Taxonomic Classification for Living Organisms Using Convolutional Neural Networks

 ,
,
Abstract
1. Introduction
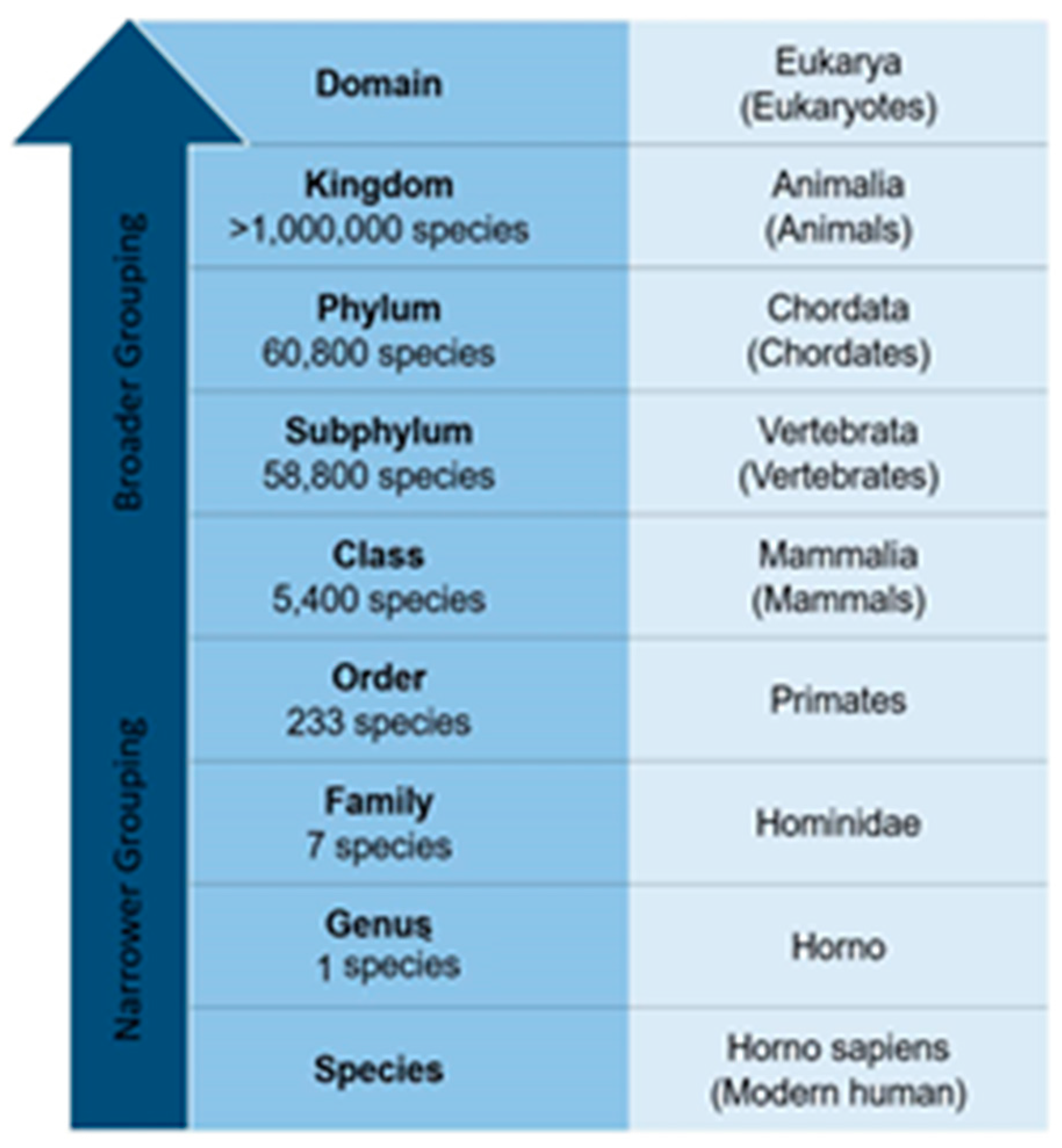
Problem Definition
2. Materials and Methods
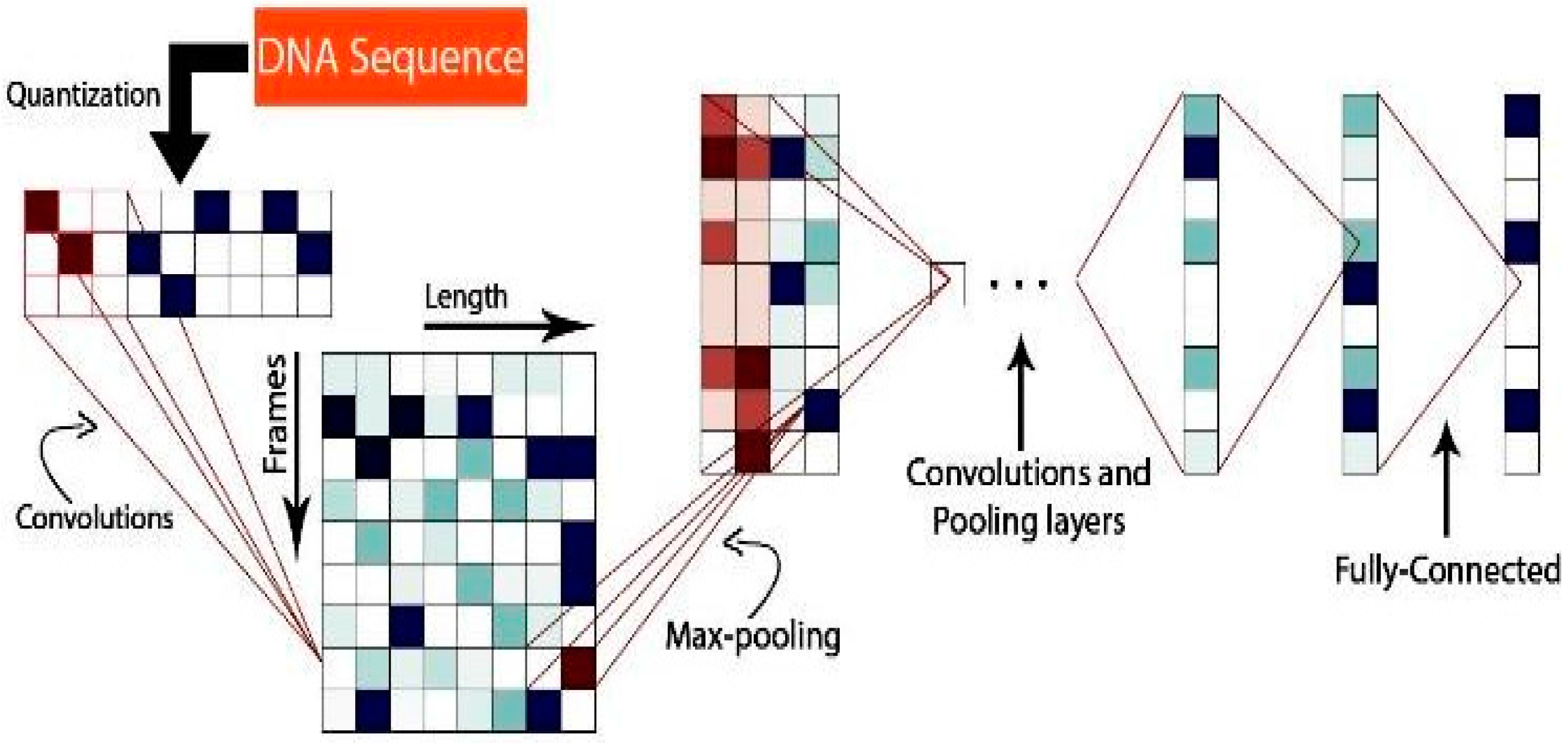
2.1. Methodology
2.2. Implementation and Experimentation
- Data preprocessing scripts that can be used to convert csv format to a Torch 7 binary format (t7b) that can be used by the training program directly: This component contains a tool called csv2t7b.lua; it is used to convert the datasets of csv format to t7b format, which can be fed to Crepe’s training component. This data preprocessing component contains two command-line parameters as shown in Table A1 in the Appendix A. The dataset t7b format can be loaded using the regular torch calls. The variable train has three members as shown in Table A2 in the Appendix A.
- The training program is a code used to process the data representing the DNA sequences: Originally, this code processed text and classified it into different classes representing the topic to which the text belongs; however, for the DNA taxonomic classification, we modified it to process DNA sequences. The reason for pursuing a nearly similar approach for processing both text and DNA data is that the two types of data are highly similar. The training subsets are fed first to the training component program in Crepe to train the classifier. Then, the model accuracy is evaluated on the remaining subsets, which were spared for testing. Table A3 shows the different training programs in Crepe.
- Training subset: including approximately 78.72% of the original dataset.
- Test subset: including approximately 21.28% of the original dataset (11.45% was used for cross-validation and 9.83% for testing).
- Experiment 1: feeding the DNA sequences to our CNN model without making any changes to them. Each DNA sequence is represented by a string, containing {’A’, ’C’, ’G’, ’T’, ’-’} characters and a label that shows to which class this DNA sequence belongs.
- Experiment 2: Encoding the DNA sequences by creating unique labels. Each unique label represents three characters in the original DNA sequence. A dictionary was generated to guide encoding the DNA sequences. The purpose of this encoding process is to ensure having a dataset similar in complexity and variety to the one used in the text classification problem.
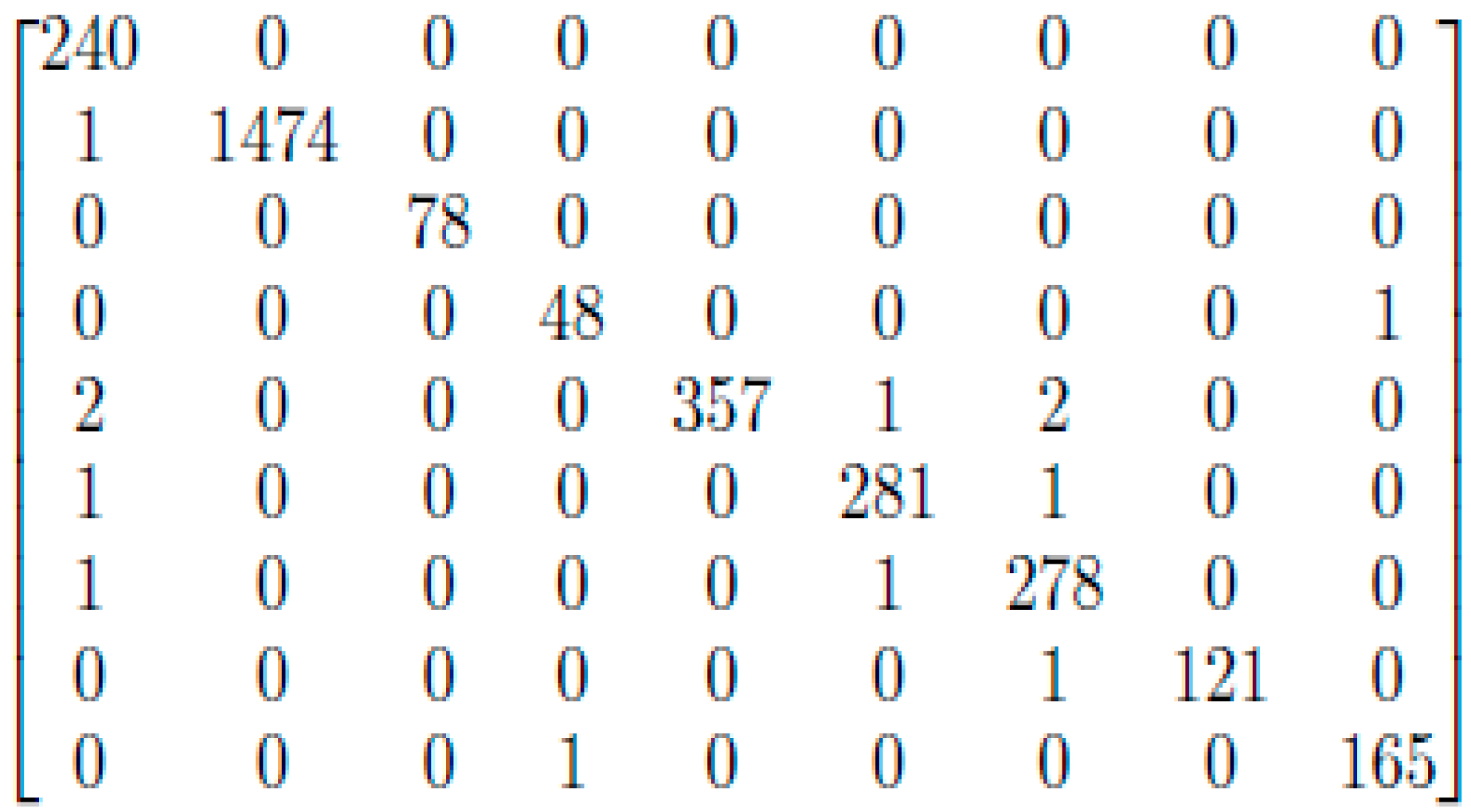
3. Results and Discussion
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| input [file] | This inputs the csv format file. The input file at this stage is in csv format where the entries of the first column of it are numbers indicating each class label, starting from 1 to ending with 9 (nine classes). Other columns are considered as text fields. |
| output [file] | This outputs the t7b format file. The output file at this stage is in torch-7-binary format (t7b). These generated files are directly used in training and validating datasets for the Crepe training program component. |
| train.content | A torch.ByteTensor that stores concatenated string data. Each string is ended with NULL (0). |
| train.index | A lua table for which train.index[i] is a 2D torch.LongTensor. The train.index[i][j][k] indicates the offset in train.content for the string in class i, j-th sample and k-th field. |
| train.length | A lua table for which train.length[i] is a 2D torch.LongTensor. The train.length[i][j][k] indicates the length for the string in class i, j-th sample and k-th field. The length does not count the ending NULL. |
| config.lua | A unified file for all configurations for the dataset, model, trainer, tester and GUI |
| data.lua | Provides a Data class; both training and validating datasets are instances of this class |
| main.lua | The main driver program |
| model.lua | Provides a Model class; it handles model creation, randomization and transformations during training |
| mui.lua | Provides a Mui class; uses the Scroll class to draw an nn.Sequential model in Qt |
| scroll.lua | Provides a Scroll class that starts a scrollable Qt window to draw text or images |
| scroll.ui | A Qt designer UI file corresponding to the scrollable Qt window |
| test.lua | Provides a Test class; handles testing, giving you losses, errors and confusion matrices |
| train.lua | Provides a Train class; handles training with Stochastic Gradient Descent (SGD) and supports things like momentum and weight decay |

References
- Zhang, Z.-Q. Contributing to the progress of descriptive taxonomy. Zootaxa 2008, 1968, 65–68. [Google Scholar]
- Padial, J.M.; Miralles, A.; De la Riva, I.; Vences, M. The integrative future of taxonomy. Front. Zool. 2010, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Fernando, S.G.S.; Perera, S.N. Empirical Analysis of Data Mining Techniques for Social Network Websites. Compusoft 2014, 3, 582. [Google Scholar]
- Huston, M.A. Biological Diversity: The Coexistence of Species; Cambridge University Press: Cambridge, UK, 1994. [Google Scholar]
- Goodman, M.; Porter, C.A.; Czelusniak, J.; Page, S.L.; Schneider, H.; Shoshani, J.; Groves, C.P. Toward a phylogenetic classification of primates based on DNA evidence complemented by fossil evidence. Mol. Phylogenet. Evol. 1998, 9, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Zhong, D.; Monteiro, A. Comparative genomics and evolution of the HSP90 family of genes across all kingdoms of organisms. BMC Genom. 2006, 7, 156. [Google Scholar] [CrossRef]
- Hebert, P.D.; Gregory, T.R. The promise of DNA barcoding for taxonomy. Systematic Biology 2005, 54, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liu, F.; Fang, L.; Wang, X.; Chou, K.C. repDNA: A Python package to generate various modes of feature vectors for DNA sequences by incorporating user-defined physicochemical properties and sequence-order effects. Bioinformatics 2014, 31, 1307–1309. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liu, F.; Fang, L.; Wang, X.; Chou, K.C. repRNA: A web server for generating various feature vectors of RNA sequences. Mol. Genet. Genom. 2016, 291, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wu, H.; Zhang, D.; Wang, X.; Chou, K.C. Pse-Analysis: A python package for DNA/RNA and protein/peptide sequence analysis based on pseudo components and kernel methods. Oncotarget 2017, 8, 13338. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liu, F.; Wang, X.; Chen, J.; Fang, L.; Chou, K.C. Pse-in-One: A web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res. 2015, 43, W65–W71. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Long, R.; Chou, K.C. iDHS-EL: Identifying DNase I hypersensitive sites by fusing three different modes of pseudo nucleotide composition into an ensemble learning framework. Bioinformatics 2016, 32, 2411–2418. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wang, S.; Long, R.; Chou, K.C. iRSpot-EL: Identify recombination spots with an ensemble learning approach. Bioinformatics 2016, 33, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Noble, W.S.; Kuehn, S.; Thurman, R.; Yu, M.; Stamatoyannopoulos, J. Predicting the in vivo signature of human gene regulatory sequences. Bioinformatics 2005, 21, i338–i343. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Jiang, N.; Liu, N. Prediction of DNase I Hypersensitive sites by using pseudo nucleotide compositions. Sci. World J. 2014, 2014, 740506. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.R.; Xiao, X.; Chou, K.C. iRSpot-TNCPseAAC: Identify recombination spots with trinucleotide composition and pseudo amino acid components. Int. J. Mol. Sci. 2014, 15, 1746–1766. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Feng, P.M.; Lin, H.; Chou, K.C. iRSpot-PseDNC: Identify recombination spots with pseudo dinucleotide composition. Nucleic Acids Res. 2013, 41, e68. [Google Scholar] [CrossRef] [PubMed]
- Weitschek, E.; Fiscon, G.; Felici, G. Supervised DNA Barcodes species classification: Analysis, comparisons and results. BioData Min. 2014, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, M.; Fiannaca, A.; Rizzo, R.; Urso, A. Probabilistic topic modeling for the analysis and classification of genomic sequences. BMC Bioinform. 2015, 16 (Suppl. 6), S2. [Google Scholar] [CrossRef] [PubMed]
- Bertolazzi, P.; Felici, G.; Weitschek, E. Learning to classify species with barcodes. BMC Bioinform. 2009, 10 (Suppl. 14), S7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, J.; LeCun, Y. Character-level convolutional networks for text classification. In Advances in Neural Information Processing Systems; MIT Press: Cambridge, UK, 2015. [Google Scholar]
- Zhang, X.; LeCun, Y. Text Understanding from Scratch. arXiv 2015, arXiv:1502.01710. [Google Scholar]
- Zeng, H.; Edwards, M.D.; Liu, G.; Gifford, D.K. Convolutional neural network architectures for predicting DNA–protein binding. Bioinformatics 2016, 32, i121–i127. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.G.; Tran, V.A.; Ngo, D.L.; Phan, D.; Lumbanraja, F.R.; Faisal, M.R.; Satou, K. DNA sequence classification by convolutional neural network. J. Biomed. Sci. Eng. 2016, 9, 280–286. [Google Scholar] [CrossRef]
- Higashihara, M.; Rebolledo-Mendez, J.D.; Yamada, Y.; Satou, K. application of a feature selection method to nucleosome data: accuracy improvement and comparison with other methods. WSEAS Trans. Biol. Biomed. 2008, 5, 153–162. [Google Scholar]
- Lee, T.K.; Nguyen, T. Protein Family Classification with Neural Networks. Stanford University, 2016. Available online: https://cs224d.stanford.edu/reports/LeeNguyen.pdf (accessed on 10 August 2017).
- Asgari, E.; Mofrad, M.R. Continuous distributed representation of biological sequences for deep proteomics and genomics. PLoS ONE 2015, 10, e0141287. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, R.; Fiannaca, A.; La Rosa, M.; Urso, A. Classification experiments of DNA sequences by using a deep neural network and chaos game representation. In Proceedings of the 17th International Conference on Computer Systems and Technologies 2016, Palermo, Italy, 23–24 June 2016; ACM: New York, NY, USA, 2016. [Google Scholar]
- Eickholt, J.; Cheng, J. DNdisorder: Predicting protein disorder using boosting and deep networks. BMC Bioinform. 2013, 14, 88. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Chen, J.; Liu, B. Protein remote homology detection based on bidirectional long short-term memory. BMC Bioinform. 2017, 18, 443. [Google Scholar] [CrossRef] [PubMed]
- Leslie, C.S.; Eskin, E.; Cohen, A.; Weston, J.; Noble, W.S. Mismatch string kernels for discriminative protein classification. Bioinformatics 2004, 20, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Håndstad, T.; Hestnes, A.J.; Sætrom, P. Motif kernel generated by genetic programming improves remote homology and fold detection. BMC Bioinform. 2007, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Saigo, H.; Vert, J.P.; Ueda, N.; Akutsu, T. Protein homology detection using string alignment kernels. Bioinformatics 2004, 20, 1682–1689. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Hu, Q.; Guo, M.; Wang, G. HAlign: Fast Multiple Similar DNA/RNA Sequence Alignment Based on the Centre Star Strategy. Bioinformatics 2015, 31, 2475–2481. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Noble, W.S. Combining pairwise sequence similarity and support vector machines for detecting remote protein evolutionary and structural relationships. J. Comput. Biol. 2003, 10, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Hochreiter, S.; Heusel, M.; Obermayer, K. Fast model-based protein homology detection without alignment. Bioinformatics 2007, 23, 1728–1736. [Google Scholar] [CrossRef] [PubMed]
- Ben-Hur, A.; Brutlag, D. Remote homology detection: A motif based approach. Bioinformatics 2003, 19 (Suppl. 1), i26–i33. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Ding, Y.; Su, R.; Tang, J.; Zou, Q. Prediction of human protein subcellular localization using deep learning. J. Parallel Distrib. Comput. 2017, in press. [Google Scholar] [CrossRef]
- Boureau, Y.L.; Bach, F.; LeCun, Y.; Ponce, J. Learning mid-level features for recognition. In Proceedings of the 2010 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), San Francisco, CA, USA, 13–18 June 2010; IEEE: Piscataway, NJ, USA, 2010; pp. 2559–2566. [Google Scholar]
- Torch | Scientific Computing for LuaJIT. Available online: http://torch.ch/ (accessed on 6 December 2016).
- GitHub-zhangxiangxiao/Crepe: Character-Level Convolutional Networks for Text Classification. Available online: https://github.com/zhangxiangxiao/Crepe (accessed on 24 November 2016).
- BOLD Systems v3. Available online: http://www.boldsystems.org/ (accessed on 10 December 2016).
- Landis, J.R.; Koch, G.G. The measurement of observer agreement for categorical data. Biometrics 1977, 33, 159–174. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Tang, J.; Zou, Q. Local-DPP: An improved DNA-binding protein prediction method by exploringg local evolutionary information. Inf. Sci. 2017, 384, 135–144. [Google Scholar] [CrossRef]
- Wei, L.; Xing, P.; Shi, G.; Ji, Z.L.; Zou, Q. Fast prediction of protein methylation sites using a sequence-based feature selection technique. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]





| Order Name | Label |
|---|---|
| Chaetothyriomycetes | 1 |
| Diptera | 2 |
| Echinoida | 3 |
| Forcipulatida | 4 |
| Lepidoptera | 5 |
| Onygenales | 6 |
| Pezizomycetes | 7 |
| Scleractinia | 8 |
| Valvatida | 9 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khawaldeh, S.; Pervaiz, U.; Elsharnoby, M.; Alchalabi, A.E.; Al-Zubi, N. Taxonomic Classification for Living Organisms Using Convolutional Neural Networks. Genes 2017, 8, 326. https://doi.org/10.3390/genes8110326
Khawaldeh S, Pervaiz U, Elsharnoby M, Alchalabi AE, Al-Zubi N. Taxonomic Classification for Living Organisms Using Convolutional Neural Networks. Genes. 2017; 8(11):326. https://doi.org/10.3390/genes8110326
Chicago/Turabian StyleKhawaldeh, Saed, Usama Pervaiz, Mohammed Elsharnoby, Alaa Eddin Alchalabi, and Nayel Al-Zubi. 2017. "Taxonomic Classification for Living Organisms Using Convolutional Neural Networks" Genes 8, no. 11: 326. https://doi.org/10.3390/genes8110326
APA StyleKhawaldeh, S., Pervaiz, U., Elsharnoby, M., Alchalabi, A. E., & Al-Zubi, N. (2017). Taxonomic Classification for Living Organisms Using Convolutional Neural Networks. Genes, 8(11), 326. https://doi.org/10.3390/genes8110326