Absence of Correlation between Chimeric RNA and Aging

Abstract
:1. Introduction
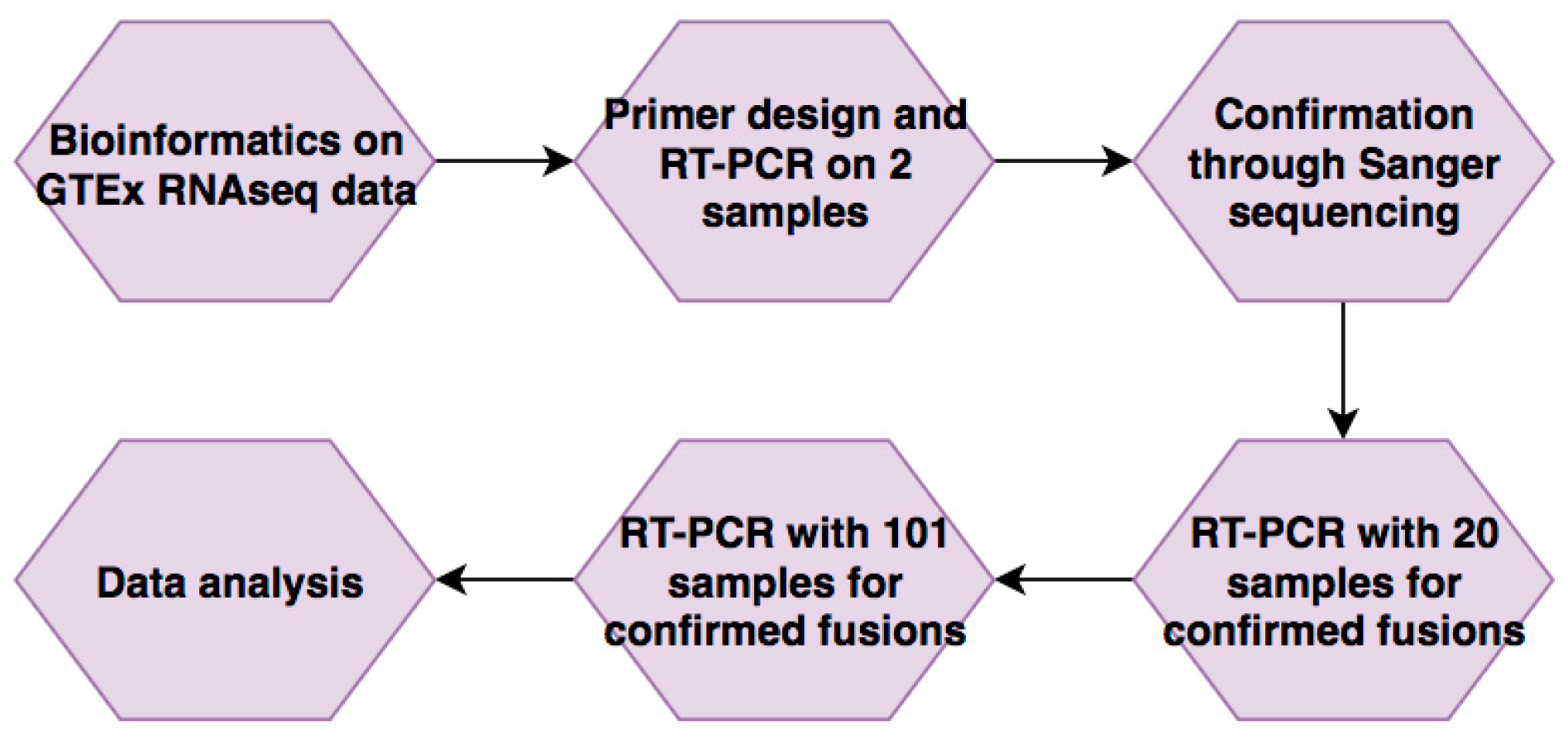
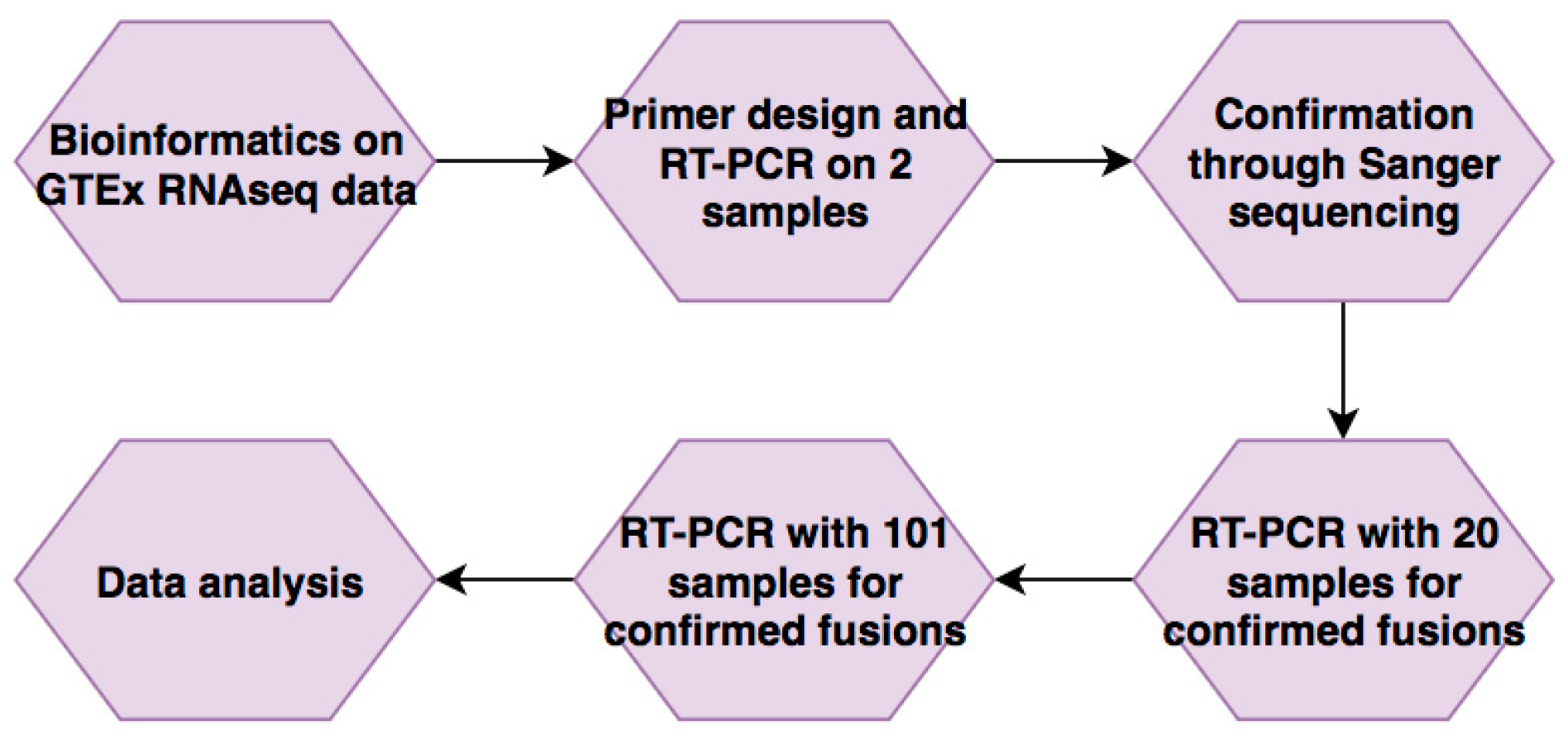
2. Materials and Methods
2.1. RNA-Seq and Bioinformatics Analyses
2.2. Sample Collection
2.3. RNA Extraction
2.4. RT-PCR and Sanger Sequencing
2.5. Statistical Analyses
3. Results
3.1. Bioinformatic Analysis of GTEx Data
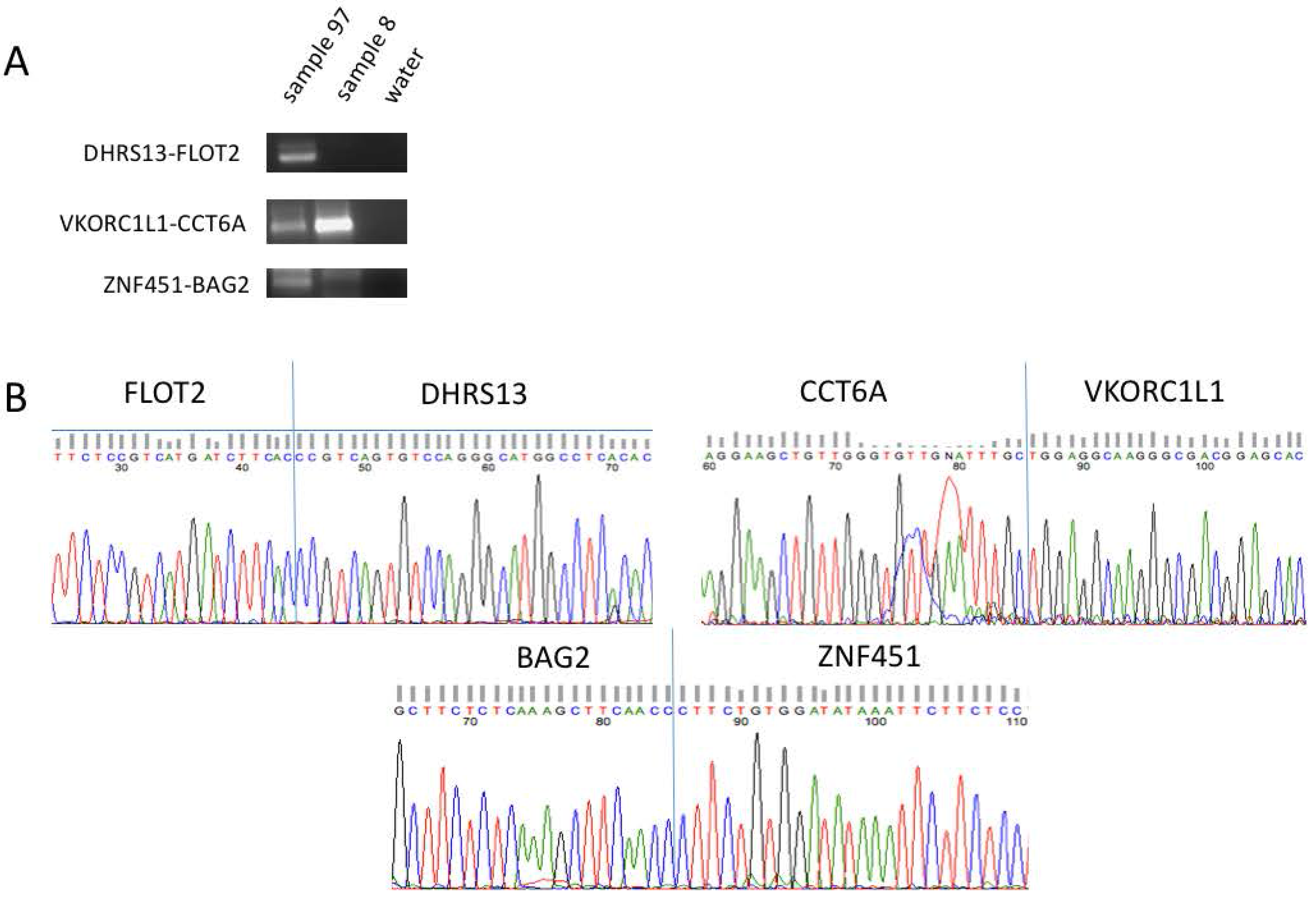
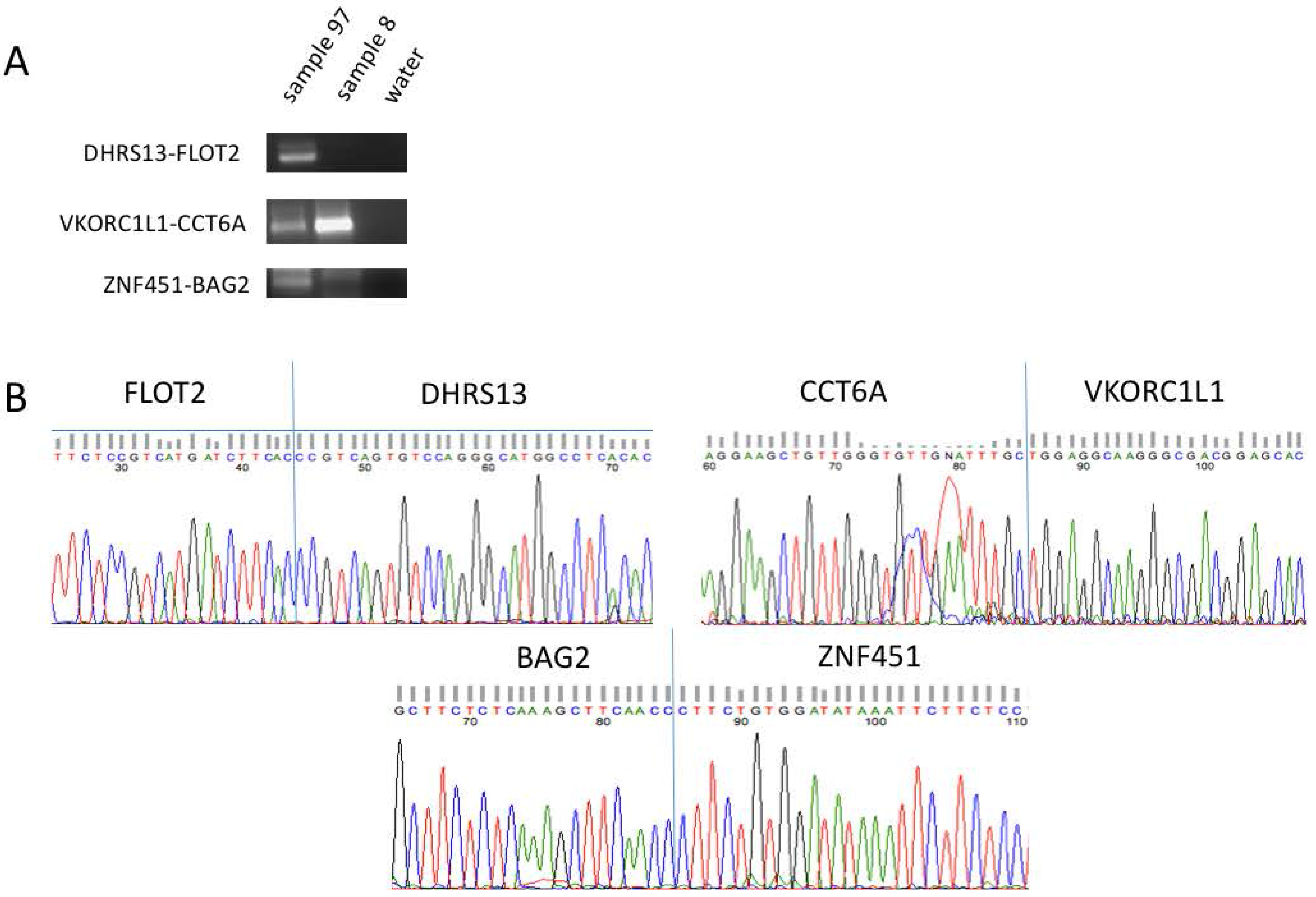
3.2. Confirmation of Five Candidates through RT-PCR and Sanger Sequencing
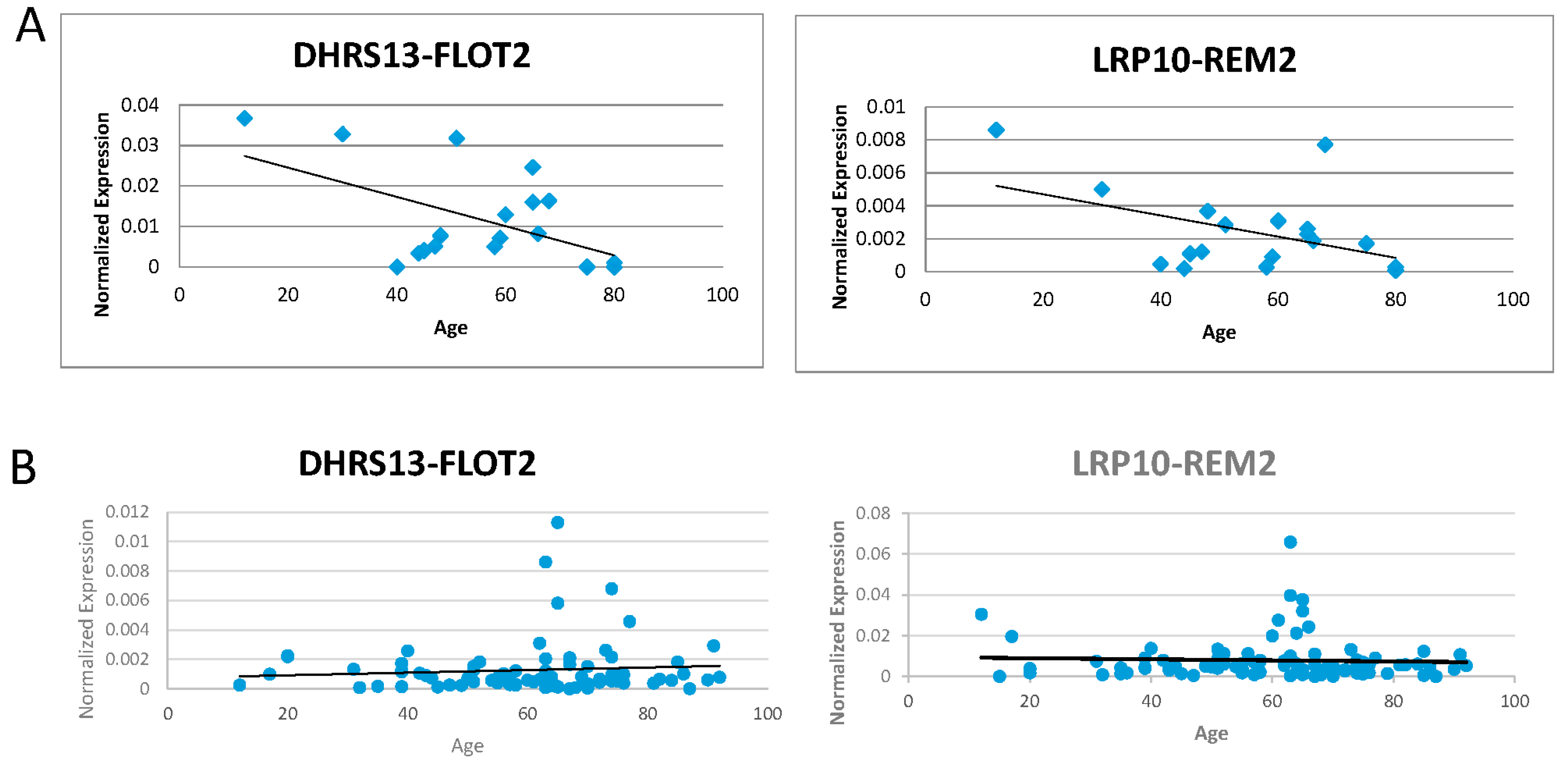
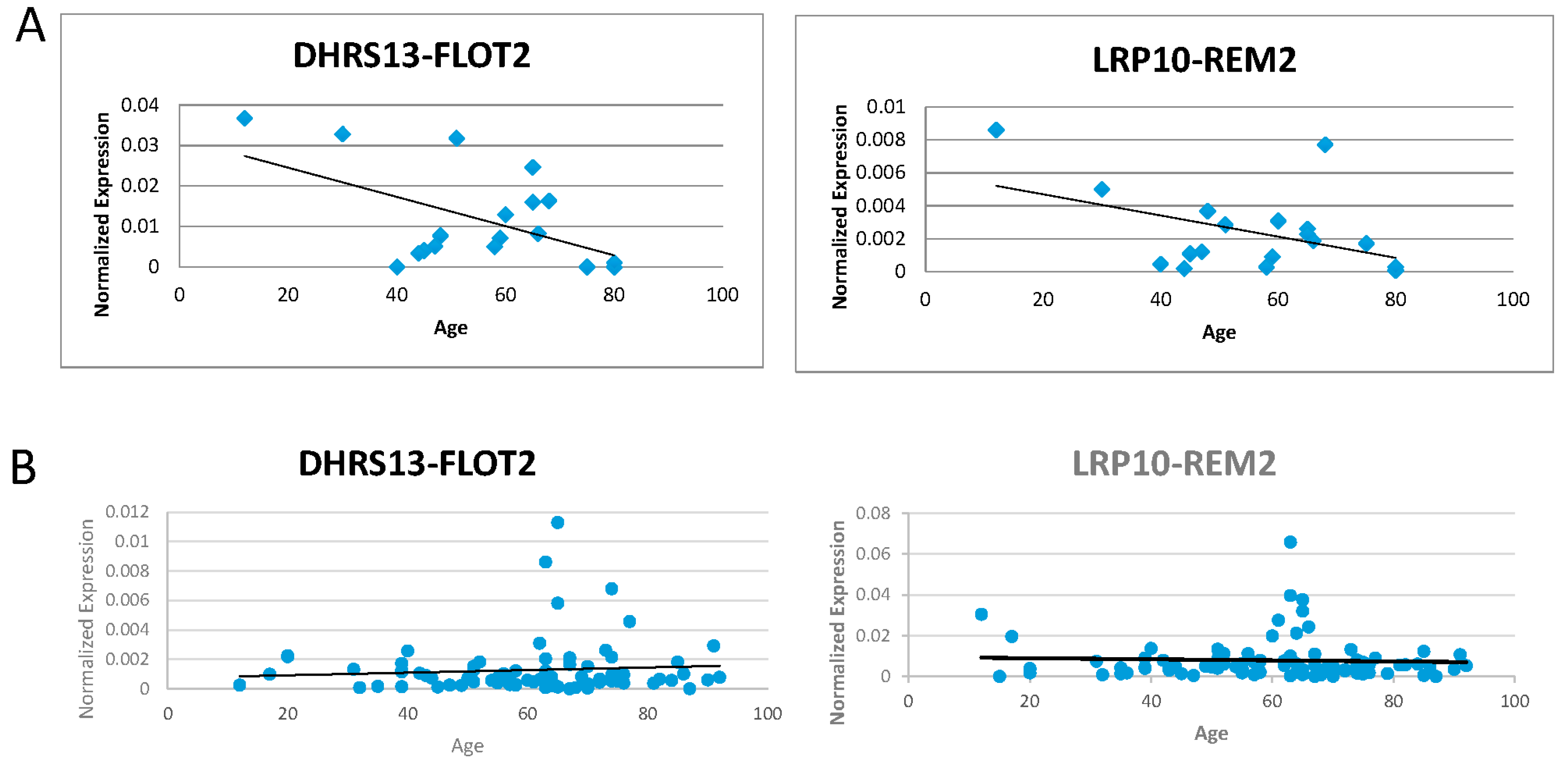
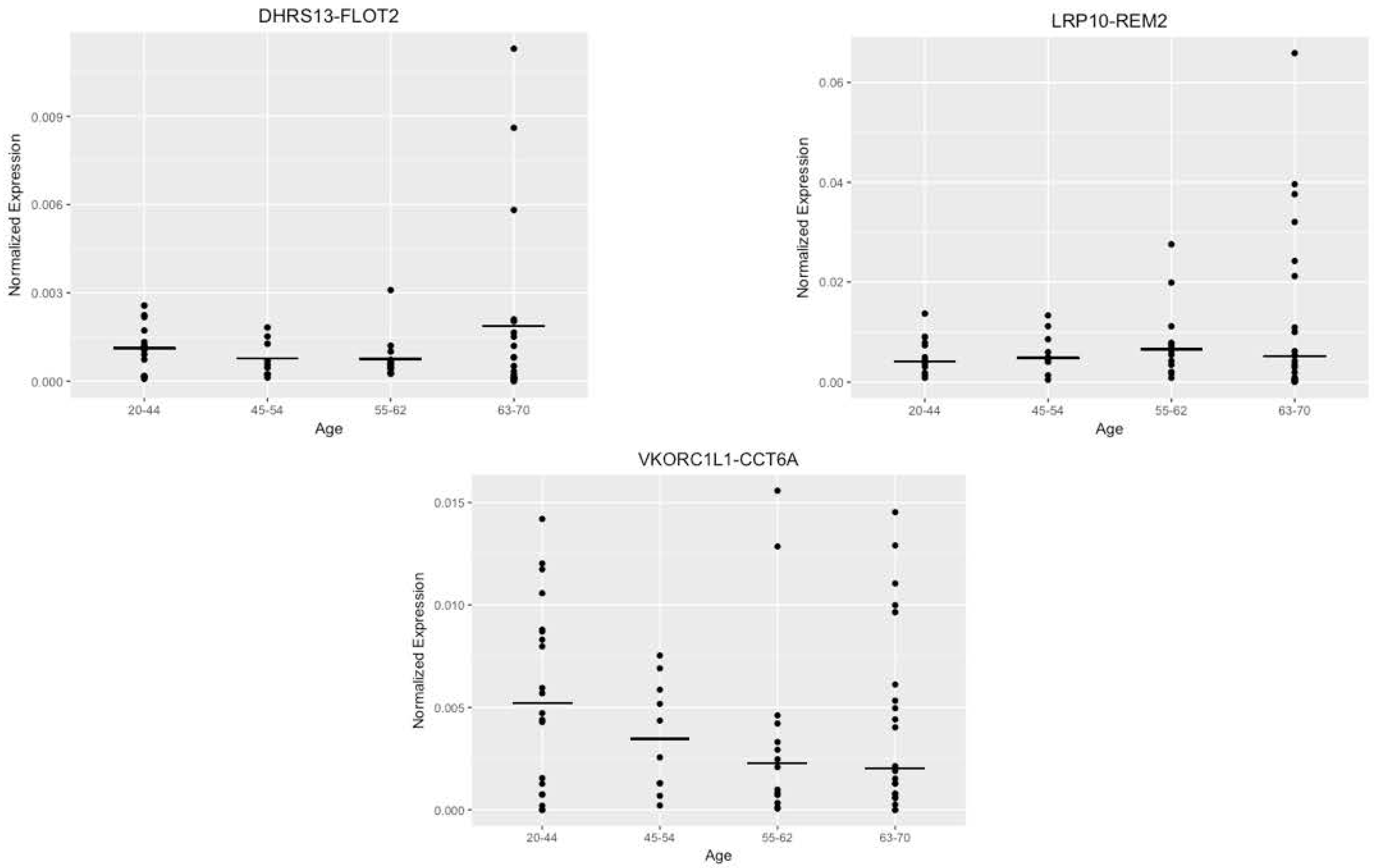
3.3. Investigation of the Correlation between the Five Candidates and Age of the Donors
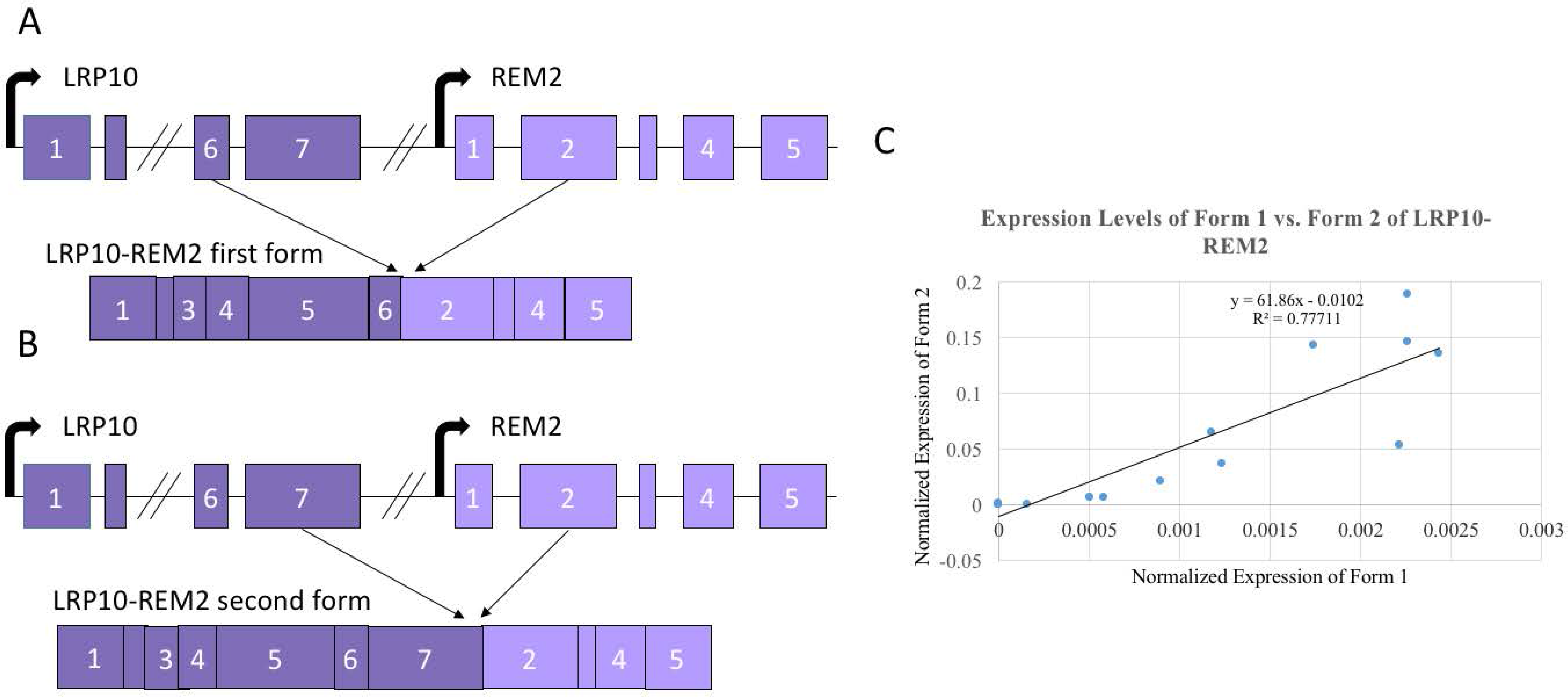
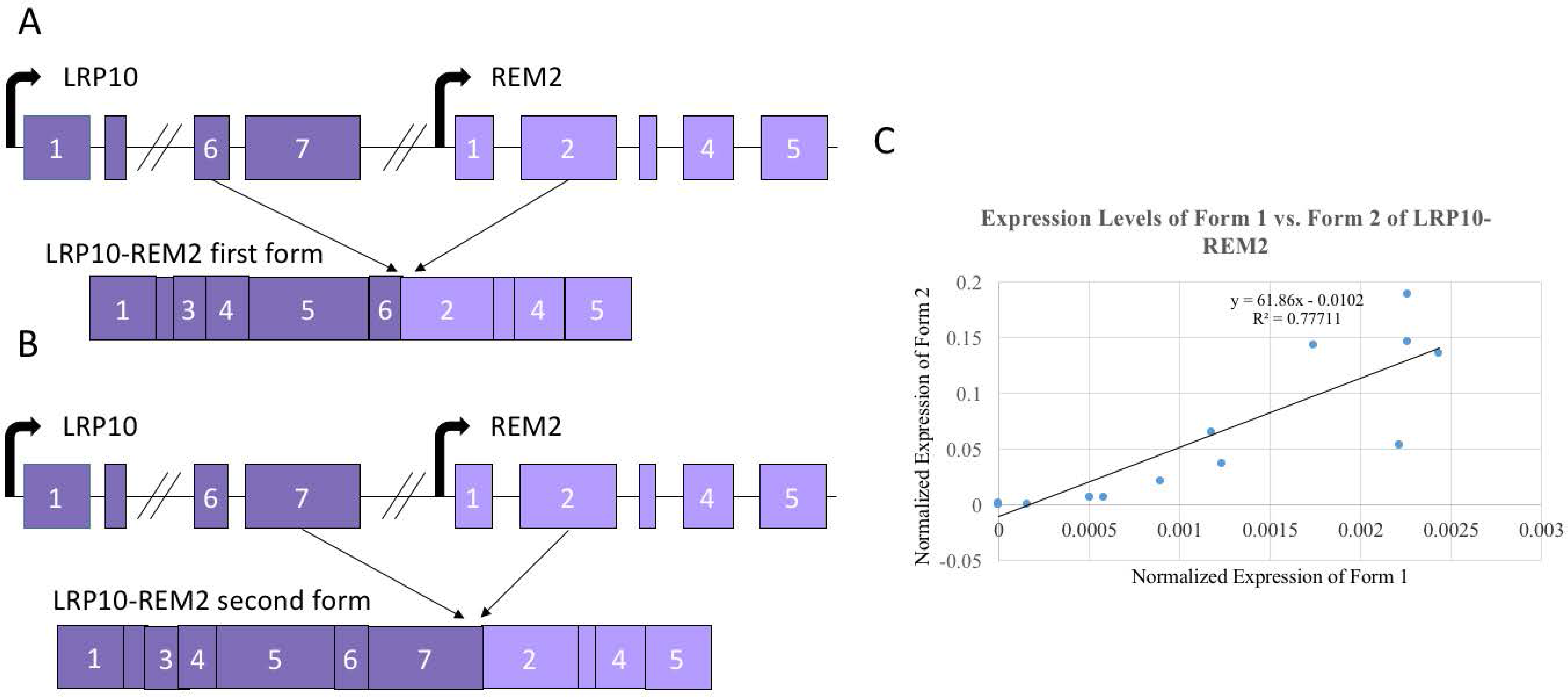
3.4. Alternate Forms of LRP10-REM2 Correlate in Expression with Each Other, But Had No Correlation with Age
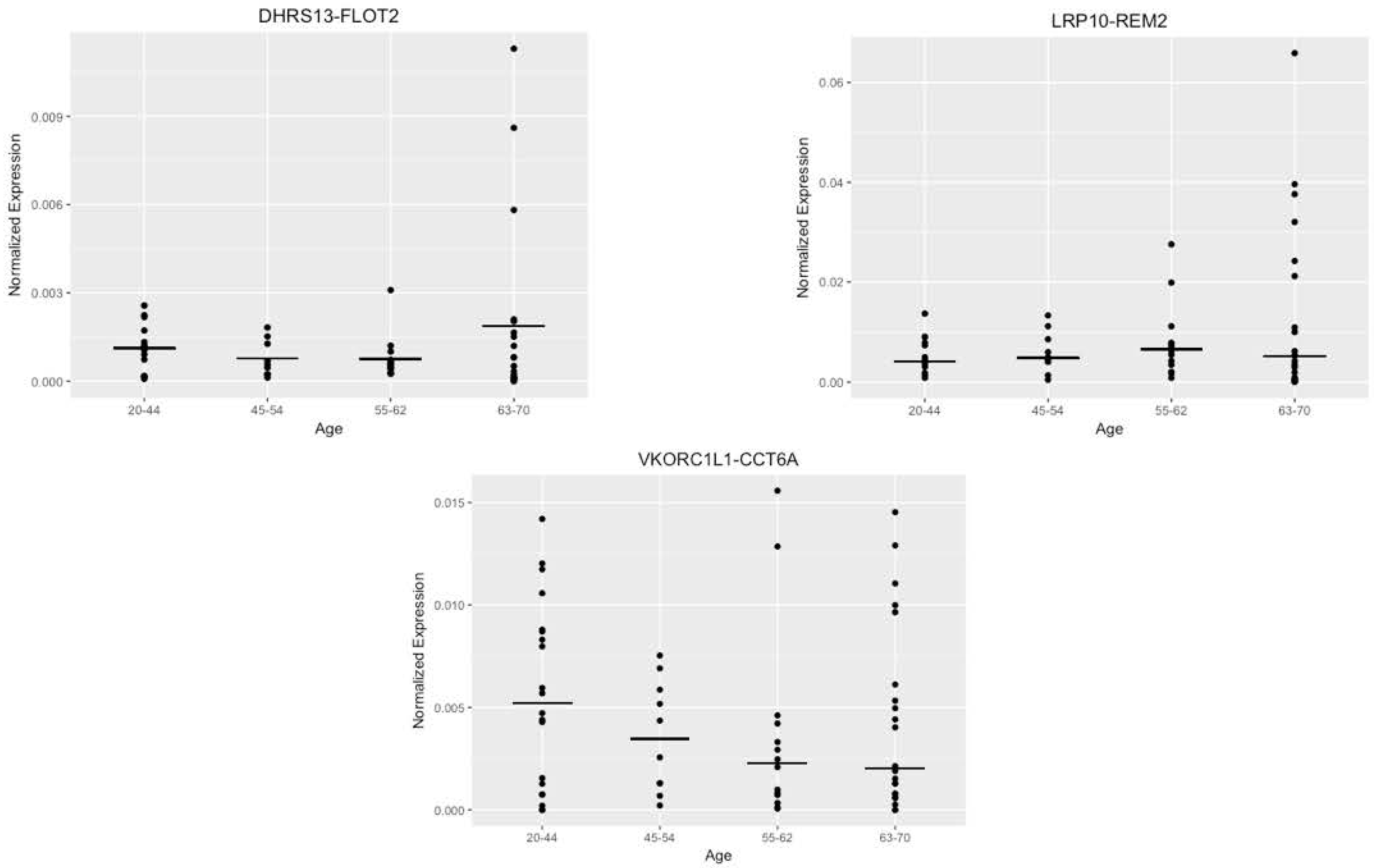
3.5. Alternate Analysis of the Data with Age Buckets also Failed to Show a Prominent Trend
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Heim, S.; Mitelman, F. Molecular screening for new fusion genes in cancer. Nat. Genet. 2008, 40, 685–686. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Qin, F.; Liu, A.; Li, H. Recurrent fusion RNA DUS4L-BCAP29 in non-cancer human tissues and cells. Oncotarget 2017, 8, 31415–31423. [Google Scholar] [CrossRef] [PubMed]
- Chwalenia, K.; Facemire, L.; Li, H. Chimeric RNAs in cancer and normal physiology. Wiley Interdiscip. Rev. RNA 2017. [Google Scholar] [CrossRef] [PubMed]
- Rabbitts, T.H. Chromosomal translocations in human cancer. Nature 1994, 372, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Qin, F.; Song, Z.; Chang, M.; Song, Y.; Frierson, H.; Li, H. Recurrent cis-SAGe chimeric RNA, D2HGDH-GAL3ST2, in prostate cancer. Cancer Lett. 2016, 380, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gong, M.; Yuan, H.; Park, H.; Frierson, H.; Li, H. Chimeric transcript generated by cis-splicing of adjacent genes regulates prostate cancer cell proliferation. Cancer Discov. 2012, 2, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Prakash, T.; Sharma, V.; Adati, N.; Ozawa, R.; Kumar, N.; Nishida, Y.; Fujikake, T.; Takeda, T.; Taylor, T. Expression of conjoined genes: Another mechanism for gene regulation in eukaryotes. PLoS ONE 2010, 5, e13284. [Google Scholar] [CrossRef] [PubMed]
- Akiva, P.; Toporik, A.; Edelheit, S.; Peretz, Y.; Diber, A.; Shemesh, R.; Novik, A.; Sorek, R. Transcription-mediated gene fusion in the human genome. Gen. Res. 2006, 16, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Babiceanu, M.; Kumar, S.; Jia, Y.; Qin, F.; Barr, F.G.; Li, H. Fusion transcriptome profiling provides insights into alveolar rhabdomyosarcoma. Proc. Natl. Acad. Sci. USA 2016, 113, 13126–13131. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Itin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.-H.; Barkho, B.; Ruiz, S.; Diep, D.; Qu, J.; Yang, S.-L.; Panapoulos, A.; Suzuki, K.; Kurian, L.; Walsh, C.; et al. Recapitulation of premature ageing with iPSCs from hutchinson-gilford progeria syndrome. Nature 2011, 472, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Arancio, W.; Pizzolanti, G.; Genovese, S.I.; Pitrone, M.; Giordano, C. Epigenetic involvement in hutchinson-gilford progeria syndrome: A mini-review. Gerontology 2014, 60, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Haithcock, E.; Dayani, Y.; Neufeld, E.; Zahand, A.; Feinstein, N.; Mattout, A.; Gruenbaum, Y.; Liu, J. Age-related changes of nuclear architecture in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2005, 102, 16690–16695. [Google Scholar] [CrossRef] [PubMed]
- Bahar, R.; Hartmann, C.H.; Rodriguez, K.; Denny, A.D.; Busuttil, R.A.; Dolle, M.E.; Calder, B.; Chisholm, G.B.; Pollock, B.; Klein, C.; et al. Increased cell-to-cell variation in gene expression in ageing mouse heart. Nature 2006, 441, 1011–1014. [Google Scholar] [CrossRef] [PubMed]
- Harries, L.W.; Hernandez, D.; Henley, W.; Wood, A.; Holly, A.; Bradley-Smith, R.; Yaghootkar, H.; Dutta, A.; Murray, A.; Frayling, T.; et al. Human aging is characterized by focused changes in gene expression and deregulation of alternative splicing. Aging Cell 2011, 10, 868–878. [Google Scholar] [CrossRef] [PubMed]
- Villborg, A.; Sabath, N.; Wiesel, Y.; Nathans, J.; Levy-Adam, F.; Yario, T.A.; Steitz, J.A.; Shalgi, R. Comparative analysis reveals genomic features of stress-induced transcriptional readthrough. Proc. Natl. Acad. Sci. USA 2017, 114, E8362–E8371. [Google Scholar] [CrossRef] [PubMed]
- Benelli, M.; Pescucci, C.; Marseglia, G.; Severgnini, M.; Torricelli, F.; Magi, A. Discovering chimeric transcripts in paired-end RNA-seq data by using EricScript. Bioinformatics 2012, 28, 3232–3239. [Google Scholar] [CrossRef] [PubMed]
- Qin, F.; Song, Z.; Babiceanu, M.; Song, Y.; Facemire, L.; Singh, R.; Adli, M.; Li, H. Discovery of CTCF-sensitive Cis-spliced fusion RNAs between adjacent genes in human prostate cells. PLoS Genet. 2015, 11, e1005001. [Google Scholar]
- Qin, F.; Song, Y.; Zhang, Y.; Facemire, L.; Frierson, H.; Li, H. Role of CTCF in regulating SLC45A3-ELK4 chimeric RNA. PLoS ONE 2016, 11, e0150382. [Google Scholar] [CrossRef] [PubMed]
- Carithers, L.J.; Moore, H.M. The genotype-tissue expression (GTEx) project. Biopreserv. Biobank. 2015, 13, 307–308. [Google Scholar] [CrossRef] [PubMed]
- Babiceanu, M.; Qin, F.; Xie, Z.; Jia, Y.; Lopez, K.; Janus, N.; Facemire, L.; Kumar, S.; Pang, Y.; Qi, Y. Recurrent chimeric fusion RNAs in non-cancer tissues and cells. Nucleic Acids Res. 2016, 44, 2859–2872. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Candidate | Frequency | Cohort | Gender | Age | Race | Ethnicity | Height | Weight | BMI |
|---|---|---|---|---|---|---|---|---|---|
| ADCK4-NUMBL | 16 | 2.4 × 10−2 | 1.90 × 10−1 | 3.60 × 10−2 | 9.39 × 10−1 | 1 | 9.90 × 10−2 | 4.40 × 10−1 | 3.01 × 10−1 |
| ATXN1L-IST1 | 7 | 1.24 × 10−1 | 1 | 3.20 × 10−2 | 9.95 × 10−1 | 1 | 2.14 × 10−1 | 5.10 × 10−2 | 9.40 × 10−2 |
| DHRS13-FLOT2 | 22 | 1.72 × 10−4 | 4.94 × 10−1 | 4.00 × 10−2 | 5.95 × 10−1 | 1 | 8.20 × 10−1 | 5.44 × 10−1 | 6.31 × 10−1 |
| LRP10-REM2 | 31 | 1.49 × 10−1 | 7.09 × 10−1 | 4.00 × 10−3 | 9.45 × 10−1 | 1 | 8.96 × 10−1 | 8.77 × 10−1 | 4.73 × 10−1 |
| AMN1-RBM28 | 51 | 1.01 × 10−5 | 6.52 × 10−1 | 2.00 × 10−2 | 5.06 × 10−1 | 7.05 × 10−1 | 5.49 × 10−1 | 1.85 × 10−1 | 3.03 × 10−1 |
| C1orf112-SMARCA4 | 23 | 9.00 × 10−4 | 1 | 6.00 × 10−3 | 7.54 × 10−1 | 1 | 9.80 × 10−1 | 9.58 × 10−1 | 5.96 × 10−1 |
| C17orf99-SYNGR2 | 7 | 1.23 × 10−1 | 9.55 × 10−1 | 4.00 × 10−3 | 7.50 × 10−1 | 1 | 9.54 × 10−1 | 3.79 × 10−1 | 9.30 × 10−1 |
| EIF4A1-SENP3-EIF4A1 | 54 | 2.20 × 10−5 | 5.00 × 10−3 | 4.50 × 10−2 | 2.55 × 10−1 | 1 | 5.70 × 10−1 | 2.94 × 10−1 | 9.02 × 10−1 |
| ARHGEF39-CCDC107 | 59 | 5.11 × 10−10 | 4.19 × 10−1 | 3.00 × 10−3 | 5.82 × 10−1 | 9.42 × 10−1 | 8.16 × 10−1 | 7.34 × 10−1 | 6.75 × 10−1 |
| Fusion | Forward Primer | Reverse Primer | Predicted Trend |
|---|---|---|---|
| ATXN1L-IST1 | AGAGGACAAGAAAGCTGGTCAC | ggctcaaagccagatcttctaa | negative |
| DHRS13-FLOT2 | ACCGAATTCAGGCTAAAGTTGA | tgatgtcctgcacattcttacc | positive |
| LRP10-REM2 | GCTACAGATCTTACGCCAGGAT | tggccagtcaagttcatctaca | positive |
| CTTGCTCCCTCGAACCAAC * | |||
| VKORC1L1-IST1 | AATCCTGCTCTCCATCTACGC | ttcagcagctctccaatgatta | positive |
| ZNF451-BAG2 | TGATAACATGGGTGCCAAAA | tctcaccgtcactgatctgc | negative |
| Fusion | 20 Sample R Value | 101 Sample R Value |
|---|---|---|
| ATXN1L-IST1 | −2.36 × 10−1 | 1.08 × 10−1 |
| DHRS13-FLOT2 | −5.25 × 10−1 | 5.25 × 10−2 |
| LRP10-REM2 | −4.54 × 10−1 | −4.64 × 10−2 |
| VKORC1L1-CCT6A | 2.36 × 10−1 | −3.04 × 10−1 |
| ZNF451-BAG2 | −8.25 × 10−2 | 3.05 × 10−2 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, R.; Kumar, S.; Li, H. Absence of Correlation between Chimeric RNA and Aging. Genes 2017, 8, 386. https://doi.org/10.3390/genes8120386
Huang R, Kumar S, Li H. Absence of Correlation between Chimeric RNA and Aging. Genes. 2017; 8(12):386. https://doi.org/10.3390/genes8120386
Chicago/Turabian StyleHuang, Reyna, Shailesh Kumar, and Hui Li. 2017. "Absence of Correlation between Chimeric RNA and Aging" Genes 8, no. 12: 386. https://doi.org/10.3390/genes8120386
APA StyleHuang, R., Kumar, S., & Li, H. (2017). Absence of Correlation between Chimeric RNA and Aging. Genes, 8(12), 386. https://doi.org/10.3390/genes8120386