
Novel EDA or EDAR Mutations Identified in Patients with X-Linked Hypohidrotic Ectodermal Dysplasia or Non-Syndromic Tooth Agenesis

 ,
, 

Abstract
1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Mutation Detection of WNT10A, EDA, EDAR, EDARADD, PAX9, MSX1, AXIN2, LRP6, and WNT10B
2.3. Whole Exome Sequencing and Data Analysis
2.4. Bioinformatics Analyses
2.5. Structural Modeling
2.6. Minigene Study
3. Results
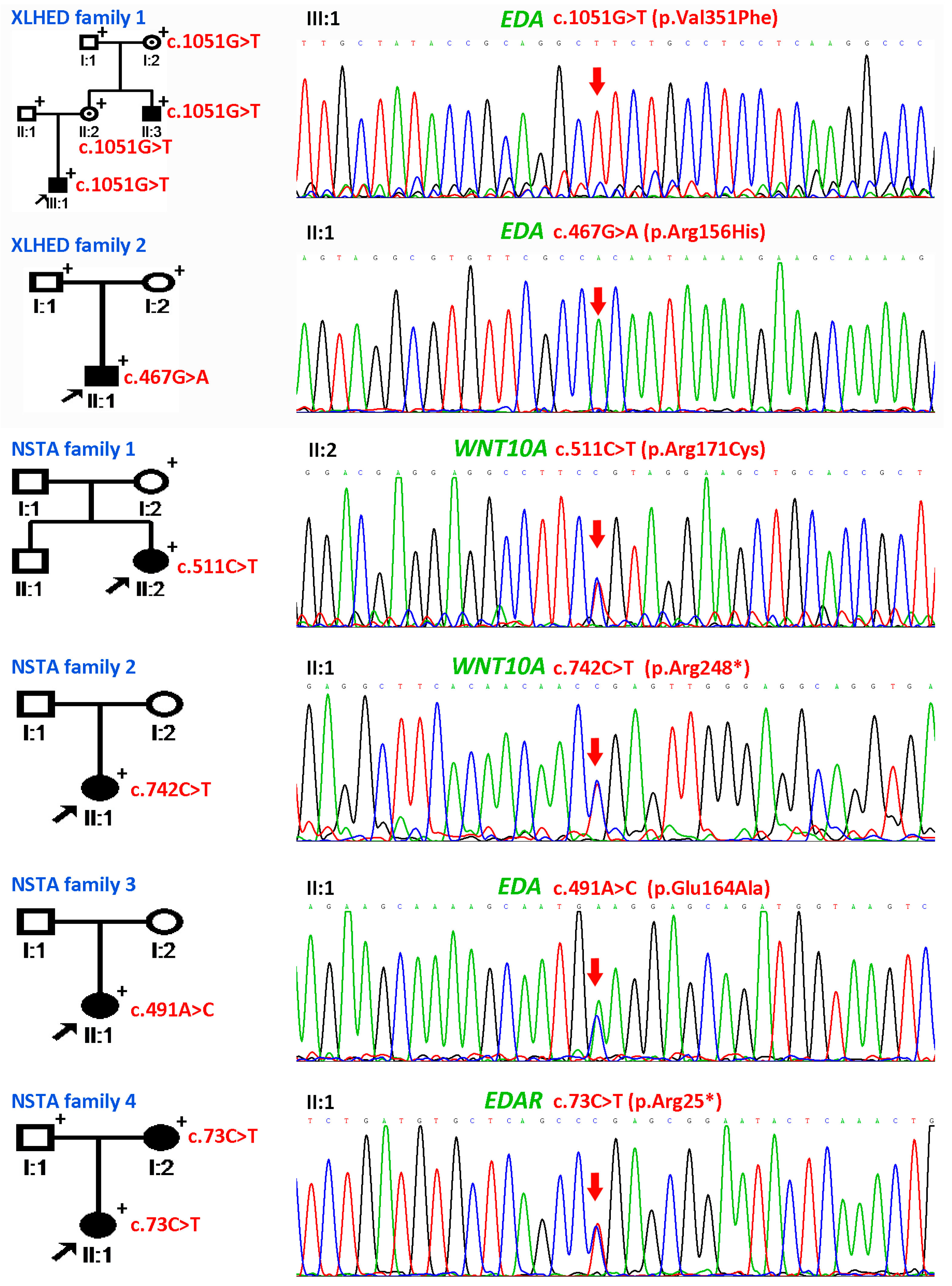
3.1. Clinical Report
3.2. Genetic Findings of WNT10A, EDA, PAX9, and MSX1
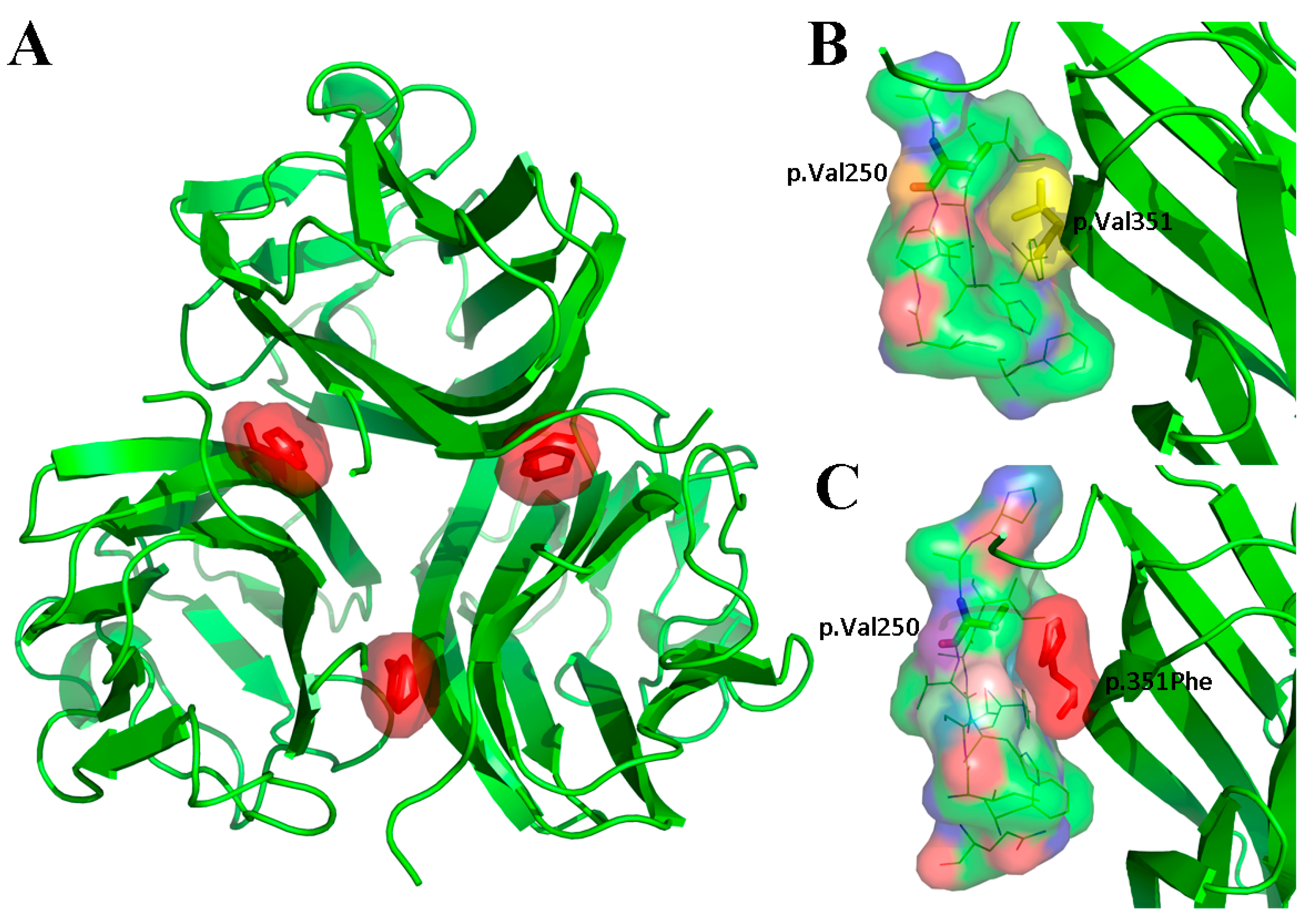
3.3. Results of Whole Exome Sequencing
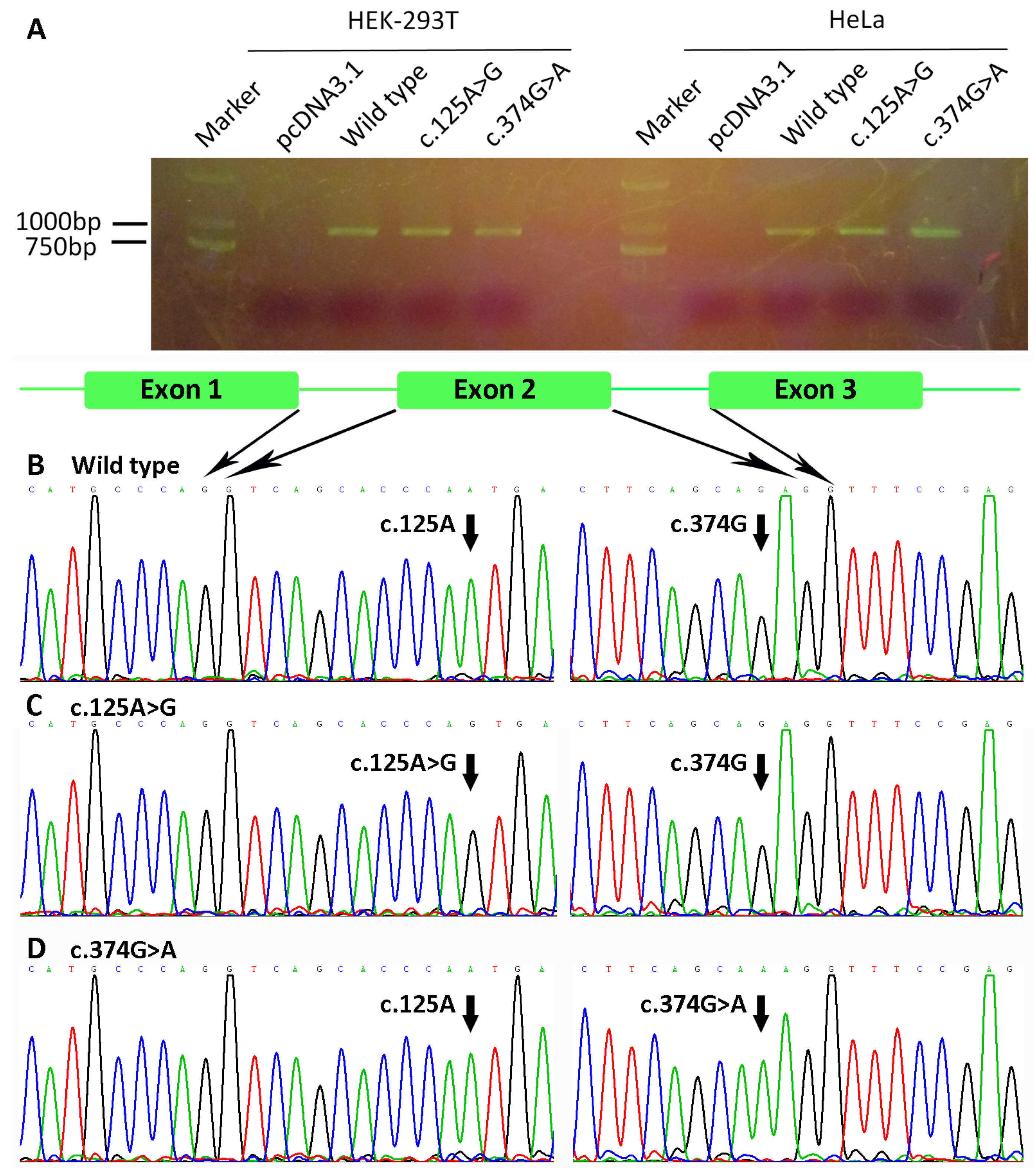
3.4. Bioinformatics Analyses and Minigene Results of WNT10A Variants c.374G>A and c.125A>G
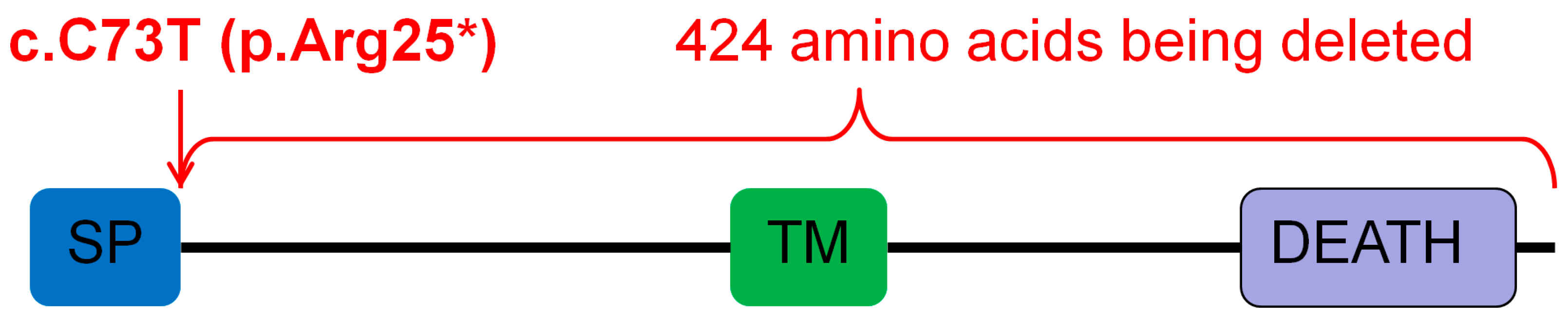
3.5. Sequencing Results of PAX9, MSX1, EDAR, EDARADD, AXIN2, LRP6, and WNT10B in NSTA Families 1 and 3
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rakhshan, V.; Rakhshan, H. Meta-analysis of congenitally missing teeth in the permanent dentition: Prevalence, variations across ethnicities, regions and time. Int. Orthod. 2015, 13, 261–273. [Google Scholar] [CrossRef] [PubMed]
- De Coster, P.J.; Marks, L.A.; Martens, L.C.; Huysseune, A. Dental agenesis: Genetic and clinical perspectives. J. Oral Pathol. Med. 2009, 38, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Qin, C.; Yue, H.; He, H.; Bian, Z. A novel initiation codon mutation of PAX9 in a family with oligodontia. Arch. Oral Biol. 2016, 61, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Pagnan, N.A.; Visinoni, A.F. Update on ectodermal dysplasias clinical classification. Am. J. Med. Genet. A 2014, 164A, 2415–2423. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Chu, E.Y.; Watt, B.; Zhang, Y.; Gallant, N.M.; Andl, T.; Yang, S.H.; Lu, M.; Piccolo, S.; Schmidt-Ullrich, R.; et al. Wnt/beta-catenin signaling directs multiple stages of tooth morphogenesis. Dev. Biol. 2008, 313, 210–224. [Google Scholar] [CrossRef] [PubMed]
- Yamashiro, T.; Zheng, L.; Shitaku, Y.; Saito, M.; Tsubakimoto, T.; Takada, K.; Takano-Yamamoto, T.; Thesleff, I. Wnt10a regulates dentin sialophosphoprotein mRNA expression and possibly links odontoblast differentiation and tooth morphogenesis. Differentiation 2007, 75, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Zhao, P.; Liu, Y.; Zhang, X.; Fu, J.; Yu, H.M.I.; Qiu, M.; Chen, Y.; Hsu, W.; Zhang, Z. Intra-epithelial Requirement of Canonical Wnt Signaling for Tooth Morphogenesis. J. Biol. Chem. 2013, 288, 12080–12089. [Google Scholar] [CrossRef] [PubMed]
- van den Boogaard, M.; Creton, M.; Bronkhorst, Y.; van der Hout, A.; Hennekam, E.; Lindhout, D.; Cune, M.; van Amstel, H.K.P. Mutations in WNT10A are present in more than half of isolated hypodontia cases. J. Med. Genet. 2012, 49, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Mostowska, A.; Biedziak, B.; Zadurska, M.; Dunin-Wilczynska, I.; Lianeri, M.; Jagodzinski, P.P. Nucleotide variants of genes encoding components of the Wnt signalling pathway and the risk of non-syndromic tooth agenesis. Clin. Genet. 2013, 84, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Arzoo, P.S.; Klar, J.; Bergendal, B.; Norderyd, J.; Dahl, N. WNT10A mutations account for (1/4) of population-based isolated oligodontia and show phenotypic correlations. Am. J. Med. Genet. A 2014, 164A, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Tardieu, C.; Jung, S.; Niederreither, K.; Prasad, M.; Hadj-Rabia, S.; Philip, N.; Mallet, A.; Consolino, E.; Sfeir, E.; Noueiri, B.; et al. Dental and extra-oral clinical features in 41 patients with, WNT10A gene mutations: A multicentric genotype-phenotype study. Clin. Genet. 2017. [Google Scholar] [CrossRef] [PubMed]
- Arte, S.; Parmanen, S.; Pirinen, S.; Alaluusua, S.; Nieminen, P. Candidate Gene Analysis of Tooth Agenesis Identifies Novel Mutations in Six Genes and Suggests Significant Role for WNT and EDA Signaling and Allele Combinations. PLoS ONE 2013, 8, e73705. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Han, D.; Feng, H.; Qu, H.; Song, S.; Bai, B.; Zhang, Z. Involvement of and interaction between WNT10A and EDA mutations in tooth agenesis cases in the Chinese population. PLoS ONE 2013, 8, e80393. [Google Scholar] [CrossRef] [PubMed]
- Tucker, A.S.; Headon, D.J.; Schneider, P.; Ferguson, B.M.; Overbeek, P.; Tschopp, J.; Sharpe, P.T. Edar/Eda interactions regulate enamel knot formation in tooth morphogenesis. Development 2000, 127, 4691–4700. [Google Scholar] [PubMed]
- Peters, H.; Neubueser, A.; Kratochwil, K.; Balling, R. Pax9-deficient mice lack pharyngeal pouch derivatives and teeth and exhibit craniofacial and limb abnormalities. Genes Dev. 1998, 12, 2735–2747. [Google Scholar] [CrossRef] [PubMed]
- Satokata, I.; Maas, R. Msx1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nat. Genet. 1994, 6, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Bian, Z. The Gene Network Underlying Hypodontia. J. Dent. Res. 2015, 94, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Ruf, S.; Klimas, D.; Hoenemann, M.; Jabir, S. Genetic background of nonsyndromic oligodontia: A systematic review and meta-analysis. J. Orofac. Orthop. 2013, 74, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Bergendal, B.; Klar, J.; Stecksen-Blicks, C.; Norderyd, J.; Dahl, N. Isolated oligodontia associated with mutations in EDARADD, AXIN2, MSX1 and PAX9 genes. Am. J. Med. Genet. A 2011, 155A, 1616–1622. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Yang, W.; Han, D.; Wang, X.; Guo, S.; Li, J.; Li, F.; Zhang, X.; Wong, S.; Bai, B.; et al. Mutations in WNT10B Are Identified in Individuals with Oligodontia. Am. J. Hum. Genet. 2016, 99, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Gaczkowska, A.; Abdalla, E.M.; Dowidar, K.M.; Elhady, G.M.; Jagodzinski, P.P.; Mostowska, A. De novo EDA mutations: Variable expression in two Egyptian families. Arch. Oral Biol. 2016, 68, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Visinoni, A.F.; Lisboa-Costa, T.; Pagnan, N.A.; Chautard-Freire-Maia, E.A. Ectodermal dysplasias: Clinical and molecular review. Am. J. Med. Genet. A 2009, 149A, 1980–2002. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xie, L. Novel nonsense mutation of the EDA gene in a Chinese family with X-linked hypohidrotic ectodermal dysplasia. J. Dermatol. 2014, 41, 1014–1016. [Google Scholar] [PubMed]
- Zeng, B.; Xiao, X.; Li, S.; Lu, H.; Lu, J.; Zhu, L.; Yu, D.; Zhao, W. Eight Mutations of Three Genes (EDA, EDAR, and WNT10A) Identified in Seven Hypohidrotic Ectodermal Dysplasia Patients. Genes 2016, 7, 65. [Google Scholar] [CrossRef] [PubMed]
- Monreal, A.W.; Ferguson, B.M.; Headon, D.J.; Street, S.L.; Overbeek, P.A.; Zonana, J. Mutations in the human homologue of mouse dl cause autosomal recessive and dominant hypohidrotic ectodermal dysplasia. Nat. Genet. 1999, 22, 366–369. [Google Scholar] [PubMed]
- Headon, D.J.; Emmal, S.A.; Ferguson, B.M.; Tucker, A.S.; Justice, M.J.; Sharpe, P.T.; Zonana, J.; Overbeek, P.A. Gene defect in ectodermal dysplasia implicates a death domain adapter in development. Nature 2001, 414, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Cluzeau, C.; Hadj-Rabia, S.; Jambou, M.; Mansour, S.; Guigue, P.; Masmoudi, S.; Bal, E.; Chassaing, N.; Vincent, M.C.; Viot, G.; et al. Only four genes (EDA1, EDAR, EDARADD, and, WNT10A) account for 90% of hypohidrotic/anhidrotic ectodermal dysplasia cases. Hum. Mutat. 2011, 32, 70–72. [Google Scholar] [CrossRef] [PubMed]
- Adaimy, L.; Chouery, E.; Megarbane, H.; Mroueh, S.; Delague, V.; Nicolas, E.; Belguith, H.; de Mazancourt, P.; Megarbane, A. Mutation in WNT10A is associated with an autosomal recessive ectodermal dysplasia: The odonto-onycho-dermal dysplasia. Am. J. Hum. Genet. 2007, 81, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Zeng, B.; Lu, H.; Xiao, X.; Zhou, L.; Lu, J.; Zhu, L.; Yu, D.; Zhao, W. Novel EDA mutation in X-linked hypohidrotic ectodermal dysplasia and genotype-phenotype correlation. Oral Dis. 2015, 21, 994–1000. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.E.; Ko, J.; Shin, T.J.; Hyun, H.K.; Lee, S.H.; Kim, J.W. Oligodontia and curly hair occur with ectodysplasin-a mutations. J. Dent. Res. 2014, 93, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Guazzarotti, L.; Tadini, G.; Mancini, G.E.; Giglio, S.; Willoughby, C.E.; Callea, M.; Sani, I.; Nannini, P.; Mameli, C.; Tenconi, A.A.; et al. Phenotypic heterogeneity and mutational spectrum in a cohort of 45 Italian males subjects with, X-linked ectodermal dysplasia. Clin. Genet. 2015, 87, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Bohring, A.; Stamm, T.; Spaich, C.; Haase, C.; Spree, K.; Hehr, U.; Hoffmann, M.; Ledig, S.; Sel, S.; Wieacker, P.; et al. WNT10A mutations are a frequent cause of a broad spectrum of ectodermal dysplasias with sex-biased manifestation pattern in heterozygotes. Am. J. Hum. Genet. 2009, 85, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Vink, C.P.; Ockeloen, C.W.; Ten, K.S.; Koolen, D.A.; Ploos, V.A.J.; Kuijpers-Jagtman, A.M.; van Heumen, C.C.; Kleefstra, T.; Carels, C.E. Variability in dentofacial phenotypes in four families with WNT10A mutations. Eur. J. Hum. Genet. 2014, 22, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e603. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1082. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Desmet, F.; Hamroun, D.; Lalande, M.; Collod-Beroud, G.; Claustres, M.; Beroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Vincent, M.C.; Biancalana, V.; Ginisty, D.; Mandel, J.L.; Calvas, P. Mutational spectrum of the ED1 gene in X-linked hypohidrotic ectodermal dysplasia. Eur. J. Hum. Genet. 2001, 9, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Mues, G.; Tardivel, A.; Willen, L.; Kapadia, H.; Seaman, R.; Frazier-Bowers, S.; Schneider, P.; D’Souza, R.N. Functional analysis of Ectodysplasin-A mutations causing selective tooth agenesis. Eur. J. Hum. Genet. 2010, 18, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Han, D.; Qu, H.; Gong, Y.; Wu, H.; Zhang, X.; Zhong, N.; Feng, H. EDA gene mutations underlie non-syndromic oligodontia. J. Dent. Res. 2009, 88, 126–131. [Google Scholar] [CrossRef] [PubMed]
- van der Hout, A.H.; Oudesluijs, G.G.; Venema, A.; Verheij, J.B.; Mol, B.G.; Rump, P.; Brunner, H.G.; Vos, Y.J.; van Essen, A.J. Mutation screening of the Ectodysplasin-A receptor gene EDAR in hypohidrotic ectodermal dysplasia. Eur. J. Hum. Genet. 2008, 16, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Trzeciak, W.H.; Koczorowski, R. Molecular basis of hypohidrotic ectodermal dysplasia: An update. J. Appl. Genet. 2016, 57, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Hosomichi, K.; Yano, K.; Kim, Y.; Nakaoka, H.; Kimura, R.; Otsuka, H.; Nonaka, N.; Haga, S.; Takahashi, M.; et al. Comprehensive genetic exploration of selective tooth agenesis of mandibular incisors by exome sequencing. Hum. Genome Var. 2017, 4, 17005. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Street, S.L.; Gaide, O.; Hertig, S.; Tardivel, A.; Tschopp, J.; Runkel, L.; Alevizopoulos, K.; Ferguson, B.M.; Zonana, J. Mutations leading to X-linked hypohidrotic ectodermal dysplasia affect three major functional domains in the tumor necrosis factor family member ectodysplasin-A. J. Biol. Chem. 2001, 276, 18819–18827. [Google Scholar] [CrossRef] [PubMed]
- Bodmer, J.L.; Schneider, P.; Tschopp, J. The molecular architecture of the TNF superfamily. Trends Biochem. Sci. 2002, 27, 19–26. [Google Scholar] [CrossRef]
- Hymowitz, S.G.; Compaan, D.M.; Yan, M.; Wallweber, H.J.; Dixit, V.M.; Starovasnik, M.A.; de Vos, A.M. The crystal structures of EDA-A1 and EDA-A2: Splice variants with distinct receptor specificity. Structure 2003, 11, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- Mues, G.; Bonds, J.; Xiang, L.; Vieira, A.R.; Seymen, F.; Klein, O.; D’Souza, R.N. The WNT10A gene in ectodermal dysplasias and selective tooth agenesis. Am. J. Med. Genet. A 2014, 164A, 2455–2460. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Ye, X.; Shi, L.; Yin, W.; Hua, B.; Song, G.; Shi, B.; Bian, Z. Mutations in the EDA gene are responsible for X-linked hypohidrotic ectodermal dysplasia and hypodontia in Chinese kindreds. Eur. J. Oral Sci. 2008, 116, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Noor, A.; Windpassinger, C.; Vitcu, I.; Orlic, M.; Rafiq, M.A.; Khalid, M.; Malik, M.N.; Ayub, M.; Alman, B.; Vincent, J.B. Oligodontia Is Caused by Mutation in LTBP3, the Gene Encoding Latent TGF-bet Binding Protein 3. Am. J. Hum. Genet. 2009, 84, 519–523. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Patient | Age and Gender | Gene | Nucleotide Change | Amino Acid Change | Number of Missing Permanent Teeth # |
|---|---|---|---|---|---|---|
| XLHED1 | III:1 | 2y, M | EDA | c.1051G>T | p.Val351Phe | ND |
| XLHED2 | II:1 | 6y, M | EDA | c.467G>A | p.Arg156His | 23 |
| NSTA1 | II:2 | 10y, F | WNT10A | c.511C>T | p.Arg171Cys | 4 |
| NSTA2 | II:1 | 6y, F | WNT10A | c.742C>T | p.Arg248* | 9 |
| NSTA3 | II:1 | 9y, F | EDA | c.491A>C | p.Glu164Ala | 2 |
| NSTA4 | II:1 | 6y, F | EDAR | c.73C>T | p.Arg25* | 6 |
| Family | Patient | MT # | Right | Left | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 8 | 7 | 6 | 5 | 4 | 3 | 2 | 1 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | ||||
| XLHED2 | II:1 | 23 | Maxillary | * | * | * | * | * | * | * | * | * | * | * | |||||
| Mandibular | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | |||
| NSTA1 | II:2 | 4 | Maxillary | * | * | * | |||||||||||||
| Mandibular | * | * | * | * | * | ||||||||||||||
| NSTA2 | II:1 | 9 | Maxillary | * | * | * | * | * | * | * | |||||||||
| Mandibular | * | * | * | * | * | * | |||||||||||||
| NSTA3 | II:1 | 2 | Maxillary | * | * | ||||||||||||||
| Mandibular | * | * | * | * | |||||||||||||||
| NSTA4 | II:1 | 6 | Maxillary | * | * | * | * | * | |||||||||||
| Mandibular | * | * | * | * | * | ||||||||||||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, B.; Zhao, Q.; Li, S.; Lu, H.; Lu, J.; Ma, L.; Zhao, W.; Yu, D. Novel EDA or EDAR Mutations Identified in Patients with X-Linked Hypohidrotic Ectodermal Dysplasia or Non-Syndromic Tooth Agenesis. Genes 2017, 8, 259. https://doi.org/10.3390/genes8100259
Zeng B, Zhao Q, Li S, Lu H, Lu J, Ma L, Zhao W, Yu D. Novel EDA or EDAR Mutations Identified in Patients with X-Linked Hypohidrotic Ectodermal Dysplasia or Non-Syndromic Tooth Agenesis. Genes. 2017; 8(10):259. https://doi.org/10.3390/genes8100259
Chicago/Turabian StyleZeng, Binghui, Qi Zhao, Sijie Li, Hui Lu, Jiaxuan Lu, Lan Ma, Wei Zhao, and Dongsheng Yu. 2017. "Novel EDA or EDAR Mutations Identified in Patients with X-Linked Hypohidrotic Ectodermal Dysplasia or Non-Syndromic Tooth Agenesis" Genes 8, no. 10: 259. https://doi.org/10.3390/genes8100259
APA StyleZeng, B., Zhao, Q., Li, S., Lu, H., Lu, J., Ma, L., Zhao, W., & Yu, D. (2017). Novel EDA or EDAR Mutations Identified in Patients with X-Linked Hypohidrotic Ectodermal Dysplasia or Non-Syndromic Tooth Agenesis. Genes, 8(10), 259. https://doi.org/10.3390/genes8100259