The Genetic Basis of Pericentral Retinitis Pigmentosa—A Form of Mild Retinitis Pigmentosa

Abstract
1. Introduction
2. Materials and Methods
2.1. Cohort
2.2. Panel-Based Exon Sequencing
2.3. Sanger Validation
2.4. Copy Number Variation (CNV) Analysis
2.5. Whole Exome Sequencing and Burden Test
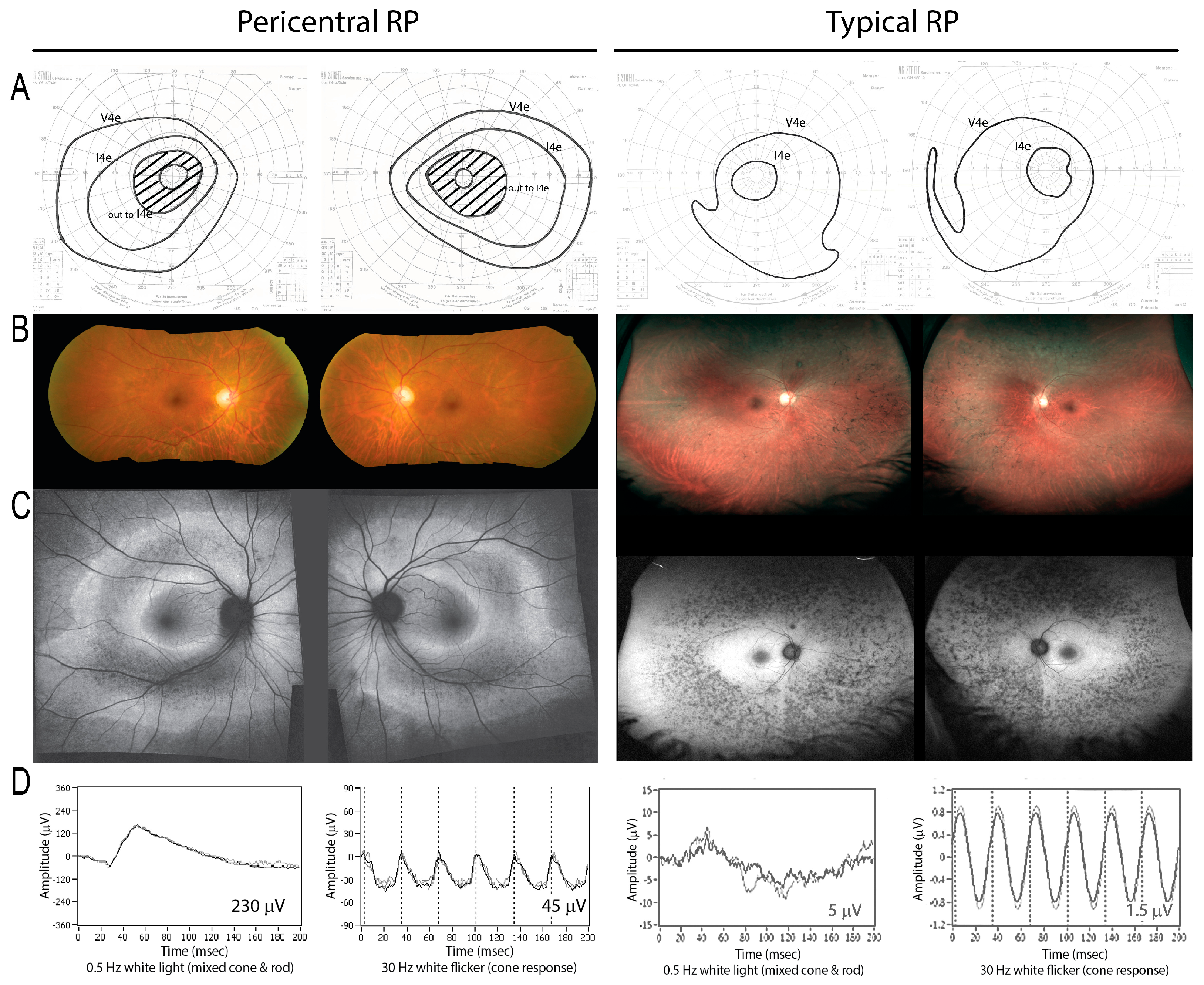
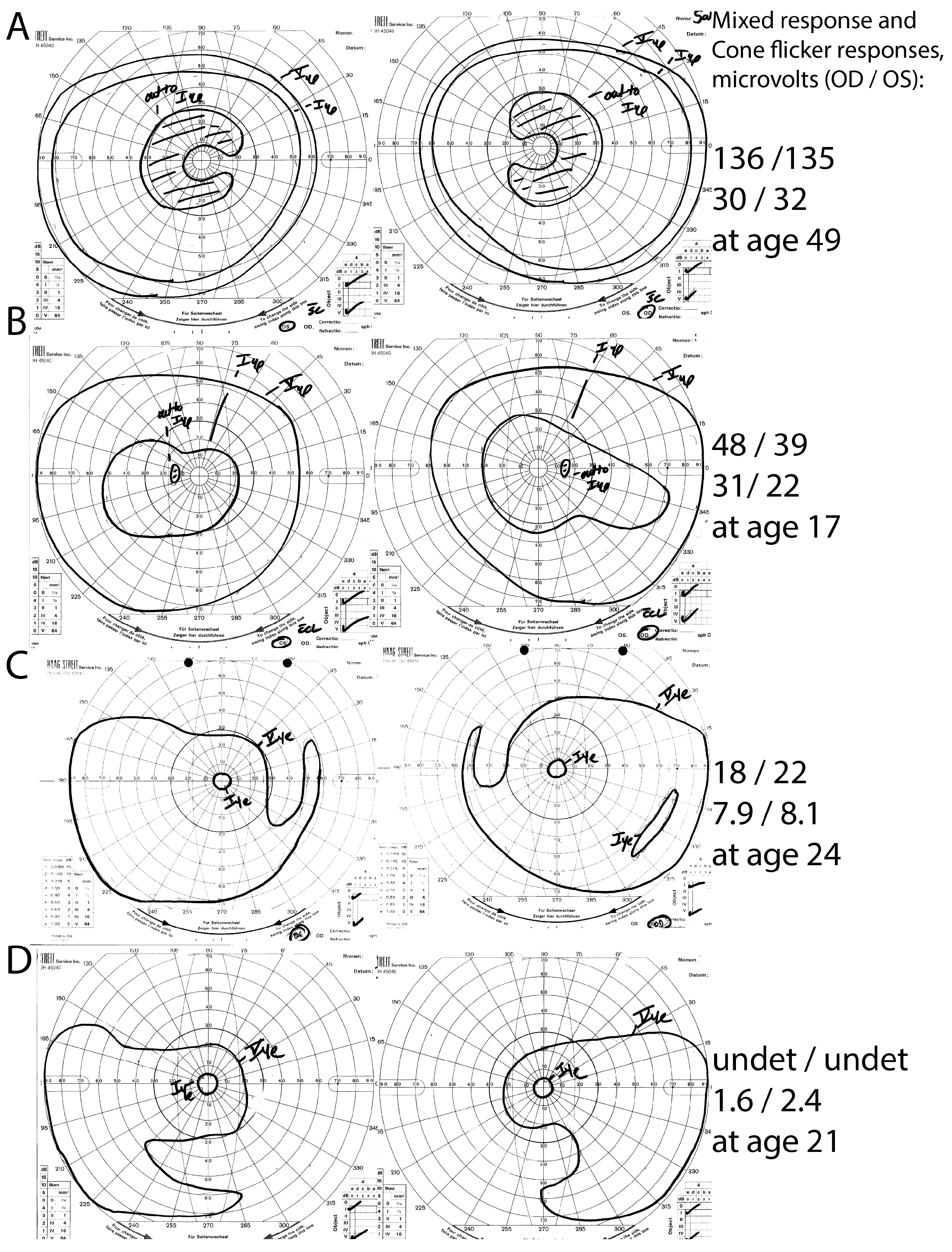
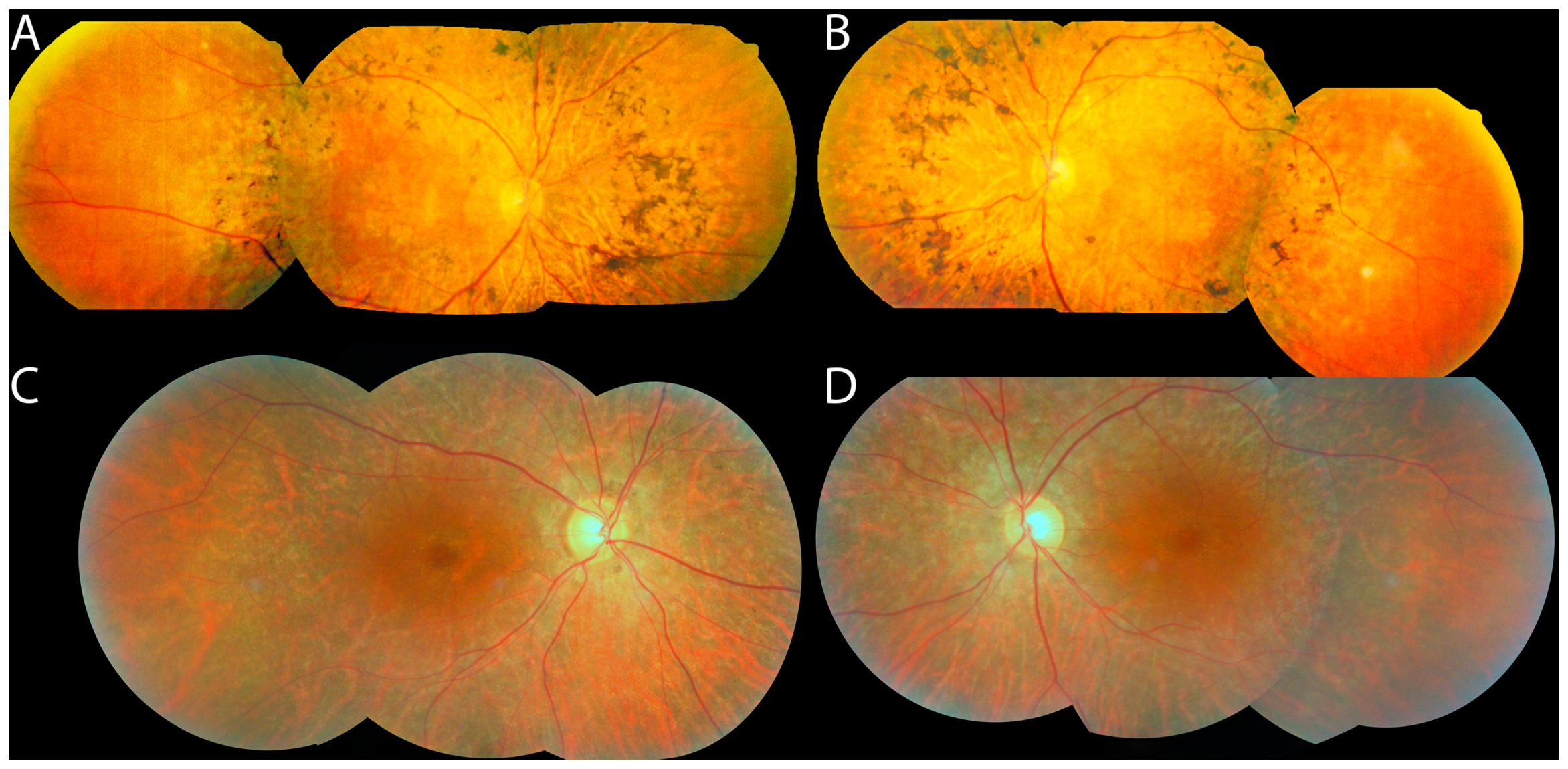
3. Results
3.1. Cohort
3.2. Panel-Based Sequencing Results
3.3. Whole Exome Sequencing Results
4. Discussion
4.1. What Genetics Reveals about the Causes of the Pericentral RP Phenotype
4.2. Whole Exome Sequencing-Based Gene Discovery and HGSNAT
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Daiger, S.P. RetNet, the Retinal Information Network. Available online: http://www.sph.uth.tmc.edu/RetNet/ (accessed on 24 June 2017).
- Grover, S.; Fishman, G.A.; Brown, J., Jr. Patterns of visual field progression in patients with retinitis pigmentosa. Ophthalmology 1998, 105, 1069–1075. [Google Scholar] [CrossRef]
- Chang, S.; Vaccarella, L.; Olatunji, S.; Cebulla, C.; Christoforidis, J. Diagnostic challenges in retinitis pigmentosa: Genotypic multiplicity and phenotypic variability. Curr. Genomics 2011, 12, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Gonin, J. Le scotome Annulaire Dans la Dégénérescence Pigmentaire de la Rétine; A. Maloine: Paris, France, 1901. [Google Scholar]
- Berson, E.L.; Howard, J. Temporal aspects of the electroretinogram in sector retinitis pigmentosa. Arch. Ophthalmol. 1971, 86, 653–665. [Google Scholar] [CrossRef] [PubMed]
- Valentine, J.A. Night Blindness: Retinitis Pigmentosa sine Pigmento. Proc. R. Soc. Med. 1923, 16, 17. [Google Scholar] [PubMed]
- Choi, J.Y.; Sandberg, M.A.; Berson, E.L. Natural course of ocular function in pigmented paravenous retinochoroidal atrophy. Am. J. Ophthalmol. 2006, 141, 763–765. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.B.; Zhang, Y.X. Pigmented paravenous retinochoroidal atrophy (Review). Exp. Ther. Med. 2014, 7, 1439–1445. [Google Scholar] [CrossRef] [PubMed]
- Francois, J.; De Rouck, A.; Cambie, E.; De Laey, J.J. Visual functions in pericentral and central pigmentary retinopathy. Ophthalmologica 1972, 165, 38–61. [Google Scholar] [CrossRef] [PubMed]
- Traboulsi, E.I.; O’Neill, J.F.; Maumenee, I.H. Autosomal recessive pericentral pigmentary retinopathy. Am. J. Ophthalmol. 1988, 106, 551–556. [Google Scholar] [CrossRef]
- Durlu, Y.K.; Burumcek, E.; Devranoglu, K.; Mudun, A.B.; Karacorlu, S.; Arslan, M.O. Associated ocular findings in pericentral pigmentary retinopathy. Acta Ophthalmol. Scand. 1997, 75, 101–103. [Google Scholar] [CrossRef] [PubMed]
- Hayasaka, S.; Fukuda, K.; Tsuchiya, M.; Mizuno, K. Pericentral pigmentary retinal degeneration. Jpn. J. Ophthalmol. 1985, 29, 161–169. [Google Scholar] [PubMed]
- Yamaguchi, K.; Kin-para, Y.; Tamai, M. Idiopathic central serous choroidopathy in a patient with pericentral pigmentary retinal degeneration. Ann. Ophthalmol. 1991, 23, 251–253. [Google Scholar] [PubMed]
- Noble, K.G. Peripapillary (pericentral) pigmentary retinal degeneration. Am. J. Ophthalmol. 1989, 108, 686–690. [Google Scholar] [CrossRef]
- Hotta, K.; Kondo, M.; Nakamura, M.; Hotta, J.; Terasaki, H.; Miyake, Y.; Hida, T. Negative electroretinograms in pericentral pigmentary retinal degeneration. Clin. Exp. Ophthalmol. 2006, 34, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Grondahl, J. Pericentral retinal dystrophy. Acta Ophthalmol. 1987, 65, 344–351. [Google Scholar] [CrossRef]
- Noble, K.G.; Carr, R.E. Peripapillary pigmentary retinal degeneration. Am. J. Ophthalmol. 1978, 86, 65–75. [Google Scholar] [CrossRef]
- Chakarova, C.F.; Papaioannou, M.G.; Khanna, H.; Lopez, I.; Waseem, N.; Shah, A.; Theis, T.; Friedman, J.; Maubaret, C.; Bujakowska, K.; et al. Mutations in TOPORS cause autosomal dominant retinitis pigmentosa with perivascular retinal pigment epithelium atrophy. Am. J. Hum. Genet. 2007, 81, 1098–1103. [Google Scholar] [CrossRef] [PubMed]
- Matsui, R.; Cideciyan, A.V.; Schwartz, S.B.; Sumaroka, A.; Roman, A.J.; Swider, M.; Huang, W.C.; Sheplock, R.; Jacobson, S.G. Molecular Heterogeneity Within the Clinical Diagnosis of Pericentral Retinal Degeneration. Invest. Ophthalmol. Vis. Sci. 2015, 56, 6007–6018. [Google Scholar] [CrossRef] [PubMed]
- Krill, A. Central and pericentral retinitis pigmentosa. In Krill’s Hereditary Retinal and Choroidal Diseases; Archer, D.B., Ed.; Blackwell Publishing: Hagerstown, MD, USA, 1977; Volume 2, pp. 617–625. [Google Scholar]
- Szamier, R.B.; Berson, E.L. Histopathologic study of an unusual form of retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 1982, 22, 559–570. [Google Scholar] [PubMed]
- Sandberg, M.A.; Gaudio, A.R.; Berson, E.L. Disease course of patients with pericentral retinitis pigmentosa. Am. J. Ophthalmol. 2005, 140, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Grondahl, J.; Riise, R.; Heiberg, A.; Leren, T.; Christoffersen, T.; Bragadottir, R. Autosomal dominant retinitis pigmentosa in Norway: A 20-year clinical follow-up study with molecular genetic analysis. Two novel rhodopsin mutations: 1003delG and I179F. Acta Ophthalmol. Scand. 2007, 85, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.G.; McGuigan, D.B., 3rd; Sumaroka, A.; Roman, A.J.; Gruzensky, M.L.; Sheplock, R.; Palma, J.; Schwartz, S.B.; Aleman, T.S.; Cideciyan, A.V. Complexity of the Class B Phenotype in Autosomal Dominant Retinitis Pigmentosa Due to Rhodopsin Mutations. Invest. Ophthalmol. Vis. Sci. 2016, 57, 4847–4858. [Google Scholar] [CrossRef] [PubMed]
- Selmer, K.K.; Grondahl, J.; Riise, R.; Brandal, K.; Braaten, O.; Bragadottir, R.; Undlien, D.E. Autosomal dominant pericentral retinal dystrophy caused by a novel missense mutation in the TOPORS gene. Acta Ophthalmol. 2010, 88, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Bowne, S.J.; Sullivan, L.S.; Gire, A.I.; Birch, D.G.; Hughbanks-Wheaton, D.; Heckenlively, J.R.; Daiger, S.P. Mutations in the TOPORS gene cause 1% of autosomal dominant retinitis pigmentosa. Mol. Vis. 2008, 14, 922–927. [Google Scholar] [PubMed]
- Schob, C.; Orth, U.; Gal, A.; Kindler, S.; Chakarova, C.F.; Bhattacharya, S.S.; Ruther, K. Mutations in TOPORS: A rare cause of autosomal dominant retinitis pigmentosa in continental Europe? Ophthalmic Genet. 2009, 30, 96–98. [Google Scholar] [CrossRef] [PubMed]
- Melles, R.B.; Marmor, M.F. Pericentral retinopathy and racial differences in hydroxychloroquine toxicity. Ophthalmology 2015, 122, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.A.; Marmor, M.F. ERG and other discriminators between advanced hydroxychloroquine retinopathy and retinitis pigmentosa. Doc. Ophthalmol. 2017, 134, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Berson, E.L.; Rosner, B.; Sandberg, M.A.; Hayes, K.C.; Nicholson, B.W.; Weigel-DiFranco, C.; Willett, W. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch. Ophthalmol. 1993, 111, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Consugar, M.B.; Navarro-Gomez, D.; Place, E.M.; Bujakowska, K.M.; Sousa, M.E.; Fonseca-Kelly, Z.D.; Taub, D.G.; Janessian, M.; Wang, D.Y.; Au, E.D.; et al. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genet. Med. 2015, 17, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.J.; Zhang, Q.; Nakamaru-Ogiso, E.; Kannabiran, C.; Fonseca-Kelly, Z.; Chakarova, C.; Audo, I.; Mackay, D.S.; Zeitz, C.; Borman, A.D.; et al. NMNAT1 mutations cause Leber congenital amaurosis. Nat. Genet. 2012, 44, 1040–1045. [Google Scholar] [CrossRef] [PubMed]
- Bujakowska, K.M.; Consugar, M.; Place, E.; Harper, S.; Lena, J.; Taub, D.G.; White, J.; Navarro-Gomez, D.; Weigel DiFranco, C.; Farkas, M.H.; et al. Targeted exon sequencing in Usher syndrome type I. Invest. Ophthalmol. Vis. Sci. 2014, 55, 8488–8496. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017, 136, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef] [PubMed]
- Gai, X.; Perin, J.C.; Murphy, K.; O’Hara, R.; D’Arcy, M.; Wenocur, A.; Xie, H.M.; Rappaport, E.F.; Shaikh, T.H.; White, P.S. CNV Workshop: An integrated platform for high-throughput copy number variation discovery and clinical diagnostics. BMC Bioinform. 2010, 11, 74. [Google Scholar] [CrossRef] [PubMed]
- GATK Best Practices. Available online: https://software.broadinstitute.org/gatk/best-practices/ (accessed on 1 October 2017).
- Purcell, S.M.; Moran, J.L.; Fromer, M.; Ruderfer, D.; Solovieff, N.; Roussos, P.; O’Dushlaine, C.; Chambert, K.; Bergen, S.E.; Kahler, A.; et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature 2014, 506, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed]
- Fedele, A.O.; Filocamo, M.; Di Rocco, M.; Sersale, G.; Lubke, T.; di Natale, P.; Cosma, M.P.; Ballabio, A. Mutational analysis of the HGSNAT gene in Italian patients with mucopolysaccharidosis IIIC (Sanfilippo C syndrome). Mutation in brief #959. Online. Hum. Mutat. 2007, 28, 523. [Google Scholar] [PubMed]
- Haer-Wigman, L.; Newman, H.; Leibu, R.; Bax, N.M.; Baris, H.N.; Rizel, L.; Banin, E.; Massarweh, A.; Roosing, S.; Lefeber, D.J.; et al. Non-syndromic retinitis pigmentosa due to mutations in the mucopolysaccharidosis type IIIC gene, heparan-alpha-glucosaminide N-acetyltransferase (HGSNAT). Hum. Mol. Genet. 2015, 24, 3742–3751. [Google Scholar] [CrossRef] [PubMed]
- Ponjavic, V.; Abrahamson, M.; Andreasson, S.; Ehinger, B.; Fex, G.; Polland, W. A mild phenotype of autosomal dominant retinitis pigmentosa is associated with the rhodopsin mutation Pro-267-Leu. Ophthalmic Genet. 1997, 18, 63–70. [Google Scholar] [PubMed]
- Berson, E.L.; Rosner, B.; Weigel-DiFranco, C.; Dryja, T.P.; Sandberg, M.A. Disease progression in patients with dominant retinitis pigmentosa and rhodopsin mutations. Invest. Ophthalmol. Vis. Sci. 2002, 43, 3027–3036. [Google Scholar] [PubMed]
- Sandberg, M.A.; Rosner, B.; Weigel-DiFranco, C.; McGee, T.L.; Dryja, T.P.; Berson, E.L. Disease course in patients with autosomal recessive retinitis pigmentosa due to the USH2A gene. Invest. Ophthalmol. Vis. Sci. 2008, 49, 5532–5539. [Google Scholar] [CrossRef] [PubMed]
- Fishman, G.A.; Stone, E.M.; Gilbert, L.D.; Sheffield, V.C. Ocular findings associated with a rhodopsin gene codon 106 mutation. Glycine-to-arginine change in autosomal dominant retinitis pigmentosa. Arch. Ophthalmol. 1992, 110, 646–653. [Google Scholar] [CrossRef] [PubMed]
- Comander, J.; Weigel-DiFranco, C.; Sandberg, M.A.; Berson, E.L. Visual Function in Carriers of X-Linked Retinitis Pigmentosa. Ophthalmology 2015, 122, 1899–1906. [Google Scholar] [CrossRef] [PubMed]
- Rose, A.M.; Shah, A.Z.; Venturini, G.; Rivolta, C.; Rose, G.E.; Bhattacharya, S.S. Dominant PRPF31 mutations are hypostatic to a recessive CNOT3 polymorphism in retinitis pigmentosa: A novel phenomenon of “linked trans-acting epistasis”. Ann. Hum. Genet. 2014, 78, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.B.; Bigham, A.W.; Buckingham, K.J.; Hannibal, M.C.; McMillin, M.J.; Gildersleeve, H.I.; Beck, A.E.; Tabor, H.K.; Cooper, G.M.; Mefford, H.C.; et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 2010, 42, 790–793. [Google Scholar] [CrossRef] [PubMed]
- Feldhammer, M.; Durand, S.; Mrazova, L.; Boucher, R.M.; Laframboise, R.; Steinfeld, R.; Wraith, J.E.; Michelakakis, H.; van Diggelen, O.P.; Hrebicek, M.; et al. Sanfilippo syndrome type C: Mutation spectrum in the heparan sulfate acetyl-CoA: alpha-glucosaminide N-acetyltransferase (HGSNAT) gene. Hum. Mutat. 2009, 30, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Van Cauwenbergh, C.; Van Schil, K.; Cannoodt, R.; Bauwens, M.; Van Laethem, T.; De Jaegere, S.; Steyaert, W.; Sante, T.; Menten, B.; Leroy, B.P.; et al. arrEYE: A customized platform for high-resolution copy number analysis of coding and noncoding regions of known and candidate retinal dystrophy genes and retinal noncoding RNAs. Genet. Med. 2017, 19, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Carss, K.J.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Hrebicek, M.; Mrazova, L.; Seyrantepe, V.; Durand, S.; Roslin, N.M.; Noskova, L.; Hartmannova, H.; Ivanek, R.; Cizkova, A.; Poupetova, H.; et al. Mutations in TMEM76* cause mucopolysaccharidosis IIIC (Sanfilippo C syndrome). Am. J. Hum. Genet. 2006, 79, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Feldhammer, M.; Durand, S.; Pshezhetsky, A.V. Protein misfolding as an underlying molecular defect in mucopolysaccharidosis III type C. PLoS ONE 2009, 4, e7434. [Google Scholar] [CrossRef] [PubMed]
- Fedele, A.O.; Hopwood, J.J. Functional analysis of the HGSNAT gene in patients with mucopolysaccharidosis IIIC (Sanfilippo C Syndrome). Hum. Mutat. 2010, 31, E1574–E1586. [Google Scholar] [CrossRef] [PubMed]
- gnomAD browser beta. Variant: 8:43054647 G/A. Available online: http://gnomad.broadinstitute.org/variant/8-43054647-G-A (accessed on 1 October 2017).




{kind=link}
{kind=link}
{kind=link}
| Family# | ID# | Sex | Age at First Visit (Years) | VA Snellen Equiv. | VA Decimal | ERG Combined Response Amplitude (µV) | ERG Cone Flicker Amplitude (µV) | V4e Total Field Area (deg²) | V4e Field Equivalent Diameter (deg) | V4e Field Description | Gene |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 003-292 | F | 42 | 20/20 | 1 | 158 | 38 | 12,793 | 128 | ring scotoma to I4e OU | PDE6B |
| 2 | 043-045 | F | 63 | 20/31 | 0.65 | 50 | 2 | 12,447 * | 126 * | pericentral scotoma | |
| 3 | 043-009 | M | 48 | 20/30 | 0.67 | 291 | 26 | NA | NA | pericentral scotoma to V4e | |
| 4 | 043-010 | F | 50 | 20/20 | 1 | 271 | 43 | 12,119 | 124 | pericentral scotoma | RHO |
| 5 | 043-011 | M | 29 | 20/27 | 0.74 | 114 | 44 | NA | NA | constricted w mid-peripheral scotoma | PDE6B |
| 6 | 043-012 | F | 67 | 20/20 | 1 | 32 | 6 | 4082 | 72 | constricted with temporal crescents | CNGA1 |
| 7 | 043-013 | F | 58 | 20/25 | 0.8 | 341 | 53 | NA | NA | full V4e OU; mid-peripheral scotoma I4e OU | RHO |
| 8 | 043-014 | M | 41 * | 20/31 | 0.65 | 136 | 34 | 9136 | 108 | mid-peripheral scotoma | RHO |
| 9 | 043-015 | M | 67 | 20/27 | 0.75 | 144 | 18 | NA | NA | constricted OD; NA OS | |
| 10 | 043-053 | M | 51 | 20/20 | 1 | 153 | 23 | NA | NA | pericentral scotoma | |
| 11 | 043-054 | M | 58 | 20/20 | 1 | 117 | 47 | 14,430 | 136 | full V4e, pericentral scotoma I4e | |
| 12 | 043-019 | F | 57 | 20/37 | 0.54 | 139 | 13 | 8368 | 103 | pericentral scotoma | USH2A |
| 13 | 231-023 | M | 35 | 20/20 | 1 | 180 | 40 | 13,083 | 129 | full to V4e, pericentral scotoma to I4e | HGSNAT? |
| 14 | 038-159 | F | 50 * | 20/20 | 1 | 249 | 56 | 12,321 | 125 | mid-peripheral scotoma | RHO |
| 15 | 043-034 | F | 55 | 20/33 | 0.6 | 251 | 33 | 15,041 | 138 | full to V4e, pericentral scotoma to I4e | HGSNAT |
| 16 | 043-027 | M | 47 | 20/20 | 1 | 71 | 18 | 10,563 | 116 | full v4e, constricted w pericentral scotoma I4e | |
| 17 | 043-032 | F | 53 | 20/24 | 0.84 | 298 | 58 | 14,375 | 135 | pericentral scotoma | NR2E3 |
| 18 | 043-043 | M | 46 | 20/20 | 1 | 182 | 27 | 14,867 | 138 | full V4e, pericentral scotoma I4e | PRPF31 |
| 19 | 038-134 | M | 30 | 20/25 | 0.8 | 159 | 68 | 12,126 | 124 | pericentral & mid-peripheral scotomas | |
| 20 | 043-002 | M | 26 | 20/133 | 0.15 | 88 | 9 | 8064 | 101 | constricted w peripheral islands | |
| 21 | 043-003 | F | 78 | 20/54 | 0.37 | 229 | 26 | 108 | 12 | constricted OU | |
| 22 | 043-005 | M | 82 | 20/59 | 0.34 | 213 | 13 | 4868 * | 79 * | pericentral scotoma | |
| 23 | 043-006 | M | 50 | 20/22 | 0.9 | 106 | 33 | NA | NA | constricted w peripheral islands | RHO |
| 24 | 043-007 | M | 55 | 20/22 | 0.9 | 140 | 28 | 4700 * | 77 * | constricted w scotoma OD; ring scotoma OS | HGSNAT? |
| 25 | 043-008 | F | 83 | 20/71 | 0.28 | 59 | 2 | NA | NA | constricted | |
| 26 | 043-016 | F | 57 * | 20/25 | 0.8 | 201 | 32 | 13,612 | 132 | ring scotoma | USH2A |
| 27 | 043-017 | F | 61 | 20/118 | 0.17 | 155 | 26 | 9673 * | 111 * | constricted with ring scotoma V4e OU | |
| 28 | 043-018 | F | 42 | 20/27 | 0.74 | 249 | 40 | 11,187 | 119 | ring scotoma | |
| 29 | 043-048 | F | 62 | 20/22 | 0.9 | 215 | 51 | 9392 | 109 | mid-peripheral scotoma V4e, ring scotoma I4e | |
| 30 | 043-049 | F | 46 | 20/20 | 1 | 125 | 46 | 12,594 * | 127 * | full V4e, pericentral scotoma I4e | USH2A |
| 31 | 043-056 | F | 49 | 20/20 | 1 | 172 | 19 | 13,369 | 130 | paracentral nasal field loss | RP2 carrier |
| 32 | 043-057 | F | 45 | 20/31 | 0.65 | 167 | 26 | 13,296 | 130 | pericentral scotoma | |
| 33 | 043-058 | F | 62 | 20/34 | 0.59 | 155 | 47 | 14,262 | 135 | constricted with ring scotoma OS, islands OD I4e | TULP1 |
| 34 | 038-162 | M | 63 | 20/333 | 0.06 | 134 | 14 | 9015 | 107 | central and pericentral scotomas | HGSNAT |
| 35 | 043-059 | F | 40 | 20/50 | 0.4 | 222 | 37 | 13,678 | 132 | fairly full V4e; pericentral scotomas I4e OU | |
| 36 | 043-060 | M | 51 | 20/20 | 1 | 192 | 35 | 10,008 | 113 | ring scotoma to V4e OU | |
| 37 | 043-061 | M | 49 | 20/20 | 1 | 186 | 45 | 12,850 | 128 | pericentral scotoma | USH2A |
| 38 | 043-062 | M | 44 | 20/20 | 1 | 138 | 34 | 13,327 | 130 | pericentral scotoma | |
| 39 | 043-063 | F | 55 | 20/25 | 0.8 | 236 | 43 | 13,546 | 131 | pericentral scotoma | |
| 40 | 043-067 | M | 26 | 20/20 | 1 | 86 | 34 | 8902 | 106 | pericentral loss OU | CNGB1 |
| 41 | 043-068 | M | 71 | 20/20 | 1 | 116 | 10 | 13,466 | 121 | bitemporal near mid-peripheral loss OU | |
| 42 | 043-069 | F | 70 | 20/30 | 0.67 | 106 | 27 | 8551 | 104 | pericentral field loss OU | |
| 43 | 043-055 | M | 49 | 20/25 | 0.8 | 99 | 23 | 12,462 | 126 | pericentral field loss OU |
| Solved by Panel-Based Sequencing | |||||
|---|---|---|---|---|---|
| Family# | Gene | Protein Variant | DNA Variant Description | Type | Sanger/Correct Segregation |
| 1 | PDE6B | p.(Cys458Tyr) | NM_000283.3:c.1373G>A | HET | proband |
| 1 | PDE6B | p.(Lys518Ile) | NM_000283.3:c.1553A>T | HET | proband |
| 4 | RHO | p.(Gly101Val) | NM_000539.3:c.302G>T | HET | |
| 5 | PDE6B | p.(Gln298*) | NM_000283.3:c.892C>T | HET | proband |
| 5 | PDE6B | p.(Arg100His) | NM_000283.3:c.299G>A | HET | proband |
| 6 | CNGA1 | p.(Thr586Serfs*17) | NM_000087.3:c.1755 _1758delAACT | HET | proband |
| 6 | CNGA1 | p.(Ser320Phe) | NM_000087.3:c.959C>T | HET | proband |
| 7 | RHO | p.(Gly18Asp) | NM_000539.3:c.53G>A | HET | proband |
| 8 | RHO | p.(Thr58Arg) | NM_000539.3:c.173C>G | HET | proband |
| 12 | USH2A | p.(Glu3448Lys) | NM_206933.2:c.10342G>A | HOM | proband, het in unaffected son |
| 14 | RHO | p.(Gly106Arg) | NM_000539.3:c.316G>A | HET | proband |
| 17 | NR2E3 | p.(Val118Met) | NM_014249.3:c.352G>A | HET | proband, affected sister |
| 18 | PRPF31 | p.? | NM_015629.3:c.421-1G>A | HET | proband, 3 affected children |
| 23 | RHO | p.(Gly51Arg) | NM_000539.3:c.151G>C | HET | proband |
| 26 | USH2A | p.(Cys870*) | NM_206933.2:c.2610C>A | HET | proband |
| 26 | USH2A | p.(Asn42Lys) | NM_206933.2:c.126C>G | HET | proband |
| 26 | USH2A | p.(Gly2313Cys) | NM_206933.2:c.6937G>T | HET | proband |
| 30 | USH2A | p.? | NM_206933.2:c.9571-2A>G | HET | proband |
| 30 | USH2A | p.? | NM_206933.2:c.7595-3C>G | HET | proband |
| 31 | RP2 carrier | p.(Gly163Glu) | NM_006915.2:c.488G>A | HET | proband |
| 33 | TULP1 | p.? | NM_003322.3:c.1496-6C>A | HET | proband |
| 33 | TULP1 | p.(Gln163*) | NM_003322.3:c.487C>T | HET | proband |
| 37 | USH2A | p.? | NM_206933.2:c.12067-2A>G | HET | proband |
| 37 | USH2A | p.(Cys3306Trp) | NM_206933.2:c.9918T>G | HET | proband |
| 37 | USH2A | p.(Ala1953Gly) | NM_206933.2:c.5858C>G | HET | proband |
| 40 | CNGB1 | p.(Arg396Gln) | NM_001297.4:c.1187G>A | HET | |
| 40 | CNGB1 | p.? | NM_001297.4:c.1801+5G>A | HET | |
| Solved by Exome Sequencing | |||||
| 15 | HGSNAT | p.(Ala615Thr) | NM_152419.2:c.1843G>A | HET | |
| 15 | HGSNAT | p.? | NG_009552.1(NM_152419.2):c.1464 + 1G>A | HET | |
| 34 | HGSNAT | p.(Ser318Asn) | NM_152419.2:c.953G>A | HOM | |
| Not Solved or Partially Solved Due to Variants of Uncertain Significance (VUS) | |||||
| 3 | ROM1 | p.(Leu238Cysfs*78) | NM_000327.3:c.708delC | HET | |
| 3 | COL11A1 | p.(Arg762Gln) | NM_001854.3:c.2285G>A | HET | |
| 13 | HGSNAT | p.(Ala615Thr) | NM_152419.2:c.1843G>A | HOM | |
| 16 | USH2A | p.(Leu1378Pro) | NM_206933.2:c.4133T>C | HET | proband, affected brother |
| 16 | USH2A | p.(Ser1369Leu) | NM_206933.2:c.4106C>T | HET | proband, affected brother |
| 20 | TRPM1 | p.(Gln1161His) | NM_002420.5:c.3483G>C | HET | |
| 20 | TRPM1 | p.(Ser157Phe) | NM_002420.5:c.470C>T | HET | |
| 24 | HGSNAT | p.(Ala615Thr) | NM_152419.2:c.1843G>A | HOM | |
| 25 | MKS1 | p.(Thr423Ile) | NM_017777.3:c.1268C>T | HOM | |
| 29 | BBS9 | p.(Pro419Ala) | NM_198428.2:c.1255C>G | HET | proband |
| 29 | BBS9 | p.(Glu753Val) | NM_198428.2:c.2258A>T | HET | proband |
| 42 | OPA1 | p.(Ala115Val) | NM_015560.2: c.344C>T | HET | |
| Gene | This Study | Matsui et al. | Grondahl et al. and Selmer et al. | Total |
|---|---|---|---|---|
| RHO | 5 | 1 | 3 | 9 |
| USH2A | 4 | 4 | ||
| HGSNAT | 2+ | 2 | ||
| PDE6B | 2 | 2 | ||
| CNGA1 | 1 | 1 | ||
| CNGB1 | 1 | 1 | ||
| NR2E3 | 1 | 1 | 2 | |
| PRPF31 | 1 | 1 | ||
| RP2 carrier | 1 | 1 | ||
| TULP1 | 1 | 1 | ||
| ABCA4 | 5 | 5 | ||
| CERKL | 3 | 3 | ||
| CRX | 1 | 1 | ||
| DHDDS | 1 | 1 | ||
| PROM1 | 1 | 1 | ||
| PRPH2 | 1 | 1 | ||
| TOPORS | 1 | 1 | ||
| Total | 22 | 14 | 4 | 37 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Comander, J.; Weigel-DiFranco, C.; Maher, M.; Place, E.; Wan, A.; Harper, S.; Sandberg, M.A.; Navarro-Gomez, D.; Pierce, E.A. The Genetic Basis of Pericentral Retinitis Pigmentosa—A Form of Mild Retinitis Pigmentosa. Genes 2017, 8, 256. https://doi.org/10.3390/genes8100256
Comander J, Weigel-DiFranco C, Maher M, Place E, Wan A, Harper S, Sandberg MA, Navarro-Gomez D, Pierce EA. The Genetic Basis of Pericentral Retinitis Pigmentosa—A Form of Mild Retinitis Pigmentosa. Genes. 2017; 8(10):256. https://doi.org/10.3390/genes8100256
Chicago/Turabian StyleComander, Jason, Carol Weigel-DiFranco, Matthew Maher, Emily Place, Aliete Wan, Shyana Harper, Michael A. Sandberg, Daniel Navarro-Gomez, and Eric A. Pierce. 2017. "The Genetic Basis of Pericentral Retinitis Pigmentosa—A Form of Mild Retinitis Pigmentosa" Genes 8, no. 10: 256. https://doi.org/10.3390/genes8100256
APA StyleComander, J., Weigel-DiFranco, C., Maher, M., Place, E., Wan, A., Harper, S., Sandberg, M. A., Navarro-Gomez, D., & Pierce, E. A. (2017). The Genetic Basis of Pericentral Retinitis Pigmentosa—A Form of Mild Retinitis Pigmentosa. Genes, 8(10), 256. https://doi.org/10.3390/genes8100256