast. Science 2011, 333, 1026–1030. [Google Scholar] [CrossRef] [PubMed]
- Solomon, D.A.; Kim, T.; Diaz-Martinez, L.A.; Fair, J.; Elkahloun, A.G.; Harris, B.T.; Toretsky, J.A.; Rosenberg, S.A.; Shukla, N.; Ladanyi, M.; et al. Mutational inactivation of stag2 causes aneuploidy in human cancer. Science 2011, 333, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Kolodner, R.D.; Cleveland, D.W.; Putnam, C.D. Cancer. Aneuploidy drives a mutator phenotype in cancer. Science 2011, 333, 942–943. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Depamphilis, M.L. Selective killing of cancer cells by suppression of geminin activity. Cancer Res. 2009, 69, 4870–4877. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Chen, Y.; Dutta, A. Rereplication by depletion of geminin is seen regardless of p53 status and activates a g2/m checkpoint. Mol. Cell. Biol. 2004, 24, 7140–7150. [Google Scholar] [CrossRef] [PubMed]
- Di Bonito, M.; Cantile, M.; Collina, F.; Scognamiglio, G.; Cerrone, M.; La Mantia, E.; Barbato, A.; Liguori, G.; Botti, G. Overexpression of cell cycle progression inhibitor geminin is associated with tumor stem-like phenotype of triple-negative breast cancer. J. Breast Cancer 2012, 15, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.E.; Kim, D.G.; Lee, K.J.; Son, J.G.; Song, M.Y.; Park, Y.M.; Kim, J.J.; Cho, S.W.; Chi, S.G.; Cheong, H.S.; et al. Frequent amplification of cenpf, gmnn and cdk13 genes in hepatocellular carcinomas. PLoS ONE 2012, 7, e43223. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, Z.; Malik, R.; Mullins, N.; Maric, C.; Luk, H.; Horio, D.; Hernandez, B.; Killeen, J.; Elshamy, W.M. Geminin overexpression induces mammary tumors via suppressing cytokinesis. Oncotarget 2011, 2, 1011–1027. [Google Scholar] [CrossRef] [PubMed]
- Adler-Wailes, D.C.; Kramer, J.A.; DePamphilis, M.L. Geminin is essential for pluripotent cell viability during teratoma formation, but not for differentiated cell viability during teratoma expansion. Stem Cells Dev. 2016. [Google Scholar] [CrossRef] [PubMed]
- DePamphilis, M.L. The ‘orc cycle’: A novel pathway for regulating eukaryotic DNA replication. Gene 2003, 310, 1–15. [Google Scholar] [CrossRef]
- Lee, K.Y.; Bang, S.W.; Yoon, S.W.; Lee, S.H.; Yoon, J.B.; Hwang, D.S. Phosphorylation of orc2 protein dissociates origin recognition complex from chromatin and replication origins. J. Biol. Chem. 2012, 287, 11891–11898. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, K.; Stillman, B. Atp-dependent assembly of the human origin recognition complex. J. Biol. Chem. 2007, 282, 32370–32383. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, K.; Vassilev, A.; Ghosh, S.; Yates, J.L.; DePamphilis, M.L. The bah domain facilitates the ability of human orc1 protein to activate replication origins in vivo. EMBO J. 2006, 25, 5372–5382. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Vassilev, A.P.; Zhang, J.; Zhao, Y.; DePamphilis, M.L. Assembly of the human origin recognition complex occurs through independent nuclear localization of its components. J. Biol. Chem. 2011, 286, 23831–23841. [Google Scholar] [CrossRef] [PubMed]
- Kara, N.; Hossain, M.; Prasanth, S.G.; Stillman, B. Orc1 binding to mitotic chromosomes precedes spatial patterning during g1 phase and assembly of the origin recognition complex in human cells. J. Biol. Chem. 2015, 290, 12355–12369. [Google Scholar] [CrossRef] [PubMed]
- Klotz-Noack, K.; McIntosh, D.; Schurch, N.; Pratt, N.; Blow, J.J. Re-replication induced by geminin depletion occurs from g2 and is enhanced by checkpoint activation. J. Cell Sci. 2012, 125, 2436–2445. [Google Scholar] [CrossRef] [PubMed]
- Ballabeni, A.; Melixetian, M.; Zamponi, R.; Masiero, L.; Marinoni, F.; Helin, K. Human geminin promotes pre-rc formation and DNA replication by stabilizing cdt1 in mitosis. EMBO J. 2004, 23, 3122–3132. [Google Scholar] [CrossRef] [PubMed]
- Mailand, N.; Diffley, J.F. Cdks promote DNA replication origin licensing in human cells by protecting cdc6 from apc/c-dependent proteolysis. Cell 2005, 122, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Kittler, R.; Pelletier, L.; Heninger, A.K.; Slabicki, M.; Theis, M.; Miroslaw, L.; Poser, I.; Lawo, S.; Grabner, H.; Kozak, K.; et al. Genome-scale rnai profiling of cell division in human tissue culture cells. Nat. Cell Biol. 2007, 9, 1401–1412. [Google Scholar] [CrossRef] [PubMed]
- Mukherji, M.; Bell, R.; Supekova, L.; Wang, Y.; Orth, A.P.; Batalov, S.; Miraglia, L.; Huesken, D.; Lange, J.; Martin, C.; et al. Genome-wide functional analysis of human cell-cycle regulators. Proc. Natl. Acad. Sci. USA 2006, 103, 14819–14824. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.L.; Compton, D.A. Chromosome missegregation in human cells arises through specific types of kinetochore-microtubule attachment errors. Proc. Natl. Acad. Sci. USA 2011, 108, 17974–17978. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.Y.; Wood, J.L.; Minter-Dykhouse, K.; Ye, L.; Saunders, T.L.; Yu, X.; Chen, J. Polo-like kinase 1 is essential for early embryonic development and tumor suppression. Mol. Cell. Biol. 2008, 28, 6870–6876. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Portoles, C.; Bird, A.W.; Hyman, A.; Canamero, M.; Perez de Castro, I.; Malumbres, M. Tpx2 controls spindle integrity, genome stability, and tumor development. Cancer Res. 2012, 72, 1518–1528. [Google Scholar] [CrossRef] [PubMed]
- Castillo, A.; Morse, H.C., 3rd; Godfrey, V.L.; Naeem, R.; Justice, M.J. Overexpression of eg5 causes genomic instability and tumor formation in mice. Cancer Res. 2007, 67, 10138–10147. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.Y.; Wood, J.L.; Ye, L.; Minter-Dykhouse, K.; Saunders, T.L.; Yu, X.; Chen, J. Aurora a is essential for early embryonic development and tumor suppression. J. Biol. Chem. 2008, 283, 31785–31790. [Google Scholar] [CrossRef] [PubMed]
- Cutts, S.M.; Fowler, K.J.; Kile, B.T.; Hii, L.L.; O'Dowd, R.A.; Hudson, D.F.; Saffery, R.; Kalitsis, P.; Earle, E.; Choo, K.H. Defective chromosome segregation, microtubule bundling and nuclear bridging in inner centromere protein gene (incenp)-disrupted mice. Hum. Mol. Genet. 1999, 8, 1145–1155. [Google Scholar] [CrossRef] [PubMed]
- Uren, A.G.; Wong, L.; Pakusch, M.; Fowler, K.J.; Burrows, F.J.; Vaux, D.L.; Choo, K.H. Survivin and the inner centromere protein incenp show similar cell-cycle localization and gene knockout phenotype. Curr. Biol. 2000, 10, 1319–1328. [Google Scholar] [CrossRef]
- Yamanaka, Y.; Heike, T.; Kumada, T.; Shibata, M.; Takaoka, Y.; Kitano, A.; Shiraishi, K.; Kato, T.; Nagato, M.; Okawa, K.; et al. Loss of borealin/dasrab leads to defective cell proliferation, p53 accumulation and early embryonic lethality. Mech. Dev. 2008, 125, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Miranda, G.; Trakala, M.; Martin, J.; Escobar, B.; Gonzalez, A.; Ghyselinck, N.B.; Ortega, S.; Canamero, M.; Perez de Castro, I.; Malumbres, M. Genetic disruption of aurora b uncovers an essential role for aurora c during early mammalian development. Development 2011, 138, 2661–2672. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Jeganathan, K.B.; Malureanu, L.; Perez-Terzic, C.; Terzic, A.; van Deursen, J.M. Early aging-associated phenotypes in bub3/rae1 haploinsufficient mice. J. Cell Biol. 2006, 172, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Babu, J.R.; Jeganathan, K.B.; Baker, D.J.; Wu, X.; Kang-Decker, N.; van Deursen, J.M. Rae1 is an essential mitotic checkpoint regulator that cooperates with bub3 to prevent chromosome missegregation. J. Cell Biol. 2003, 160, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Jeganathan, K.B.; Cameron, J.D.; Thompson, M.; Juneja, S.; Kopecka, A.; Kumar, R.; Jenkins, R.B.; de Groen, P.C.; Roche, P.; et al. Bubr1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat. Genet. 2004, 36, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Homer, H.A.; McDougall, A.; Levasseur, M.; Yallop, K.; Murdoch, A.P.; Herbert, M. Mad2 prevents aneuploidy and premature proteolysis of cyclin b and securin during meiosis i in mouse oocytes. Genes Dev. 2005, 19, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Niault, T.; Hached, K.; Sotillo, R.; Sorger, P.K.; Maro, B.; Benezra, R.; Wassmann, K. Changing mad2 levels affects chromosome segregation and spindle assembly checkpoint control in female mouse meiosis i. PLoS ONE 2007, 2, e1165. [Google Scholar] [CrossRef] [PubMed]
- Michel, L.S.; Liberal, V.; Chatterjee, A.; Kirchwegger, R.; Pasche, B.; Gerald, W.; Dobles, M.; Sorger, P.K.; Murty, V.V.; Benezra, R. Mad2 haplo-insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature 2001, 409, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Chi, Y.H.; Ward, J.M.; Cheng, L.I.; Yasunaga, J.; Jeang, K.T. Spindle assembly checkpoint and p53 deficiencies cooperate for tumorigenesis in mice. Int. J. Cancer 2009, 124, 1483–1489. [Google Scholar] [CrossRef] [PubMed]
- Foijer, F.; Xie, S.Z.; Simon, J.E.; Bakker, P.L.; Conte, N.; Davis, S.H.; Kregel, E.; Jonkers, J.; Bradley, A.; Sorger, P.K. Chromosome instability induced by mps1 and p53 mutation generates aggressive lymphomas exhibiting aneuploidy-induced stress. Proc. Natl. Acad. Sci. USA 2014, 111, 13427–13432. [Google Scholar] [CrossRef] [PubMed]
- Yamada, H.Y.; Yao, Y.; Wang, X.; Zhang, Y.; Huang, Y.; Dai, W.; Rao, C.V. Haploinsufficiency of sgo1 results in deregulated centrosome dynamics, enhanced chromosomal instability and colon tumorigenesis. Cell Cycle 2012, 11, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Wirth, K.G.; Wutz, G.; Kudo, N.R.; Desdouets, C.; Zetterberg, A.; Taghybeeglu, S.; Seznec, J.; Ducos, G.M.; Ricci, R.; Firnberg, N.; et al. Separase: A universal trigger for sister chromatid disjunction but not chromosome cycle progression. J. Cell Biol. 2006, 172, 847–860. [Google Scholar] [CrossRef] [PubMed]
- Di Fiore, B.; Pines, J. Defining the role of emi1 in the DNA replication-segregation cycle. Chromosoma 2008, 117, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Musacchio, A. The molecular biology of spindle assembly checkpoint signaling dynamics. Curr. Biol. 2015, 25, R1002–1018. [Google Scholar] [CrossRef] [PubMed]
- Foley, E.A.; Kapoor, T.M. Microtubule attachment and spindle assembly checkpoint signalling at the kinetochore. Nat. Rev. Mol. Cell Biol. 2013, 14, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chow, J.P.; Poon, R.Y. Inhibition of eg5 acts synergistically with checkpoint abrogation in promoting mitotic catastrophe. Mol. Cancer Res. 2012, 10, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Carmena, M.; Wheelock, M.; Funabiki, H.; Earnshaw, W.C. The chromosomal passenger complex (cpc): From easy rider to the godfather of mitosis. Nat. Rev. Mol. Cell Biol. 2012, 13, 789–803. [Google Scholar] [CrossRef] [PubMed]
- Remeseiro, S.; Losada, A. Cohesin, a chromatin engagement ring. Curr. Opin. Cell Biol. 2013, 25, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, S.; Yuan, J.; Wang, Z.B.; Sun, S.C.; Schatten, H.; Sun, Q.Y. Bub3 is a spindle assembly checkpoint protein regulating chromosome segregation during mouse oocyte meiosis. PLoS ONE 2009, 4, e7701. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Liang, X.W.; Zhang, Q.H.; Li, M.; Yuan, J.; Li, S.; Sun, S.C.; Ouyang, Y.C.; Schatten, H.; Sun, Q.Y. Bubr1 is a spindle assembly checkpoint protein regulating meiotic cell cycle progression of mouse oocyte. Cell Cycle 2010, 9, 1112–1121. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Vousden, K.H. P53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- DePamphilis, M.L. (Ed.) Mammalian Preimplantation Development; Elsevier Inc.: New York, NY, USA, 2016.
- Visvader, J.E.; Clevers, H. Tissue-specific designs of stem cell hierarchies. Nat. Cell Biol. 2016, 18, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Condic, M.L. Totipotency: What it is and what it is not. Stem Cells Dev. 2014, 23, 796–812. [Google Scholar] [CrossRef] [PubMed]
- Gage, F.H.; Temple, S. Neural stem cells: Generating and regenerating the brain. Neuron 2013, 80, 588–601. [Google Scholar] [CrossRef] [PubMed]
- Eaves, C.J. Hematopoietic stem cells: Concepts, definitions, and the new reality. Blood 2015, 125, 2605–2613. [Google Scholar] [CrossRef] [PubMed]
- Barker, N. Adult intestinal stem cells: Critical drivers of epithelial homeostasis and regeneration. Nat. Rev. Mol. Cell Biol. 2014, 15, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Niakan, K.K.; Schrode, N.; Cho, L.T.; Hadjantonakis, A.K. Derivation of extraembryonic endoderm stem (xen) cells from mouse embryos and embryonic stem cells. Nat. Protoc. 2013, 8, 1028–1041. [Google Scholar] [CrossRef] [PubMed]
- Cross, J.C. How to make a placenta: Mechanisms of trophoblast cell differentiation in mice—A review. Placenta 2005, 26 (Suppl A), S3–S9. [Google Scholar] [CrossRef] [PubMed]
- Kunath, T.; Strumpf, D.; Rossant, J. Early trophoblast determination and stem cell maintenance in the mouse—A review. Placenta 2004, 25 (Suppl A), S32–S38. [Google Scholar] [CrossRef] [PubMed]
- Garg, V.; Morgani, S.; Hadjantonakis, A.K. Capturing identity and fate ex vivo: Stem cells from the mouse blastocyst. Curr. Top. Dev. Biol. 2016, 120, 361–400. [Google Scholar] [PubMed]
- Boroviak, T.; Loos, R.; Bertone, P.; Smith, A.; Nichols, J. The ability of inner-cell-mass cells to self-renew as embryonic stem cells is acquired following epiblast specification. Nat. Cell Biol. 2014, 16, 516–528. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Barbacioru, C.; Bao, S.; Lee, C.; Nordman, E.; Wang, X.; Lao, K.; Surani, M.A. Tracing the derivation of embryonic stem cells from the inner cell mass by single-cell rna-seq analysis. Cell Stem Cell 2010, 6, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Martello, G.; Smith, A. The nature of embryonic stem cells. Annu. Rev. Cell Dev. Biol. 2014, 30, 647–675. [Google Scholar] [CrossRef] [PubMed]
- Saitou, M.; Yamaji, M. Primordial germ cells in mice. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Ko, K.; Tapia, N.; Wu, G.; Kim, J.B.; Bravo, M.J.; Sasse, P.; Glaser, T.; Ruau, D.; Han, D.W.; Greber, B.; et al. Induction of pluripotency in adult unipotent germline stem cells. Cell Stem Cell 2009, 5, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Orkin, S.H. Embryonic stem cell-specific signatures in cancer: Insights into genomic regulatory networks and implications for medicine. Genome Med. 2011, 3, 75. [Google Scholar] [CrossRef] [PubMed]
- Palmer, N.P.; Schmid, P.R.; Berger, B.; Kohane, I.S. A gene expression profile of stem cell pluripotentiality and differentiation is conserved across diverse solid and hematopoietic cancers. Genome Biol. 2012, 13, R71. [Google Scholar] [CrossRef] [PubMed]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Benvenisty, N. The tumorigenicity of human embryonic and induced pluripotent stem cells. Nat. Rev. Cancer 2011, 11, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Bonnet, D.; De Maria, R.; Lapidot, T.; Copland, M.; Melo, J.V.; Chomienne, C.; Ishikawa, F.; Schuringa, J.J.; Stassi, G.; et al. Cancer stem cell definitions and terminology: The devil is in the details. Nat. Rev. Cancer 2012, 12, 767–775. [Google Scholar] [CrossRef] [PubMed]
- De Miguel, M.P.; Alcaina, Y.; de la Maza, D.S.; Lopez-Iglesias, P. Cell metabolism under microenvironmental low oxygen tension levels in stemness, proliferation and pluripotency. Curr. Mol. Med. 2015, 15, 343–359. [Google Scholar] [CrossRef] [PubMed]
- Rebuzzini, P.; Zuccotti, M.; Redi, C.A.; Garagna, S. Achilles’ heel of pluripotent stem cells: Genetic, genomic and epigenetic variations during prolonged culture. Cell. Mol. Life Sci. 2016, 73, 2453–2466. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, K.; Zambelli, F.; Mertzanidou, A.; Smolders, I.; Geens, M.; Nguyen, H.T.; Barbe, L.; Sermon, K.; Spits, C. Higher-density culture in human embryonic stem cells results in DNA damage and genome instability. Stem Cell Rep. 2016, 6, 330–341. [Google Scholar] [CrossRef] [PubMed]
- Yeo, D.; Kiparissides, A.; Cha, J.M.; Aguilar-Gallardo, C.; Polak, J.M.; Tsiridis, E.; Pistikopoulos, E.N.; Mantalaris, A. Improving embryonic stem cell expansion through the combination of perfusion and bioprocess model design. PLoS ONE 2013, 8, e81728. [Google Scholar]
- Brons, I.G.; Smithers, L.E.; Trotter, M.W.; Rugg-Gunn, P.; Sun, B.; Chuva de Sousa Lopes, S.M.; Howlett, S.K.; Clarkson, A.; Ahrlund-Richter, L.; Pedersen, R.A.; et al. Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature 2007, 448, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Joo, J.Y.; Choi, H.W.; Kim, M.J.; Zaehres, H.; Tapia, N.; Stehling, M.; Jung, K.S.; Do, J.T.; Scholer, H.R. Establishment of a primed pluripotent epiblast stem cell in fgf4-based conditions. Sci. Rep. 2014, 4, 7477. [Google Scholar] [CrossRef] [PubMed]
- Bernemann, C.; Greber, B.; Ko, K.; Sterneckert, J.; Han, D.W.; Arauzo-Bravo, M.J.; Scholer, H.R. Distinct developmental ground states of epiblast stem cell lines determine different pluripotency features. Stem Cells 2011, 29, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Bulic-Jakus, F.; Katusic Bojanac, A.; Juric-Lekic, G.; Vlahovic, M.; Sincic, N. Teratoma: From spontaneous tumors to the pluripotency/malignancy assay. Wiley Interdiscip. Rev. Dev. Biol. 2016, 5, 186–209. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, J.J.; Ulbright, T.M.; Pera, M.F.; Looijenga, L.H. Lessons from human teratomas to guide development of safe stem cell therapies. Nat. Biotechnol. 2012, 30, 849–857. [Google Scholar] [CrossRef] [PubMed]
- De Los Angeles, A.; Ferrari, F.; Xi, R.; Fujiwara, Y.; Benvenisty, N.; Deng, H.; Hochedlinger, K.; Jaenisch, R.; Lee, S.; Leitch, H.G.; et al. Hallmarks of pluripotency. Nature 2015, 525, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Solter, D. From teratocarcinomas to embryonic stem cells and beyond: A history of embryonic stem cell research. Nat. Rev. Genet. 2006, 7, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Papaioannou, V.E.; McBurney, M.W.; Gardner, R.L.; Evans, M.J. Fate of teratocarcinoma cells injected into early mouse embryos. Nature 1975, 258, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Kahan, B.W.; Ephrussi, B. Developmental potentialities of clonal in vitro cultures of mouse testicular teratoma. J. Natl. Cancer Inst. 1970, 44, 1015–1036. [Google Scholar] [PubMed]
- Economou, C.; Tsakiridis, A.; Wymeersch, F.J.; Gordon-Keylock, S.; Dewhurst, R.E.; Fisher, D.; Medvinsky, A.; Smith, A.J.; Wilson, V. Intrinsic factors and the embryonic environment influence the formation of extragonadal teratomas during gestation. BMC Dev. Biol. 2015, 15, 35. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.A.; Schiesser, J.; Stanley, E.G.; Elefanty, A.G. Differentiating embryonic stem cells pass through ‘temporal windows’ that mark responsiveness to exogenous and paracrine mesendoderm inducing signals. PLoS ONE 2010, 5, e10706. [Google Scholar] [CrossRef] [PubMed]
- Thomson, M.; Liu, S.J.; Zou, L.N.; Smith, Z.; Meissner, A.; Ramanathan, S. Pluripotency factors in embryonic stem cells regulate differentiation into germ layers. Cell 2011, 145, 875–889. [Google Scholar] [CrossRef] [PubMed]
- Trosko, J.E. Commentary: “Re-programming or selecting adult stem cells?”. Stem Cell Rev. 2008, 4, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.Y.; Kaneko, K.J.; Pan, H.; DePamphilis, M.L. Geminin is essential to prevent DNA re-replication-dependent apoptosis in pluripotent cells, but not in differentiated cells. Stem Cells 2015, 33, 3239–3253. [Google Scholar] [CrossRef] [PubMed]
- Nakuci, E.; Xu, M.; Pujana, M.A.; Valls, J.; Elshamy, W.M. Geminin is bound to chromatin in g2/m phase to promote proper cytokinesis. Int. J. Biochem. Cell Biol. 2006, 38, 1207–1220. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Cicalese, A.; Fumagalli, M.; Dobreva, M.; Verrecchia, A.; Pelicci, P.G.; di Fagagna, F. DNA damage response activation in mouse embryonic fibroblasts undergoing replicative senescence and following spontaneous immortalization. Cell Cycle 2008, 7, 3601–3606. [Google Scholar] [CrossRef] [PubMed]
- Iliou, M.S.; Kotantaki, P.; Karamitros, D.; Spella, M.; Taraviras, S.; Lygerou, Z. Reduced geminin levels promote cellular senescence. Mech. Ageing Dev. 2013, 134, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Oosterhuis, J.W.; Stoop, H.; Honecker, F.; Looijenga, L.H. Why human extragonadal germ cell tumours occur in the midline of the body: Old concepts, new perspectives. Int. J. Androl. 2007, 30, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Elf, S.E.; Miyata, Y.; Sashida, G.; Liu, Y.; Huang, G.; Di Giandomenico, S.; Lee, J.M.; Deblasio, A.; Menendez, S.; et al. P53 regulates hematopoietic stem cell quiescence. Cell Stem Cell 2009, 4, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Williamson, S.R.; Delahunt, B.; Magi-Galluzzi, C.; Algaba, F.; Egevad, L.; Ulbright, T.M.; Tickoo, S.K.; Srigley, J.R.; Epstein, J.I.; Berney, D.M. The WHO 2016 classification of testicular germ cell tumours: A review and update from the ISUP testis consultation panel. Histopathology 2017, 70, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Sekita, Y.; Nakamura, T.; Kimura, T. Reprogramming of germ cells into pluripotency. World J. Stem Cells 2016, 8, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Nettersheim, D.; Jostes, S.; Schneider, S.; Schorle, H. Elucidating human male germ cell development by studying germ cell cancer. Reproduction 2016, 152, R101–R113. [Google Scholar] [CrossRef] [PubMed]
- Blow, J.J.; Gillespie, P.J. Replication licensing and cancer--a fatal entanglement? Nat. Rev. Cancer 2008, 8, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Lutzmann, M.; Maiorano, D.; Mechali, M. A cdt1-geminin complex licenses chromatin for DNA replication and prevents rereplication during s phase in xenopus. EMBO J. 2006, 25, 5764–5774. [Google Scholar] [CrossRef] [PubMed]
- Kroll, K.L. Geminin in embryonic development: Coordinating transcription and the cell cycle during differentiation. Front. Biosci. 2007, 12, 1395–1409. [Google Scholar] [CrossRef] [PubMed]
- Kerns, S.L.; Schultz, K.M.; Barry, K.A.; Thorne, T.M.; McGarry, T.J. Geminin is required for zygotic gene expression at the xenopus mid-blastula transition. PLoS ONE 2012, 7, e38009. [Google Scholar] [CrossRef] [PubMed]
- Takada, S.; Kwak, S.; Koppetsch, B.S.; Theurkauf, W.E. Grp (chk1) replication-checkpoint mutations and DNA damage trigger a chk2-dependent block at the drosophila midblastula transition. Development 2007, 134, 1737–1744. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.W.; Hummert, P.; Mills, J.C.; Kroll, K.L. Geminin cooperates with polycomb to restrain multi-lineage commitment in the early embryo. Development 2011, 138, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.A.; Tachibana, K.E.; Adams, D.J.; van der Weyden, L.; Hemberger, M.; Coleman, N.; Bradley, A.; Laskey, R.A. Geminin is essential to prevent endoreduplication and to form pluripotent cells during mammalian development. Genes Dev. 2006, 20, 1880–1884. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Nakayama, K.I.; Nakayama, K. Geminin is essential for the development of preimplantation mouse embryos. Genes Cells 2006, 11, 1281–1293. [Google Scholar] [CrossRef] [PubMed]
- Patterson, E.S.; Waller, L.E.; Kroll, K.L. Geminin loss causes neural tube defects through disrupted progenitor specification and neuronal differentiation. Dev. Biol. 2014, 393, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Schultz, K.M.; Banisadr, G.; Lastra, R.O.; McGuire, T.; Kessler, J.A.; Miller, R.J.; McGarry, T.J. Geminin-deficient neural stem cells exhibit normal cell division and normal neurogenesis. PLoS ONE 2011, 6, e17736. [Google Scholar] [CrossRef] [PubMed]
- Shinnick, K.M.; Eklund, E.A.; McGarry, T.J. Geminin deletion from hematopoietic cells causes anemia and thrombocytosis in mice. J. Clin. Investig. 2010, 120, 4303–4315. [Google Scholar] [CrossRef] [PubMed]
- Spella, M.; Kyrousi, C.; Kritikou, E.; Stathopoulou, A.; Guillemot, F.; Kioussis, D.; Pachnis, V.; Lygerou, Z.; Taraviras, S. Geminin regulates cortical progenitor proliferation and differentiation. Stem Cells 2011, 29, 1269–1282. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, G.A.; Bose, K.; Reimann, Y.; Kessel, M. Geminin is required for the maintenance of pluripotency. PLoS ONE 2013, 8, e73826. [Google Scholar] [CrossRef] [PubMed]
- Yang, V.S.; Carter, S.A.; Hyland, S.J.; Tachibana-Konwalski, K.; Laskey, R.A.; Gonzalez, M.A. Geminin escapes degradation in g1 of mouse pluripotent cells and mediates the expression of oct4, sox2, and nanog. Curr. Biol. 2011, 21, 692–699. [Google Scholar] [CrossRef] [PubMed]
- Yellajoshyula, D.; Patterson, E.S.; Elitt, M.S.; Kroll, K.L. Geminin promotes neural fate acquisition of embryonic stem cells by maintaining chromatin in an accessible and hyperacetylated state. Proc. Natl. Acad. Sci. USA 2011, 108, 3294–3299. [Google Scholar] [CrossRef] [PubMed]
- Slawny, N.; O'Shea, K.S. Geminin promotes an epithelial-to-mesenchymal transition in an embryonic stem cell model of gastrulation. Stem Cells Dev. 2013, 22, 1177–1189. [Google Scholar] [CrossRef] [PubMed]
- Barry, K.A.; Schultz, K.M.; Payne, C.J.; McGarry, T.J. Geminin is required for mitotic proliferation of spermatogonia. Dev. Biol. 2012, 371, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Jeong, J.; Kang, H.; Lim, J.; Heo, J.; Ratajczak, J.; Ratajczak, M.Z.; Shin, D.M. The molecular nature of very small embryonic-like stem cells in adult tissues. Int. J. Stem Cells 2014, 7, 55–62. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Function |
|---|---|
| Origin Licensing Block | |
| FBXO5/Emi1 | inhibits APC/C |
| GMNN/Geminin | inhibits Cdt1 |
| CUL1/Cullin 1 | CRL1 E3-ubiquitin ligase subunit |
| NEDD8 | |
| RBX1/ROC1 | |
| DTL/Cdt2/DCAF2 | CRL4 E3-ubiquitin ligase subunit |
| DDB1 | |
| Chromatin Untangling | |
| TOP2A/Topoisomerase IIα | resolves catenated intertwines |
| Mitotic Entry & Maintenance | |
| LIN54 | regulates G2→M transition |
| CCNB1/Cyclin B1 | initiates and maintains mitosis |
| MASTL/Greatwall | accelerates entry into mitosis and blocks exit from mitosis |
| PLK1/Polo-like kinase 1 | mitotic entry, centrosome maturation, microtubule nucleation |
| SMC2 | condensin subunits, chromosome condensation during mitosis |
| SMC4 | |
| Mitotic Spindle Assembly | |
| TPX2 | promotes spindle assembly |
| KIF11/Eg5/Kinesin-11 | required for bipolar spindle formation |
| CEP192 | required for centriole duplication |
| AURKA/Aurora kinase A | builds bipolar spindle, regulates centrosome separation and microtubule dynamics |
| POC1A/WDR51A | ensures centriole integrity |
| Spindle Assembly Checkpoint | |
| INCENP | Chromosome Passenger Complex (CPC) |
| BIRC5/Survivin | |
| CDCA8/Borealin | |
| AURKB/Aurora kinase B | |
| CASC5/D40/KNL1 | KMN network component, ensures MCC assembly |
| BUB3 | recruits SAC proteins to kinetochore |
| BUB1B | Mitotic Checkpoint Complex (MCC) |
| MAD2L1/MAD2 | |
| TTK/Mps1 | stimulates CPC and MCC |
| NUF2 | NDC80 kinetochore complex subunit |
| Sister Chromatid Cohesion | |
| CDCA5/Sororin | inhibits cohesin dissociation |
| PPP2R1A/PP2A-alpha | prevents cohesin phosphorylation |
| SGOL1/Sgo1/Shugoshin-like 1 | targets PPA2 to centromeric cohesin |
| Chromosome Segregation | |
| ESPL1/Separase | cleaves cohesin |
| CDC16/APC6 | Anaphase Promoting Complex (APC/C) |
| CDC26/APC12 | |
| CDC27/APC3 | |
| Cytokinesis | |
| ANLN/Anillin | crosslinks filaments in contractile ring |
| PRC1 | midzone formation |
| RACGAP1 | Centralspindlin |
| ECT2 | |
| KIF23/MKLP1/Kinesin-23 | |
| CHMP4B | component of the ESCRTIII complex |
| Genes | Cell Cycle Event | *Aneuploidy | *Tumors | Ref. |
|---|---|---|---|---|
| PLK1/ Polo-like Kinase 1 | Mitotic Entry & Maintenance | Yes | Yes | [104] |
| TPX2 | Mitotic Spindle Assembly | Yes | Yes | [105] |
| KIF11/Eg5/Kinesin-11 | Yes | Yes | [106] | |
| AURKA/Aurora Kinase A | Yes | Yes | [107] | |
| INCENP/ Inner Centromere Protein | Spindle Assembly Checkpoint | Yes | [108,109] | |
| BIRC5/Survivin | Yes | [109] | ||
| CDCA8/Borealin | Yes | [110] | ||
| AURKB/Aurora Kinase B | Yes | [111] | ||
| BUB3 | Yes | [112,113] | ||
| BUB1B | Yes | [114] | ||
| MAD2L1/MAD2/ Mitotic Arrest Deficient | Yes | Yes | [115,116,117,118] | |
| TTK/Mps1 | Yes | Yes | [119] | |
| SGOL1/Sgo1/ Shugoshin-like 1 | Sister Chromatid Cohesion | Yes | Yes | [120] |
| ESPL1/Separase | Chromosome Segregation | Yes | Yes | [121] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vassilev, A.; DePamphilis, M.L. Links between DNA Replication, Stem Cells and Cancer. Genes 2017, 8, 45. https://doi.org/10.3390/genes8020045
Vassilev A, DePamphilis ML. Links between DNA Replication, Stem Cells and Cancer. Genes. 2017; 8(2):45. https://doi.org/10.3390/genes8020045
Chicago/Turabian StyleVassilev, Alex, and Melvin L. DePamphilis. 2017. "Links between DNA Replication, Stem Cells and Cancer" Genes 8, no. 2: 45. https://doi.org/10.3390/genes8020045
APA StyleVassilev, A., & DePamphilis, M. L. (2017). Links between DNA Replication, Stem Cells and Cancer. Genes, 8(2), 45. https://doi.org/10.3390/genes8020045