Optimized mtDNA Control Region Primer Extension Capture Analysis for Forensically Relevant Samples and Highly Compromised mtDNA of Different Age and Origin



Abstract
1. Introduction
2. Materials and Methods
2.1. Samples and DNA Extraction and Quantification
2.2. Control Sample Types and Preparation
2.3. Library Preparation
2.4. PEC Primer Design
2.5. Primer Extension Capture Workflow and Reaction Details
- The reaction was stopped by adding 10 µL of 0.5 M EDTA to inhibit further polymerase activity as the reaction is cooling down.
- The MinElute purification step was excluded.
- Thirty microliters (30 µL) of Dynabeads MyOne Streptavidin C1 were used in 60 µL 2× BW buffer.
- Washing steps were modified to 3 × 200 µL of 1× BW Buffer at room temperature (RT); 2× 200 µL of 2× SCC (+0.1% SDS, 0.01% Tween 20) at RT; and 1× 2× SCC (+0.1% SDS, 0.01% Tween 20) at 65 °C for 2 min while shaking mildly.
- Elution was done in 25 µL.
- After amplification, the reaction was cleaned up using Ampure XP beads (Agencourt).
2.6. Massively Parallel Sequencing
2.7. Data Analysis
2.8. Sanger-Type Sequencing
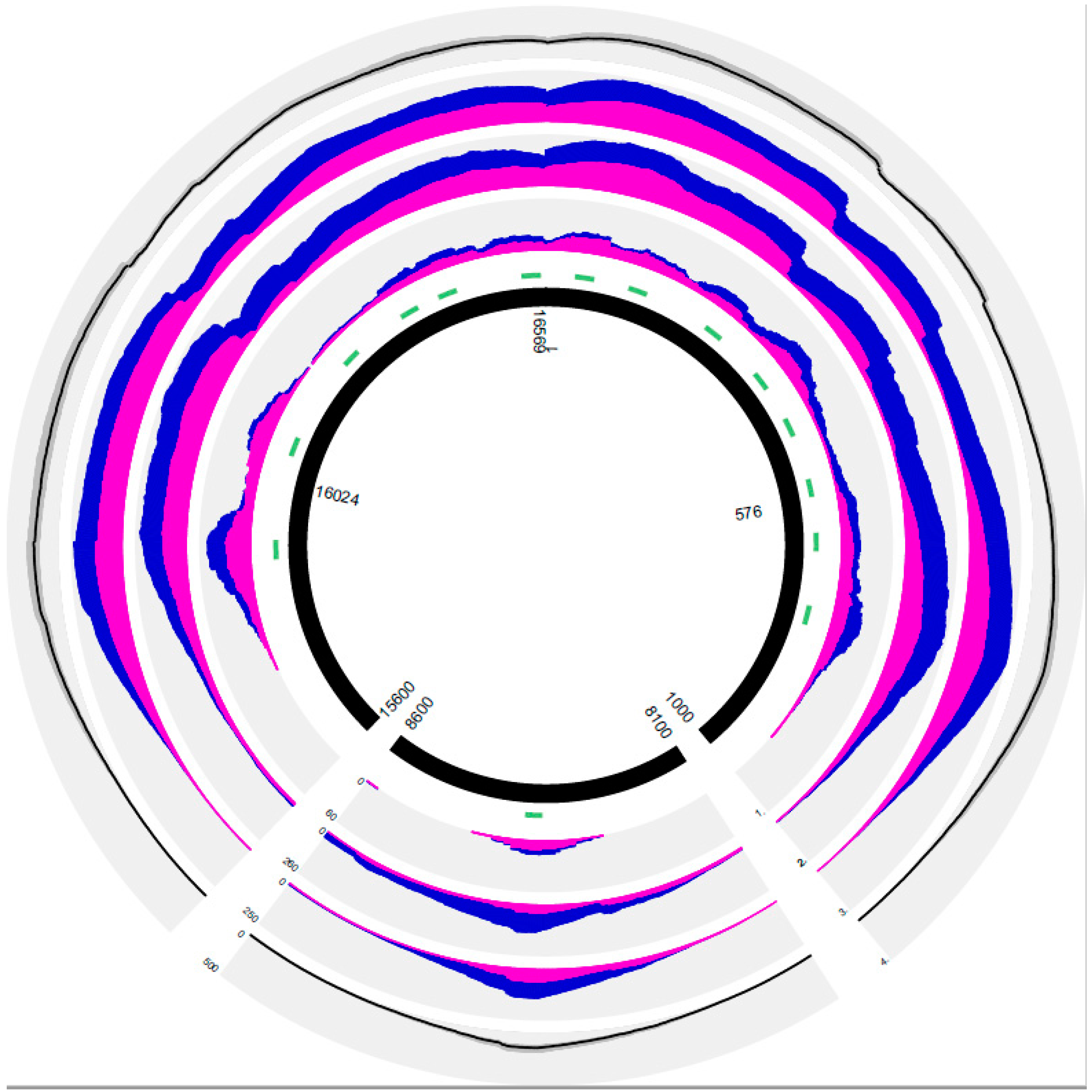
3. Results
3.1. Characterization of the PEC Method Based on the Results of Positive and Negative Controls Subsection
3.2. General Results of PEC MPS in the Forensic and Solid Tissue Samples
3.3. PEC MPS Results of the Forensic Samples
3.4. PEC MPS Results of the Solid Tissue Samples
4. Discussion
4.1. Positive Controls and PEC Analyses
4.2. Regime and Results of Negative Controls
- (i)
- extraction blank (EX0) that came with each batch of DNA extracted samples,
- (ii)
- no template control (NTC) that included E. coli to increase the complexity of background DNA,
- (iii)
- no primer control (NPC), and
- (iv)
- no template library preparation control (NTC-LP) consisting of a sheared E. coli DNA template for monitoring correct adapter annealing.
4.3. Selection Criteria for and Results of Samples Included in This Study
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Parson, W.; Parsons, T.J.; Scheithauer, R.; Holland, M.M. Population data for 101 austrian caucasian mitochondrial DNA d-loop sequences: Application of mtdna sequence analysis to a forensic case. Int. J. Leg. Med. 1998, 111, 124–132. [Google Scholar] [CrossRef]
- Zietkiewicz, E.; Witt, M.; Daca, P.; Zebracka-Gala, J.; Goniewicz, M.; Jarzab, B.; Witt, M. Current genetic methodologies in the identification of disaster victims and in forensic analysis. J. Appl. Genet. 2012, 53, 41–60. [Google Scholar] [CrossRef] [PubMed]
- Coble, M.D.; Loreille, O.M.; Wadhams, M.J.; Edson, S.M.; Maynard, K.; Meyer, C.E.; Niederstatter, H.; Berger, C.; Berger, B.; Falsetti, A.B.; et al. Mystery solved: The identification of the two missing romanov children using DNA analysis. PLoS ONE 2009, 4, e4838. [Google Scholar] [CrossRef] [PubMed]
- King, T.E.; Fortes, G.G.; Balaresque, P.; Thomas, M.G.; Balding, D.; Delser, P.M.; Neumann, R.; Parson, W.; Knapp, M.; Walsh, S.; et al. Identification of the remains of king richard III. Nat. Commun. 2014, 5, 5631. [Google Scholar] [CrossRef] [PubMed]
- Melton, T.; Dimick, G.; Higgins, B.; Lindstrom, L.; Nelson, K. Forensic mitochondrial DNA analysis of 691 casework hairs. J. Forensic Sci. 2005, 50, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.R.; DiZinno, J.A.; Polanskey, D.; Replogle, J.; Budowle, B. Validation of mitochondrial DNA sequencing for forensic casework analysis. Int. J. Leg. Med. 1995, 108, 68–74. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Salas, A.; Lutz-Bonengel, S. Artificial recombination in forensic mtdna population databases. Int. J. Leg. Med. 2004, 118, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Brandstatter, A.; Peterson, C.T.; Irwin, J.A.; Mpoke, S.; Koech, D.K.; Parson, W.; Parsons, T.J. Mitochondrial DNA control region sequences from Nairobi (Kenya): Inferring phylogenetic parameters for the establishment of a forensic database. Int. J. Leg. Med. 2004, 118, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Parson, W.; Gusmao, L.; Hares, D.R.; Irwin, J.A.; Mayr, W.R.; Morling, N.; Pokorak, E.; Prinz, M.; Salas, A.; Schneider, P.M.; et al. DNA commission of the international society for forensic genetics: Revised and extended guidelines for mitochondrial DNA typing. Forensic Sci. Int. Genet. 2014, 13, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Berger, C.; Parson, W. Mini-midi-mito: Adapting the amplification and sequencing strategy of mtdna to the degradation state of crime scene samples. Forensic Sci. Int. Genet. 2009, 3, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Eichmann, C.; Parson, W. ‘Mitominis’: Multiplex pcr analysis of reduced size amplicons for compound sequence analysis of the entire mtDNA control region in highly degraded samples. Int. J. Leg. Med. 2008, 122, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, M.N.; Huffine, E.F.; Ryan, J.H.; Holland, M.M.; Parsons, T.J. Improved mtdna sequence analysis of forensic remains using a “mini-primer set” amplification strategy. J. Forensic Sci. 2001, 46, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Chaitanya, L.; Ralf, A.; van Oven, M.; Kupiec, T.; Chang, J.; Lagace, R.; Kayser, M. Simultaneous whole mitochondrial genome sequencing with short overlapping amplicons suitable for degraded DNA using the ion torrent personal genome machine. Hum. Mutat. 2015, 36, 1236–1247. [Google Scholar] [CrossRef] [PubMed]
- Parson, W.; Huber, G.; Moreno, L.; Madel, M.B.; Brandhagen, M.D.; Nagl, S.; Xavier, C.; Eduardoff, M.; Callaghan, T.C.; Irwin, J.A. Massively parallel sequencing of complete mitochondrial genomes from hair shaft samples. Forensic Sci. Int. Genet. 2015, 15, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Hofreiter, M.; Serre, D.; Poinar, H.N.; Kuch, M.; Paabo, S. Ancient DNA. Nat. Rev. Genet. 2001, 2, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Knapp, M.; Hofreiter, M. Next generation sequencing of ancient DNA: Requirements, strategies and perspectives. Genes 2010, 1, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.T.; Tomsho, L.P.; Rendulic, S.; Packard, M.; Drautz, D.I.; Sher, A.; Tikhonov, A.; Dalen, L.; Kuznetsova, T.; Kosintsev, P.; et al. Whole-genome shotgun sequencing of mitochondria from ancient hair shafts. Science 2007, 317, 1927–1930. [Google Scholar] [CrossRef] [PubMed]
- Hofreiter, M.; Paijmans, J.L.; Goodchild, H.; Speller, C.F.; Barlow, A.; Fortes, G.G.; Thomas, J.A.; Ludwig, A.; Collins, M.J. The future of ancient DNA: Technical advances and conceptual shifts. BioEssays 2015, 37, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Dabney, J.; Knapp, M.; Glocke, I.; Gansauge, M.T.; Weihmann, A.; Nickel, B.; Valdiosera, C.; Garcia, N.; Paabo, S.; Arsuaga, J.L.; et al. Complete mitochondrial genome sequence of a middle pleistocene cave bear reconstructed from ultrashort DNA fragments. Proc. Natl. Acad. Sci. USA 2013, 110, 15758–15763. [Google Scholar] [CrossRef] [PubMed]
- Krause, J.; Fu, Q.; Good, J.M.; Viola, B.; Shunkov, M.V.; Derevianko, A.P.; Paabo, S. The complete mitochondrial DNA genome of an unknown hominin from southern Siberia. Nature 2010, 464, 894–897. [Google Scholar] [CrossRef] [PubMed]
- Maricic, T.; Whitten, M.; Paabo, S. Multiplexed DNA sequence capture of mitochondrial genomes using pcr products. PLoS ONE 2010, 5, e14004. [Google Scholar] [CrossRef] [PubMed]
- Templeton, J.E.; Brotherton, P.M.; Llamas, B.; Soubrier, J.; Haak, W.; Cooper, A.; Austin, J.J. DNA capture and next-generation sequencing can recover whole mitochondrial genomes from highly degraded samples for human identification. Investig. Genet. 2013, 4, 26. [Google Scholar] [CrossRef] [PubMed]
- Enk, J.; Rouillard, J.M.; Poinar, H. Quantitative PCR as a predictor of aligned ancient DNA read counts following targeted enrichment. BioTechniques 2013, 55, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Kihana, M.; Mizuno, F.; Sawafuji, R.; Wang, L.; Ueda, S. Emulsion pcr-coupled target enrichment: An effective fishing method for high-throughput sequencing of poorly preserved ancient DNA. Gene 2013, 528, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Briggs, A.W.; Good, J.M.; Green, R.E.; Krause, J.; Maricic, T.; Stenzel, U.; Paabo, S. Primer extension capture: Targeted sequence retrieval from heavily degraded DNA sources. J. Vis. Exp. 2009, 31, 1573. [Google Scholar] [CrossRef] [PubMed]
- Loreille, O.; Koshinsky, H.; Fofanov, V.Y.; Irwin, J.A. Application of next generation sequencing technologies to the identification of highly degraded unknown soldiers’ remains. Forensic Sci. Int. Suppl. Ser. 2011, 3, e540–e541. [Google Scholar] [CrossRef]
- Bauer, C.M.; Niederstatter, H.; McGlynn, G.; Stadler, H.; Parson, W. Comparison of morphological and molecular genetic sex-typing on mediaeval human skeletal remains. Forensic Sci. Int. Genet. 2013, 7, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Niederstatter, H.; Kochl, S.; Grubwieser, P.; Pavlic, M.; Steinlechner, M.; Parson, W. A modular real-time pcr concept for determining the quantity and quality of human nuclear and mitochondrial DNA. Forensic Sci. Int. Genet. 2007, 1, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Bauer, C.M.; Bodner, M.; Niederstatter, H.; Niederwieser, D.; Huber, G.; Hatzer-Grubwieser, P.; Holubar, K.; Parson, W. Molecular genetic investigations on austria’s patron saint leopold iii. Forensic Sci. Int. Genet. 2013, 7, 313–315. [Google Scholar] [CrossRef] [PubMed]
- Koressaar, T.; Remm, M. Enhancements and modifications of primer design program primer3. Bioinformatics 2007, 23, 1289–1291. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.M.; Kubacka, I.; Chinnery, P.F.; Lightowlers, R.N.; Turnbull, D.M.; Howell, N. Reanalysis and revision of the cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 1999, 23, 147. [Google Scholar] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Group, E.D.W. DNA Database Management Review and Recommendations. Available online: http://enfsi.eu/wp-content/uploads/2016/09/final_version_enfsi_2016_document_on_dna-database_management_0.pdf (accessed on 20 September 2017).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and samtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis toolkit: A mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Ginolhac, A.; Rasmussen, M.; Gilbert, M.T.; Willerslev, E.; Orlando, L. Mapdamage: Testing for damage patterns in ancient DNA sequences. Bioinformatics 2011, 27, 2153–2155. [Google Scholar] [CrossRef] [PubMed]
- Van Oven, M.; Kayser, M. Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum. Mutat. 2009, 30, E386–E394. [Google Scholar] [CrossRef] [PubMed]
- Rock, A.W.; Dur, A.; van Oven, M.; Parson, W. Concept for estimating mitochondrial DNA haplogroups using a maximum likelihood approach (EMMA). Forensic Sci. Int. Genet. 2013, 7, 601–609. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2008; ISBN 3-900051-07-0. Available online: http://www.R-project.org (accessed on 20 September 2017).
- Eduardoff, M.; Santos, C.; de la Puente, M.; Gross, T.E.; Fondevila, M.; Strobl, C.; Sobrino, B.; Ballard, D.; Schneider, P.M.; Carracedo, A.; et al. Inter-laboratory evaluation of SNP-based forensic identification by massively parallel sequencing using the ion PGM. Forensic Sci. Int. Genet. 2015, 17, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Der Sarkissian, C.; Ermini, L.; Jonsson, H.; Alekseev, A.N.; Crubezy, E.; Shapiro, B.; Orlando, L. Shotgun microbial profiling of fossil remains. Mol. Ecol. 2014, 23, 1780–1798. [Google Scholar] [CrossRef] [PubMed]
- Poinar, H.N.; Schwarz, C.; Qi, J.; Shapiro, B.; Macphee, R.D.; Buigues, B.; Tikhonov, A.; Huson, D.H.; Tomsho, L.P.; Auch, A.; et al. Metagenomics to paleogenomics: Large-scale sequencing of mammoth DNA. Science 2006, 311, 392–394. [Google Scholar] [CrossRef] [PubMed]
- Parson, W.; Dur, A. Empop—A forensic mtdna database. Forensic Sci. Int. Genet. 2007, 1, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Butler, J.M.; Gusmao, L.; Linacre, A.; Parson, W.; Roewer, L.; Schneider, P.M. Update of the guidelines for the publication of genetic population data. Forensic Sci. Int. Genet. 2014, 10, A1–A2. [Google Scholar] [CrossRef] [PubMed]
- Casas-Vargas, A.; Gomez, A.; Briceno, I.; Diaz-Matallana, M.; Bernal, J.E.; Rodriguez, J.V. High genetic diversity on a sample of pre-columbian bone remains from guane territories in northwestern Colombia. Am. J. Phys. Anthropol. 2011, 146, 637–649. [Google Scholar] [CrossRef] [PubMed]
- Briggs, A.W.; Stenzel, U.; Johnson, P.L.; Green, R.E.; Kelso, J.; Prufer, K.; Meyer, M.; Krause, J.; Ronan, M.T.; Lachmann, M.; et al. Patterns of damage in genomic DNA sequences from a neandertal. Proc. Natl. Acad. Sci. USA 2007, 104, 14616–14621. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.T.; Willerslev, E.; Hansen, A.J.; Barnes, I.; Rudbeck, L.; Lynnerup, N.; Cooper, A. Distribution patterns of postmortem damage in human mitochondrial DNA. Am. J. Hum. Genet. 2003, 72, 32–47. [Google Scholar] [CrossRef] [PubMed]
- Picard. Available online: http://broadinstitute.github.io/picard/ (accessed on 20 September 2017).
- Green, R.E.; Briggs, A.W.; Krause, J.; Prufer, K.; Burbano, H.A.; Siebauer, M.; Lachmann, M.; Paabo, S. The Neandertal genome and ancient DNA authenticity. EMBO J. 2009, 28, 2494–2502. [Google Scholar] [CrossRef] [PubMed]
- Krause, J.; Briggs, A.W.; Kircher, M.; Maricic, T.; Zwyns, N.; Derevianko, A.; Paabo, S. A complete mtDNA genome of an early modern human from Kostenki, Russia. Curr. Biol. 2010, 20, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.D.; Watanabe, C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B. Aligning short sequencing reads with Bowtie. Curr. Protoc. Bioinform. 2010. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Troyanskaya, O.G.; Arbell, O.; Koren, Y.; Landau, G.M.; Bolshoy, A. Sequence complexity profiles of prokaryotic genomic sequences: a fast algorithm for calculating linguistic complexity. Bioinformatics 2002, 18, 679–688. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Sample | mtGE/µL | Volume | Analysis Method | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ID | No. | TYPE | AGE | TISSUE | EXTR. METHOD | 143 bp | PEC | PEC-CR | MT | STS-CR |
| Positive controls | ||||||||||
| PC 1-6 | 1–6 | Control | recent | blood | Qiagen Blood Maxi | ~1,700,000 | 1 µL in 25 µL H2O | full | full | full |
| Forensic samples | ||||||||||
| Hair 1 | 7 | Forensic | recent | hair | QIAamp DNA Investigator | 972 | 27 | full | - | full |
| Hair 2 | 8 | Forensic | recent | hair | QIAamp DNA Investigator | 5 | 42 | full | - | - |
| Ancient solid tissue samples | ||||||||||
| g52 | 9 | aDNA | ~1 kyrs | molar | PCI | 60 | 30 | full | - | partial |
| g27 | 10 | aDNA | ~1 kyrs | molar | SF | NA | 25 | full | - | full |
| g43 | 11 | aDNA | ~1 kyrs | femur | SF | 0 | 30 | full | - | - |
| g121 | 12 | aDNA | ~1 kyrs | femur | PCl | 0 | 30 | partial | - | - |
| g17 | 13 | aDNA | ~1 kyrs | costa | PCI | 0 | 30 | partial | - | - |
| g81 | 14 | aDNA | ~1 kyrs | tubular bone | PCI | 0 | 28 | fail | - | - |
| g7 | 15 | aDNA | ~1 kyrs | humerus | PCI | 0 | 28 | fail | - | - |
| DT08 | 16 | aDNA | ~2 kyrs | anterior tooth | SF | 149 | 25 | full | partial | - |
| DT11-1 | 17 | aDNA | ~2 kyrs | femur | SF | 216 | 28 | full | fail | - |
| F98E-1 | 18 | aDNA | ~8 kyrs | anterior tooth | SF | 11,722 | 28 | partial | fail | - |
| F98E-4 | 19 | aDNA | ~8 kyrs | fragment anterior tooth | SF | 2005 | 30 | partial | - | - |
| F98F-2 | 20 | aDNA | ~8 kyrs | molar | SF | 736 | 30 | partial | - | - |
| F98A-2 | 21 | aDNA | ~8 kyrs | skull fragment | SF | 1633 | 30 | fail | - | - |
| F98B-1 | 22 | aDNA | ~8 kyrs | skull fragment | SF | 2272 | 30 | fail | - | - |
| F98C-2 | 23 | aDNA | ~8 kyrs | skull fragment | SF | 12,543 | 30 | fail | - | - |
| Sample Number | Total Reads | % mtDNA Reads of Total Reads | % Unique mtDNA Reads/Total Reads | No. of unique mtDNA reads | % CR Reads/Total mtDNA Reads | % Human Genome Aligned Reads/Total Reads | % Unaligned Reads/Total Reads | Mean Strand Bias | % Coverage of Whole Mitogenome (min 2×) | % CR Coverage (min 2×) | Minimal CR Coverage | Mean CR Coverage | Mean SD of CR Coverage | Maximal CR Coverage | Minimal RL | Mean RL | SD of Mean RL | Maximal RL | Mean RL of Aligned Reads | Mean RL Other Reads | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Positive controls | |||||||||||||||||||||
| PC 1 | 1 | 102,534 | 3.11 | 1.57 | 1605 | 67.66 | 57.55 | 39.34 | 0.43 | 35.74 | 100.00 | 57 | 110.33 | 27.13 | 171 | 34 | 148.20 | 51.76 | 308 | 134.64 | 56.89 |
| PC 2 | 2 | 233,078 | 3.50 | 1.07 | 2495 | 66.33 | 56.08 | 40.42 | 0.40 | 48.96 | 100.00 | 90 | 176.01 | 41.71 | 249 | 30 | 154.69 | 54.66 | 326 | 133.98 | 51.94 |
| PC 3 | 3 | 842,114 | 2.40 | 0.40 | 3346 | 61.78 | 38.31 | 59.29 | 0.39 | 70.90 | 100.00 | 112 | 215.50 | 54.49 | 301 | 30 | 150.84 | 53.56 | 331 | 123.71 | 32.74 |
| PC 4 | 4 | 125,099 | 3.22 | 1.42 | 1777 | 66.18 | 54.03 | 42.75 | 0.40 | 34.16 | 100.00 | 60 | 114.85 | 25.00 | 168 | 33 | 135.68 | 43.69 | 311 | 122.34 | 50.64 |
| PC 5 | 5 | 197,285 | 2.89 | 1.11 | 2191 | 66.77 | 58.97 | 38.14 | 0.40 | 48.30 | 100.00 | 81 | 150.50 | 37.15 | 228 | 30 | 146.81 | 51.57 | 330 | 129.78 | 54.29 |
| PC 6 | 6 | 348,593 | 3.87 | 0.85 | 2957 | 62.09 | 63.09 | 33.04 | 0.39 | 60.70 | 100.00 | 108 | 191.57 | 43.81 | 275 | 31 | 146.26 | 47.11 | 316 | 123.06 | 64.72 |
| mean | 308,117.2 | 3.16 (0.4) | 1.07 | 2395.17 | 65.14 | 54.67 (7.83) | 42.16 (8.21) | 0.40 (0.01) | 49.79 | 100.00 | 84.67 | 159.79 (42.26) | 38.22 | 232.00 | 31.33 | 147.08 (6.39) | 50.39 | 320.33 | 127.92 | 51.87 | |
| Forensic samples | |||||||||||||||||||||
| Hair 1 | 7 | 256,877 | 88.84 | 1.39 | 3565 | 49.14 | 2.05 | 9.11 | 0.36 | 78.77 | 100.00 | 108 | 183.62 | 39.05 | 258 | 30 | 139.82 | 58.87 | 333 | 114.94 | 39.28 |
| Hair 2 | 8 | 346,147 | 65.51 | 0.24 | 819 | 44.93 | 5.58 | 28.91 | 0.42 | 54.13 | 100.00 | 2 | 26.85 | 10.28 | 49 | 32 | 112.60 | 38.72 | 241 | 119.60 | 43.63 |
| Ancient solid tissue samples | |||||||||||||||||||||
| 1 kyrs | |||||||||||||||||||||
| g52 | 9 | 7230 | 14.18 | 8.40 | 607 | 78.25 | 44.04 | 41.78 | 0.15 | 10.40 | 100.00 | 17 | 46.16 | 17.93 | 98 | 31 | 138.57 | 44.52 | 288 | 130.90 | 58.46 |
| g27 | 10 | 829,116 | 0.38 | 0.09 | 749 | 60.88 | 54.77 | 44.85 | 0.42 | 35.95 | 100.00 | 21 | 45.29 | 14.97 | 91 | 35 | 138.76 | 49.88 | 309 | 136.47 | 80.81 |
| g43 | 11 | 72,886 | 1.38 | 0.84 | 609 | 57.96 | 36.64 | 61.98 | 0.46 | 31.36 | 100.00 | 12 | 33.51 | 11.17 | 61 | 33 | 142.22 | 53.96 | 325 | 141.97 | 115.87 |
| g121 | 12 | 88,249 | 0.06 | 0.04 | 37 | 48.65 | 1.00 | 98.95 | 0.53 | 6.73 | 39.13 | 1 | 1.95 | 0.96 | 4 | 70 | 136.30 | 37.11 | 283 | 131.08 | 121.58 |
| g17 | 13 | 185,894 | 0.03 | 0.02 | 44 | 68.18 | 0.33 | 99.64 | 0.24 | 10.17 | 94.56 | 1 | 3.08 | 1.48 | 7 | 32 | 141.91 | 58.50 | 279 | 112.27 | 119.91 |
| g81 | 14 | 317,785 | 0.00 | 0.00 | 8 | 0.00 | 3.50 | 96.50 | 0.00 | 0.00 | 0.36 | 0 | 0.00 | 0.00 | 0 | 48 | 188.38 | 91.77 | 290 | 95.64 | 111.02 |
| g7 | 15 | 317,487 | 1.33 | 0.03 | 90 | 51.11 | 2.30 | 96.37 | 0.47 | 8.47 | 63.19 | 1 | 4.35 | 2.76 | 10 | 52 | 132.80 | 43.99 | 279 | 133.24 | 100.04 |
| 2 kyrs | |||||||||||||||||||||
| DT08 | 16 | 540,060 | 3.41 | 0.56 | 3048 | 56.53 | 51.42 | 45.18 | 0.39 | 71.80 | 100.00 | 59 | 171.61 | 47.51 | 253 | 31 | 142.85 | 53.37 | 311 | 130.19 | 114.19 |
| DT11-1 | 17 | 201,917 | 16.09 | 0.91 | 1829 | 53.31 | 45.88 | 38.03 | 0.40 | 68.45 | 100.00 | 37 | 96.37 | 27.00 | 145 | 31 | 142.08 | 56.34 | 315 | 138.45 | 94.66 |
| 8 kyrs | |||||||||||||||||||||
| F98E-1 | 18 | 879,757 | 0.06 | 0.01 | 69 | 55.07 | 0.03 | 99.91 | 0.52 | 8.05 | 76.29 | 1 | 2.99 | 1.60 | 8 | 37 | 96.45 | 40.30 | 342 | 104.01 | 142.54 |
| F98E-4 | 19 | 532,948 | 0.14 | 0.04 | 206 | 52.91 | 0.11 | 99.76 | 0.49 | 10.52 | 96.43 | 1 | 8.10 | 4.22 | 18 | 38 | 100.65 | 34.30 | 244 | 94.57 | 124.41 |
| F98F-2 | 20 | 534,508 | 0.84 | 0.06 | 318 | 61.32 | 0.90 | 98.26 | 0.47 | 12.36 | 99.20 | 1 | 14.42 | 6.67 | 32 | 37 | 102.75 | 37.63 | 246 | 108.52 | 128.40 |
| F98A-2 | 21 | 73 | 0.00 | 0.00 | 0 | 0.00 | 0.00 | 100.00 | 0.00 | 0.00 | 0.36 | 0 | 0.00 | 0.00 | 0 | 0 | 0.00 | 0.00 | 0 | NaN | 173.97 |
| F98B-1 | 22 | 451 | 0.22 | 0.22 | 1 | 100.00 | 0.00 | 99.78 | 1.00 | 0.00 | 0.00 | 1 | 1.00 | 0.00 | 1 | 98 | 98.00 | NA | 98 | NaN | 125.79 |
| F98C-2 | 23 | 910 | 1.21 | 0.33 | 3 | 100.00 | 0.00 | 98.79 | 0.38 | 0.88 | 12.92 | 2 | 2.70 | 0.46 | 3 | 101 | 130.33 | 25.40 | 145 | NaN | 95.68 |
| Negtive Controls | |||||||||||||||||||||
| NPC | 4146 | 0.12 | 0.12 | 5 | 20.00 | 75.01 | 24.87 | 0.00 | 0.40 | 0.00 | 1 | 1.00 | 0.00 | 1 | 76 | 152.60 | 54.00 | 226 | 151.06 | 77.85 | |
| NTC-LP | 48,396 | 0.00 | 0.00 | 0 | 0.00 | 8.41 | 91.59 | 0.00 | 0.00 | 0.36 | 0 | 0.00 | 0.00 | 0 | 0 | 0.00 | 0.00 | 0 | 162.85 | 40.23 | |
| NTC-LP | 37,769 | 0.00 | 0.00 | 1 | 100.00 | 18.35 | 81.65 | 1.00 | 0.00 | 0.00 | 1 | 1.00 | 0.00 | 1 | 135 | 135.00 | NA | 135 | 142.74 | 27.95 | |
| NTC_LP | 81,476 | 0.01 | 0.00 | 2 | 50.00 | 9.99 | 90.01 | 1.00 | 0.00 | 0.00 | 1 | 1.00 | 0.00 | 1 | 66 | 98.50 | 45.96 | 131 | 134.78 | 26.94 | |
| EX0 | 31,886 | 0.00 | 0.00 | 0 | 0.00 | 24.81 | 75.19 | 0.00 | 0.00 | 0.36 | 0 | 0.00 | 0.00 | 0 | 0 | 0.00 | 0.00 | 0 | 126.05 | 60.15 | |
| EX0 | 158,827 | 0.00 | 0.00 | 1 | 0.00 | 1.35 | 98.65 | 0.00 | 0.00 | 0.36 | 0 | 0.00 | 0.00 | 0 | 70 | 70.00 | NA | 70 | 127.15 | 42.96 | |
| mean | 60,417 | 0.02 | 0.02 | 2 | 28.33 | 22.99 | 76.99 | 0.33 | 0.07 | 0.18 | 0.50 | 0.50 | 0.00 | 0.50 | 57.83 | ||||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eduardoff, M.; Xavier, C.; Strobl, C.; Casas-Vargas, A.; Parson, W. Optimized mtDNA Control Region Primer Extension Capture Analysis for Forensically Relevant Samples and Highly Compromised mtDNA of Different Age and Origin. Genes 2017, 8, 237. https://doi.org/10.3390/genes8100237
Eduardoff M, Xavier C, Strobl C, Casas-Vargas A, Parson W. Optimized mtDNA Control Region Primer Extension Capture Analysis for Forensically Relevant Samples and Highly Compromised mtDNA of Different Age and Origin. Genes. 2017; 8(10):237. https://doi.org/10.3390/genes8100237
Chicago/Turabian StyleEduardoff, Mayra, Catarina Xavier, Christina Strobl, Andrea Casas-Vargas, and Walther Parson. 2017. "Optimized mtDNA Control Region Primer Extension Capture Analysis for Forensically Relevant Samples and Highly Compromised mtDNA of Different Age and Origin" Genes 8, no. 10: 237. https://doi.org/10.3390/genes8100237
APA StyleEduardoff, M., Xavier, C., Strobl, C., Casas-Vargas, A., & Parson, W. (2017). Optimized mtDNA Control Region Primer Extension Capture Analysis for Forensically Relevant Samples and Highly Compromised mtDNA of Different Age and Origin. Genes, 8(10), 237. https://doi.org/10.3390/genes8100237