
A DNA Structural Alphabet Distinguishes Structural Features of DNA Bound to Regulatory Proteins and in the Nucleosome Core Particle


Abstract
1. Introduction
2. Methods
2.1. Selection of Structures
2.2. DNA Conformer Classes NtC and the Structural Alphabet CANA
2.3. CANA/Sequence Matrices
2.4. Statistical Treatment of the Data
3. Results and Discussion
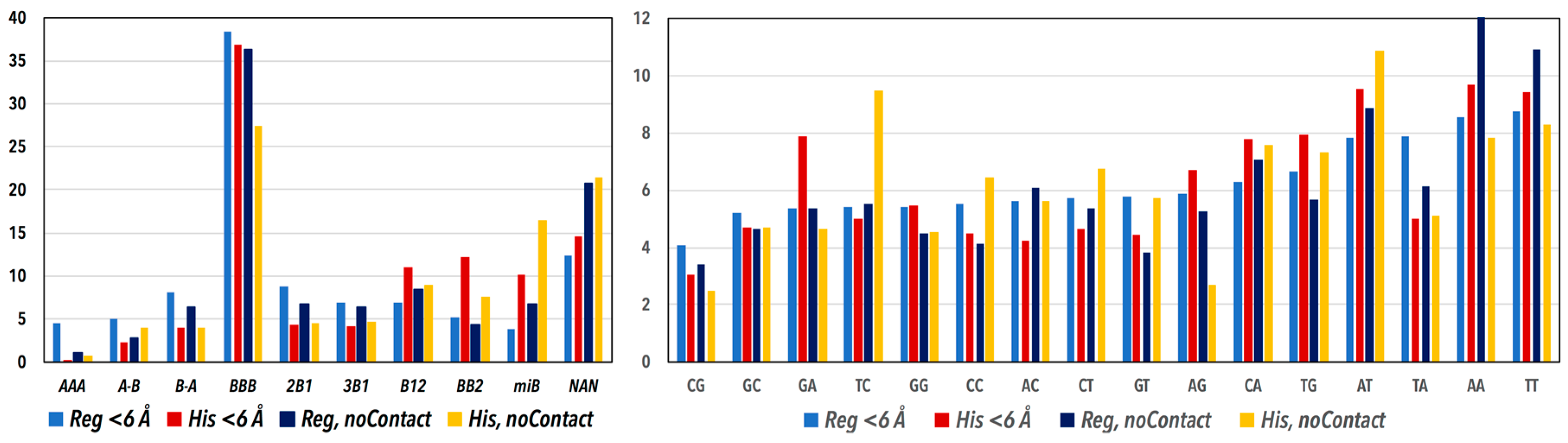
3.1. CANA and Sequence Distributions
3.2. CANA—Sequence Associations
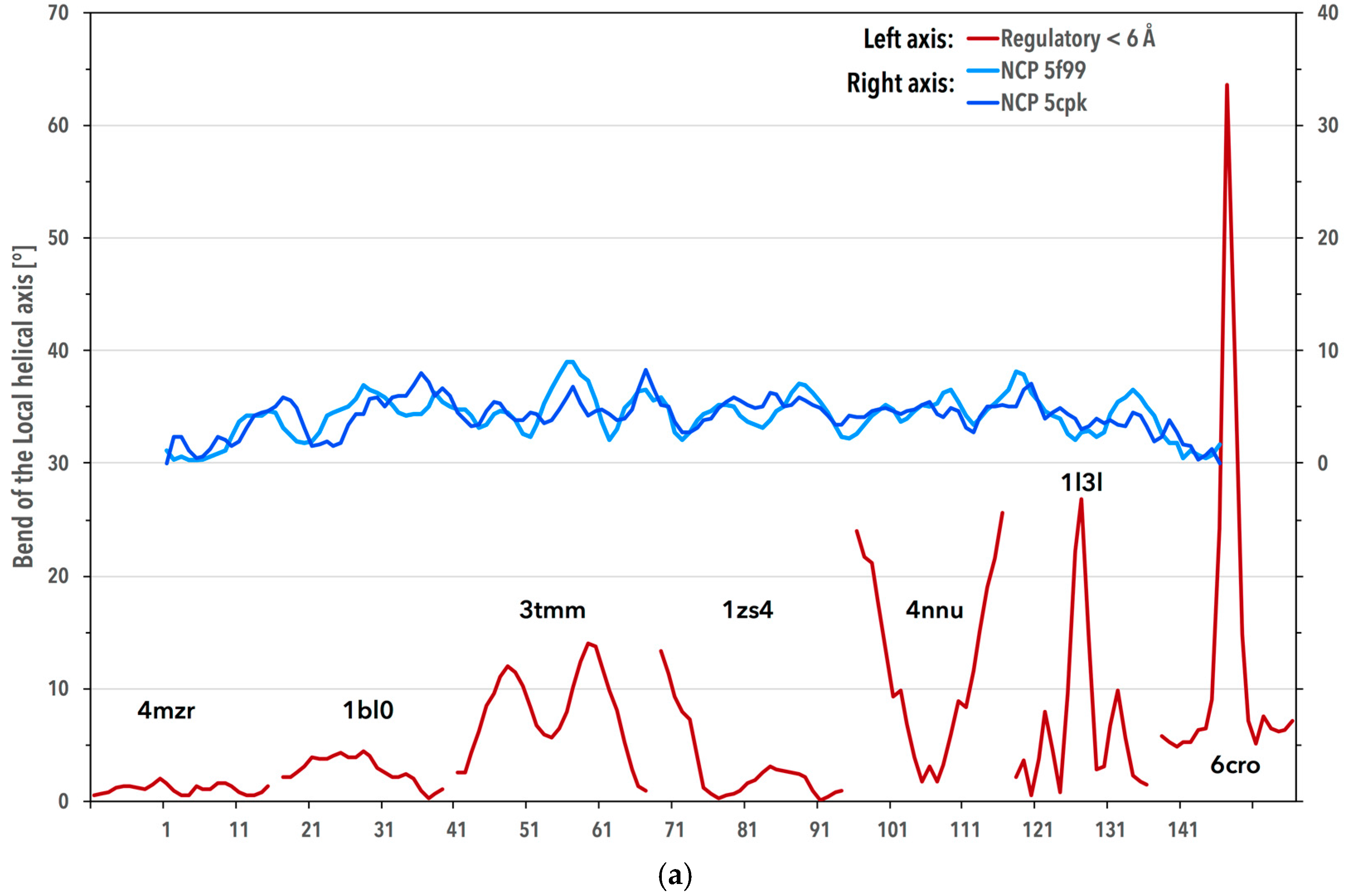
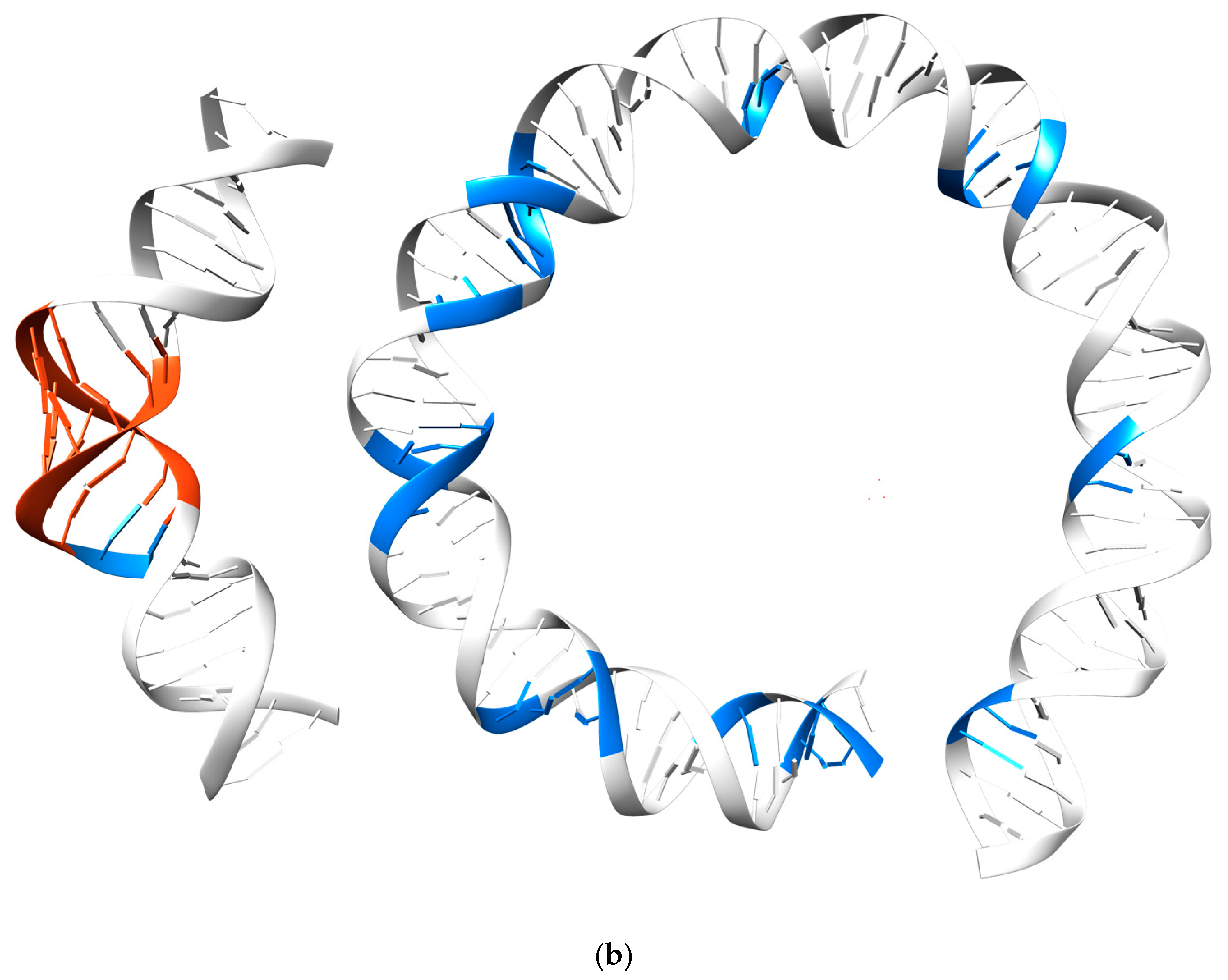
3.3. Periodicity of the Structural Behavior of DNA in the Nucleosome Core Particle
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Watson, J.D.; Crick, F.H.C. A structure for deoxyribose nucleic acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Rohs, R.; Jin, X.; West, S.M.; Joshi, R.; Honig, B.; Mann, R.S. Origins of specificity in protein-DNA recognition. Annu. Rev. Biochem. 2010, 79, 233–269. [Google Scholar] [CrossRef] [PubMed]
- Parker, S.C.; Hansen, L.; Abaan, H.O.; Tullius, T.D.; Margulies, E.H. Local DNA topography correlates with functional noncoding regions of the human genome. Science 2009, 324, 389–392. [Google Scholar] [CrossRef] [PubMed]
- Unger, R.; Harel, D.; Wherland, S.; Sussman, J.L. A 3D building blocks approach to analyzing and predicting structure of proteins. Proteins 1989, 5, 355–373. [Google Scholar] [CrossRef] [PubMed]
- Levitt, M. Accurate modeling of protein conformation by automatic segment matching. J. Mol. Biol. 1992, 226, 507–533. [Google Scholar] [CrossRef]
- Joseph, A.P.; Agarwal, G.; Mahajan, S.; Gelly, J.-C.; Swapna, L.S.; Offmann, B.; Cadet, F.; Bornot, A.; Tyagi, M.; Valadié, H.; et al. A short survey on protein blocks. Biophys. Rev. 2010, 2, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Cech, P.; Kukal, J.; Cerny, J.; Schneider, B.; Svozil, D. Automatic workflow for the classification of local DNA conformations. BMC Bioinform. 2013, 14, 205. [Google Scholar] [CrossRef] [PubMed]
- Černý, J.; Božíková, P.; Schneider, B. DNATCO: Assignment of DNA conformers at dnatco.org. Nucleic Acids Res. 2016, 44, W284–W287. [Google Scholar] [CrossRef] [PubMed]
- Schneider, B.; Cerny, J.; Svozil, D.; Cech, P.; Gelly, J.C.; de Brevern, A.G. Bioinformatic analysis of the protein/DNA interface. Nucleic Acids Res. 2014, 42, 3381–3394. [Google Scholar] [CrossRef] [PubMed]
- Patikoglou, G.; Burley, S.K. Eukaryotic transcription factor-DNA complexes. Annu. Rev. Biophys. Biomol. Struct. 1997, 26, 289–325. [Google Scholar] [CrossRef] [PubMed]
- Cui, F.; Zhurkin, V.B. Rotational positioning of nucleosomes facilitates selective binding of p53 to response elements associated with cell cycle arrest. Nucleic Acids Res. 2014, 42, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Laptenko, O.; Beckerman, R.; Freulich, E.; Prives, C. P53 binding to nucleosomes within the p21 promoter in vivo leads to nucleosome loss and transcriptional activation. Proc. Natl. Acad. Sci. USA 2011, 108, 10385–10390. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.R.; Pálfy, M.; Hilbert, L.; Kumar, M.; Karschau, J.; Zaburdaev, V.; Shevchenko, A.; Vastenhouw, N.L. Competition between histone and transcription factor binding regulates the onset of transcription in zebrafish embryos. eLife 2017, 6, e23326. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Iype, L.; Schneider, B.; Zardecki, C. The nucleic acid database. Acta Crystallogr. D 2002, 58, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Richmond, T.J.; Davey, C.A. The structure of DNA in the nucleosome core. Nature 2003, 423, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Schneider, B.; Gelly, J.C.; de Brevern, A.G.; Cerny, J. Local dynamics of proteins and DNA evaluated from crystallographic b factors. Acta Crystallogr. D 2014, 70, 2413–2419. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A language and environment for statistical computing. 2016. [Google Scholar]
- Agresti, A. An Introduction to Categorical Data Analysis; Wiley: Hoboken, NJ, USA, 2007. [Google Scholar]
- Xu, F.; Olson, W.K. DNA architecture, deformability, and nucleosome positioning. J. Biomol. Struct. Dyn. 2010, 27, 725–739. [Google Scholar] [CrossRef] [PubMed]
- Cui, F.; Zhurkin, V.B. Structure-based analysis of DNA sequence patterns guiding nucleosome positioning in vitro. J. Biomol. Struct. Dyn. 2010, 27, 821–841. [Google Scholar] [CrossRef] [PubMed]
- Gouge, J.; Satia, K.; Guthertz, N.; Widya, M.; Thompson, A.J.; Cousin, P.; Dergai, O.; Hernandez, N.; Vannini, A. Redox signaling by the RNA polymerase III TFIIB-related factor Brf2. Cell 2015, 163, 1375–1387. [Google Scholar] [CrossRef] [PubMed]
- Olson, W.K.; Zhurkin, V.B. Working the kinks out of nucleosomal DNA. Curr. Opin. Struct. Biol. 2011, 21, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Blanchet, C.; Pasi, M.; Zakrzewska, K.; Lavery, R. Curves+ web server for analyzing and visualizing the helical, backbone and groove parameters of nucleic acid structures. Nucleic Acids Res. 2011, 39, W68–W73. [Google Scholar] [CrossRef] [PubMed]
- Zaret, K.S.; Caravaca, J.M.; Tulin, A.; Sekiya, T. Nuclear mobility and mitotic chromosome binding: Similarities between pioneer transcription factor foxa and linker histone H1. Cold Spring Harb. Symp. Quant. Biol. 2010, 75, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Magnani, L.; Eeckhoute, J.; Lupien, M. Pioneer factors: Directing transcriptional regulators within the chromatin environment. Trends Genet. 2011, 27, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.L.; Halay, E.D.; Lai, E.; Burley, S.K. Co-crystal structure of the HNF-3/fork head DNA-recognition motif resembles histone H5. Nature 1993, 364, 412–420. [Google Scholar] [CrossRef] [PubMed]
- LaRonde-LeBlanc, N.A.; Wolberger, C. Structure of Hoxa9 and Pbx1 bound to DNA: Hox hexapeptide and DNA recognition anterior to posterior. Genes Dev. 2003, 17, 2060–2072. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, X.; Dantas Machado, A.C.; Ding, Y.; Chen, Z.; Qin, P.Z.; Rohs, R.; Chen, L. Structure of p53 binding to the bax response element reveals DNA unwinding and compression to accommodate base-pair insertion. Nucleic Acids Res. 2013, 41, 8368–8376. [Google Scholar] [CrossRef] [PubMed]
- Frouws, T.D.; Duda, S.C.; Richmond, T.J. X-ray structure of the MMTV-A nucleosome core. Proc. Natl. Acad. Sci. USA 2016, 113, 1214–1219. [Google Scholar] [CrossRef] [PubMed]
- Osakabe, A.; Adachi, F.; Arimura, Y.; Maehara, K.; Ohkawa, Y.; Kurumizaka, H. Influence of DNA methylation on positioning and DNA flexibility of nucleosomes with pericentric satellite DNA. Open Biol. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Emamzadah, S.; Tropia, L.; Vincenti, I.; Falquet, B.; Halazonetis, T.D. Reversal of the DNA-binding-induced loop L1 conformational switch in an engineered human p53 protein. J. Mol. Biol. 2014, 426, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.; Martin, R.G.; Rosner, J.L.; Davies, D.R. A novel DNA-binding motif in MarA: The first structure for an AraC family transcriptional activator. Proc. Natl. Acad. Sci. USA 1998, 95, 10413–10418. [Google Scholar] [CrossRef] [PubMed]
- Ngo, H.B.; Kaiser, J.T.; Chan, D.C. The mitochondrial transcription and packaging factor tfam imposes a U-turn on mitochondrial DNA. Nat. Struct. Mol. Biol. 2011, 18, 1290–1296. [Google Scholar] [CrossRef] [PubMed]
- Jain, D.; Kim, Y.; Maxwell, K.L.; Beasley, S.; Zhang, R.; Gussin, G.N.; Edwards, A.M.; Darst, S.A. Crystal structure of bacteriophage λcII and its DNA complex. Mol. Cell. 2005, 19, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Ngo, H.B.; Lovely, G.A.; Phillips, R.; Chan, D.C. Distinct structural features of tfam drive mitochondrial DNA packaging versus transcriptional activation. Nat. Commun. 2014, 5, 3077. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.G.; Pappas, K.M.; Brace, J.L.; Miller, P.C.; Oulmassov, T.; Molyneaux, J.M.; Anderson, J.C.; Bashkin, J.K.; Winans, S.C.; Joachimiak, A. Structure of a bacterial quorum-sensing transcription factor complexed with pheromone and DNA. Nature 2002, 417, 971–974. [Google Scholar] [CrossRef] [PubMed]
- Albright, R.A.; Matthews, B.W. Crystal structure of lambda-Cro bound to a consensus operator at 3.0 a resolution. J. Mol. Biol. 1998, 280, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. Phenix: A comprehensive python-based system for macromolecular structure solution. Acta Crystallogr. D 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D 1997, 53, 240–255. [Google Scholar] [CrossRef] [PubMed]
- Tickle, I.J. Statistical quality indicators for electron-density maps. Acta Crystallogr. D 2012, 68, 454–467. [Google Scholar] [CrossRef] [PubMed]
- Branden, C.; Jones, T. Between objectivity and subjectivity. Nature 1990, 343, 687–689. [Google Scholar] [CrossRef]
- Kleywegt, G.J.; Harris, M.R.; Zou, J.-Y.; Taylor, T.C.; Wahlby, A.; Jones, T.A. The uppsala electron-density server. Acta Crystallogr. D 2004, 60, 2240–2249. [Google Scholar] [CrossRef] [PubMed]
- Virstedt, J.; Berge, T.; Henderson, R.M.; Waring, M.J.; Travers, A.A. The influence of DNA stiffness upon nucleosome formation. J. Struct. Biol. 2004, 148, 66–85. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xi, Z.; Hegde, R.S.; Shakked, Z.; Crothers, D.M. Predicting indirect readout effects in protein–DNA interactions. Proc. Natl. Acad. Sci. USA 2004, 101, 8337–8341. [Google Scholar] [CrossRef] [PubMed]
- Lowary, P.T.; Widom, J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 1998, 276, 19–42. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Blocker, A.W.; Airoldi, E.M.; O’Shea, E.K. A computational approach to map nucleosome positions and alternative chromatin states with base pair resolution. eLife 2016, 5, e16970. [Google Scholar] [CrossRef] [PubMed]
- Satchwell, S.C.; Drew, H.R.; Travers, A.A. Sequence periodicities in chicken nucleosome core DNA. J. Mol. Biol. 1986, 191, 659–675. [Google Scholar] [CrossRef]
- Segal, E.; Fondufe-Mittendorf, Y.; Chen, L.; Thastrom, A.; Field, Y.; Moore, I.K.; Wang, J.P.; Widom, J. A genomic code for nucleosome positioning. Nature 2006, 442, 772–778. [Google Scholar] [CrossRef] [PubMed]
- Trifonov, E.N.; Nibhani, R. Review fifteen years of search for strong nucleosomes. Biopolymers 2015, 103, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Ong, M.S.; Richmond, T.J.; Davey, C.A. DNA stretching and extreme kinking in the nucleosome core. J. Mol. Biol. 2007, 368, 1067–1074. [Google Scholar] [CrossRef] [PubMed]
- Chua, E.Y.; Vasudevan, D.; Davey, G.E.; Wu, B.; Davey, C.A. The mechanics behind DNA sequence-dependent properties of the nucleosome. Nucleic Acids Res. 2012, 40, 6338–6352. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| The Main Structural Features of the CANA Letters | CANA Letter | Regulatory | NCP | ||
|---|---|---|---|---|---|
| # | % | # | % | ||
| A-form conformers | AAA | 802 | 4 | 20 | 0.4 |
| conformers bridging A- to B-form | A-B | 925 | 4.7 | 133 | 2.9 |
| conformers bridging B- to A-form | B-A | 1564 | 7.9 | 184 | 4 |
| the most frequent “canonical” B-form | BBB | 7559 | 38.1 | 1548 | 33.7 |
| less populated BI conformer | 2B1 | 1692 | 8.5 | 204 | 4.4 |
| less populated BI conformers with switched values of torsions α and γ | 3B1 | 1346 | 6.8 | 201 | 4.4 |
| conformer bridging BI- to BII-form | B12 | 1380 | 6.9 | 475 | 10.3 |
| BII conformers | BB2 | 1005 | 5.1 | 490 | 10.7 |
| various minor B conformers | miB | 827 | 4.2 | 565 | 12.3 |
| conformers with bases in syn orientation, may occur in quadruplexes, other non-duplexes | SQX | 69 | 0.3 | 0 | 0 |
| non-Assigned Steps | NAN | 2688 | 13.5 | 778 | 16.9 |
| All Steps | ASt | 19,857 | 100 | 4598 | 100 |
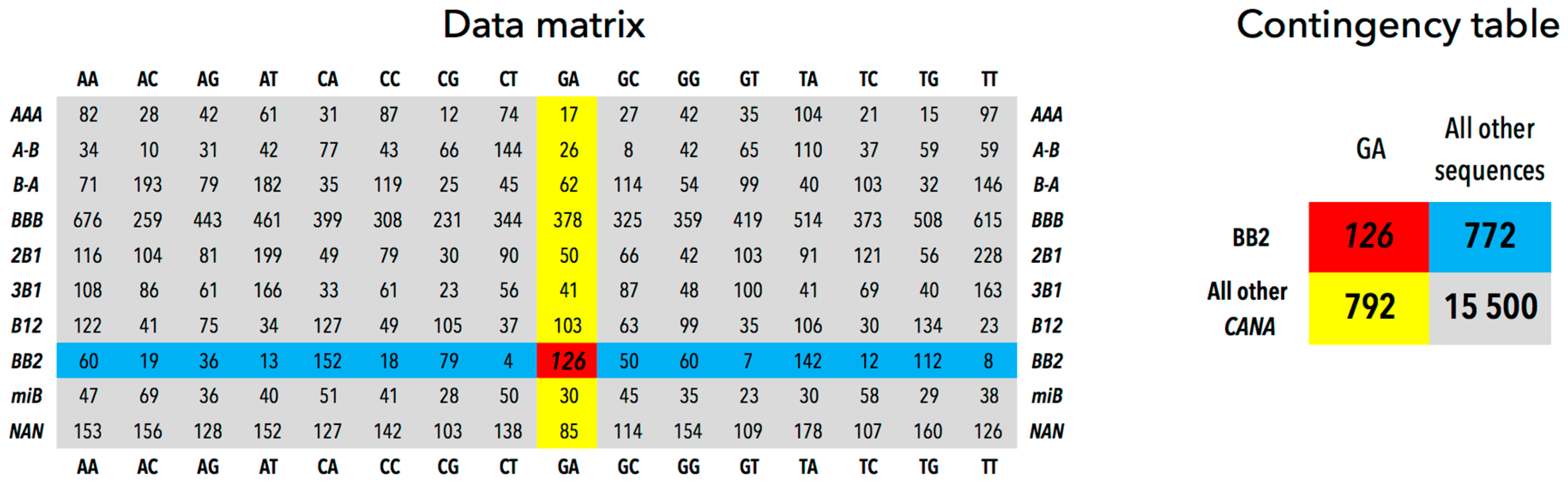
| GA | All Other Sequences | |
|---|---|---|
| BB2 | 126 | 772 |
| All other CANA | 792 | 15,500 |
| GA | All Other Sequences | |
|---|---|---|
| Regulatory < 6 Å BB2 | 126 | 772 |
| Histone < 6 Å BB2 | 38 | 332 |
| BB2 | All Other CANA | |
|---|---|---|
| Regulatory < 6 Å GA | 126 | 792 |
| Histone < 6 Å GA | 38 | 201 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schneider, B.; Božíková, P.; Čech, P.; Svozil, D.; Černý, J. A DNA Structural Alphabet Distinguishes Structural Features of DNA Bound to Regulatory Proteins and in the Nucleosome Core Particle. Genes 2017, 8, 278. https://doi.org/10.3390/genes8100278
Schneider B, Božíková P, Čech P, Svozil D, Černý J. A DNA Structural Alphabet Distinguishes Structural Features of DNA Bound to Regulatory Proteins and in the Nucleosome Core Particle. Genes. 2017; 8(10):278. https://doi.org/10.3390/genes8100278
Chicago/Turabian StyleSchneider, Bohdan, Paulína Božíková, Petr Čech, Daniel Svozil, and Jiří Černý. 2017. "A DNA Structural Alphabet Distinguishes Structural Features of DNA Bound to Regulatory Proteins and in the Nucleosome Core Particle" Genes 8, no. 10: 278. https://doi.org/10.3390/genes8100278
APA StyleSchneider, B., Božíková, P., Čech, P., Svozil, D., & Černý, J. (2017). A DNA Structural Alphabet Distinguishes Structural Features of DNA Bound to Regulatory Proteins and in the Nucleosome Core Particle. Genes, 8(10), 278. https://doi.org/10.3390/genes8100278