
Translesion Synthesis: Insights into the Selection and Switching of DNA Polymerases

Abstract
:1. Introduction
2. Selection and Switching of Specialized DNA Polymerases
2.1. Selection of the “Right” TLS Pol for Benzo[a]pyrene-Derived DNA Lesions: A Case Study
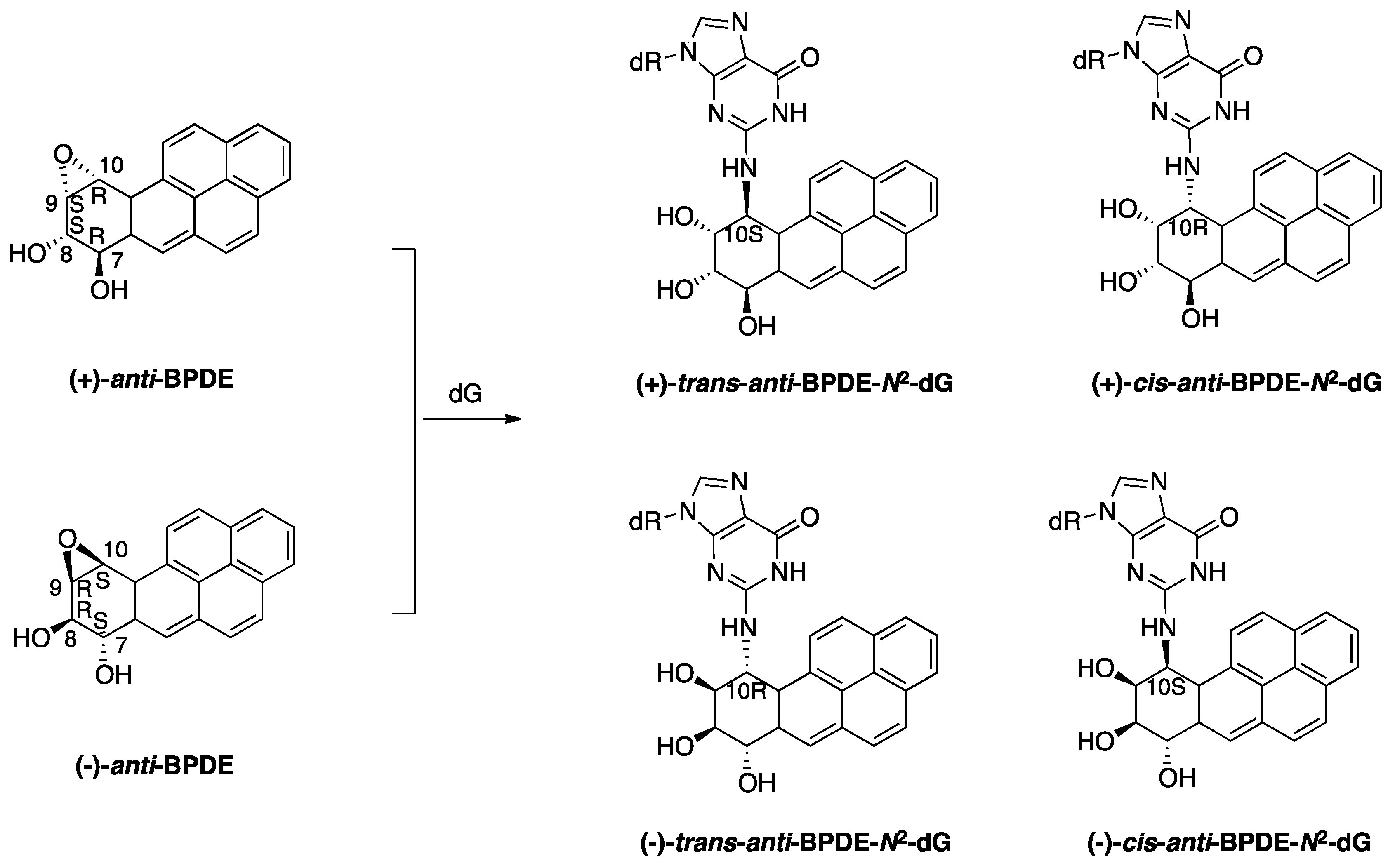
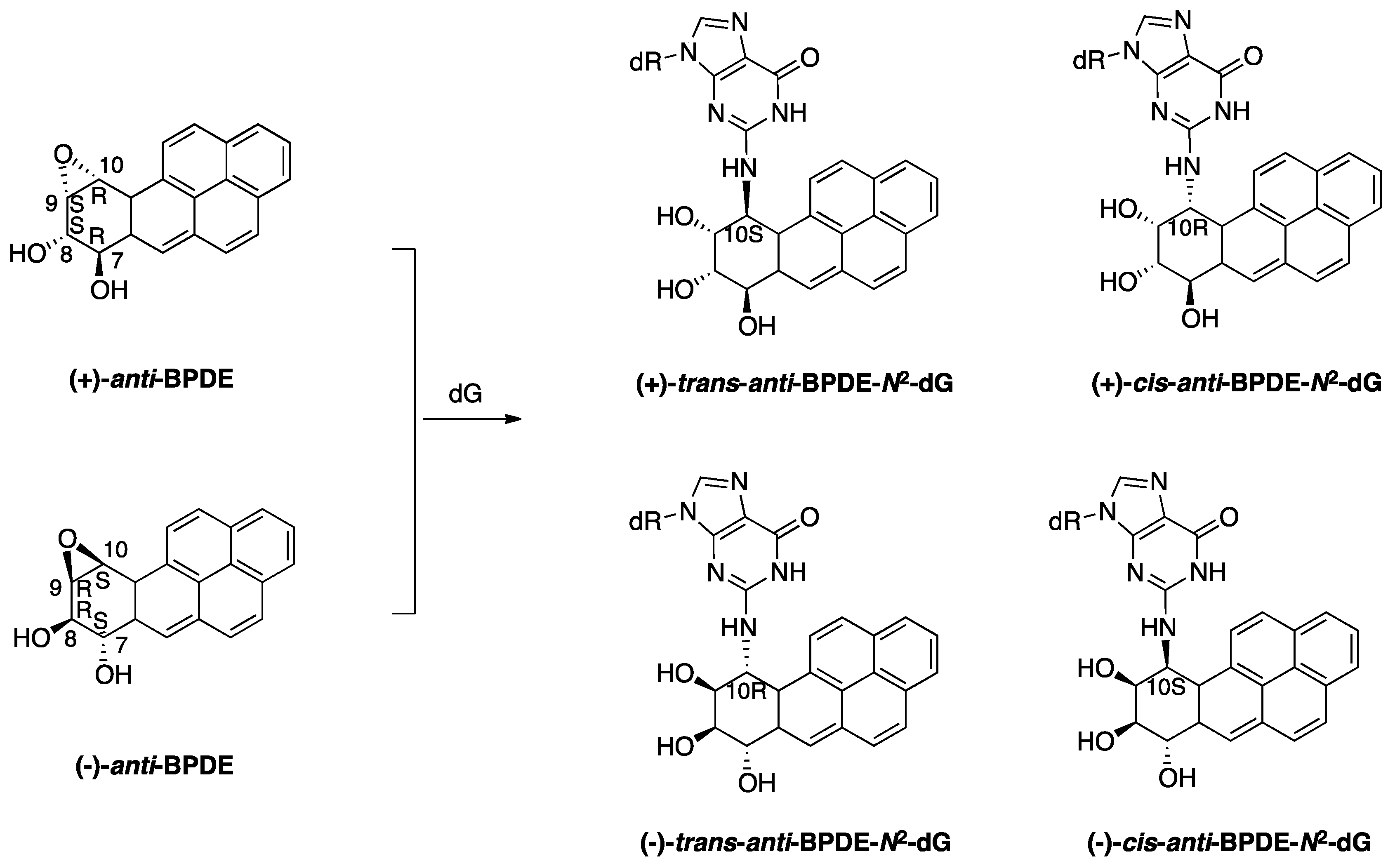
2.1.1. BaP-Induced DNA Damage
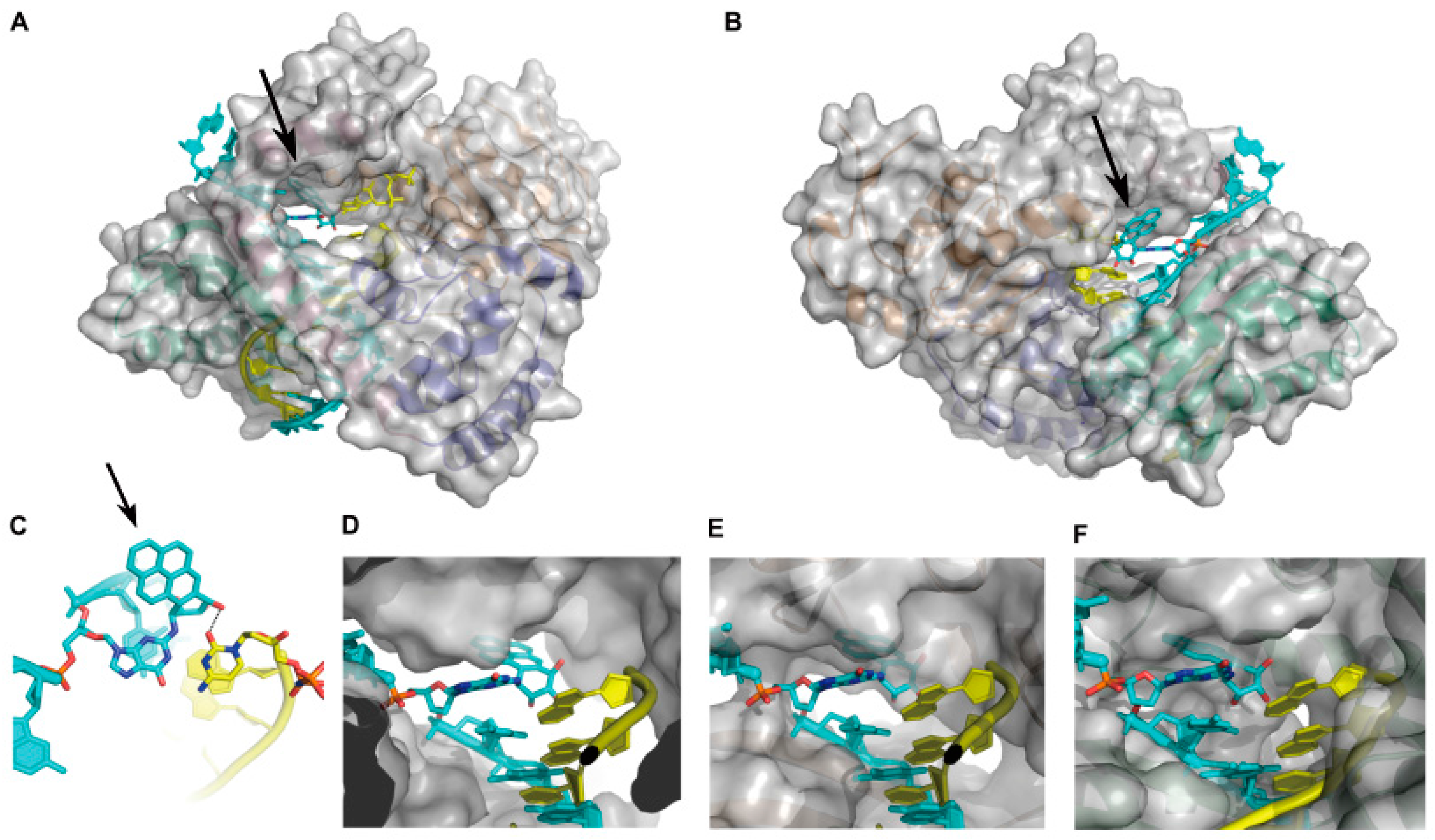
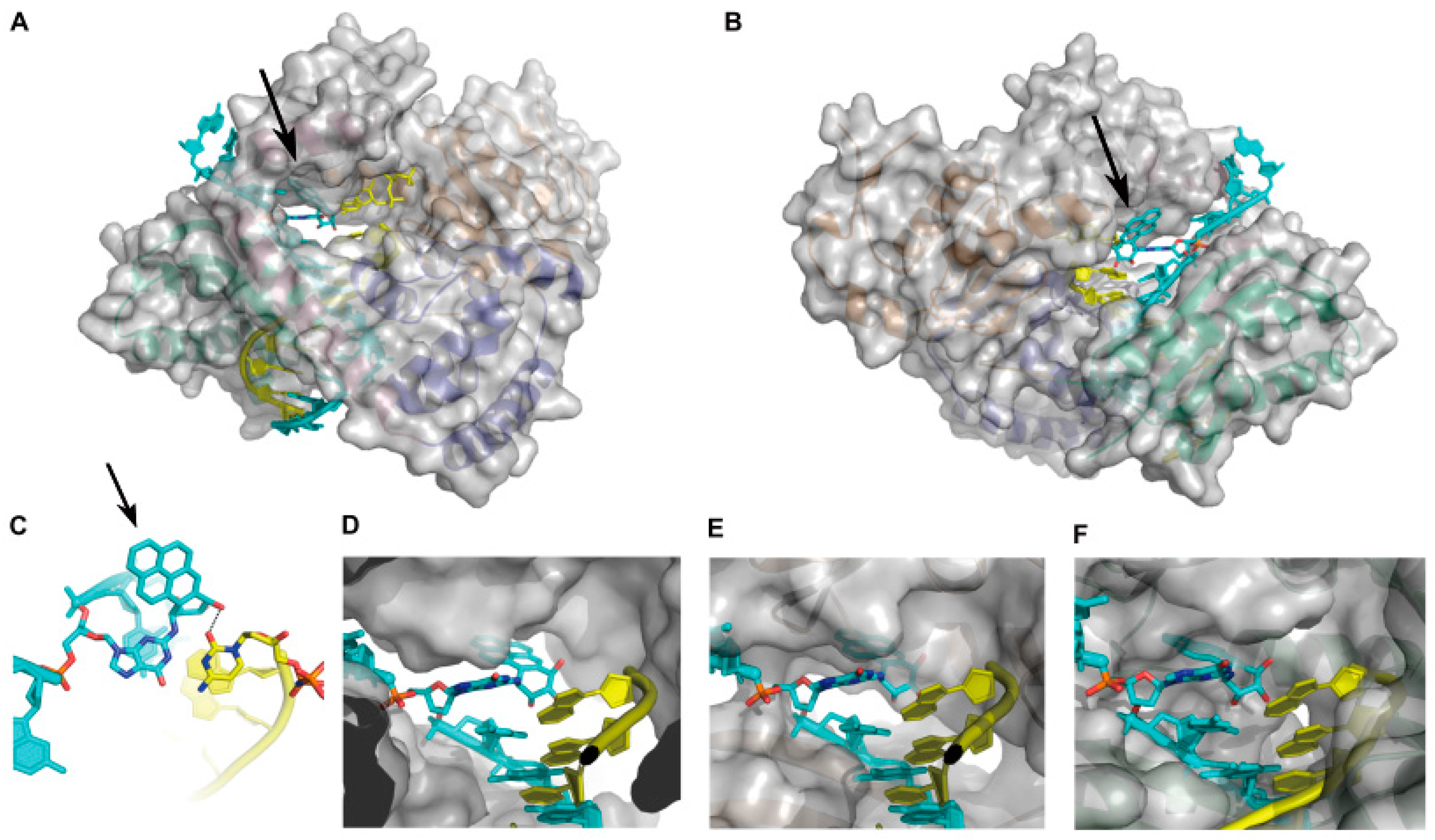
2.1.2. Accurate Bypass of BaP-Derived DNA Lesions
2.1.3. Error-Prone Bypass of BaP-Derived DNA Lesions
2.2. PCNA: An Interaction Hub for Many Partners
2.2.1. Interactions between PCNA and DNA Polymerases
2.2.2. Biochemical Basis of PCNA-Pol Interactions
2.2.3. Ubiquitination of PCNA
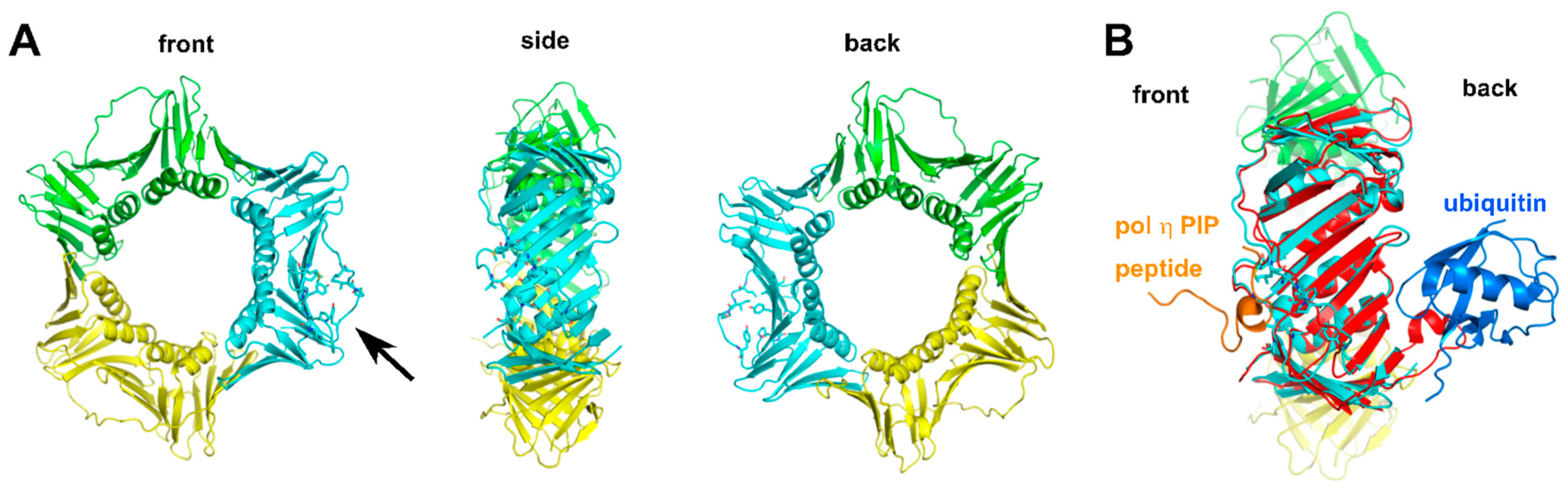
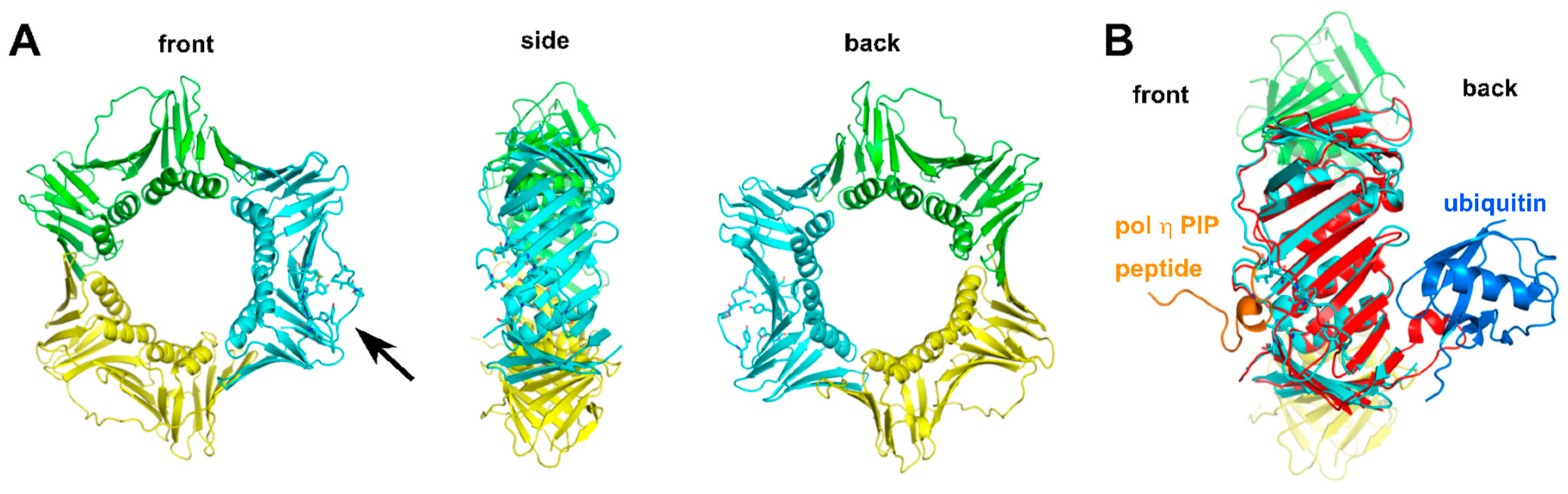
2.2.4. Structure of Monoubiquitinated PCNA
2.2.5. Additional Structural Motifs for Stabilizing PCNA-Pol Complexes
2.3. Rev1: A Scaffold Protein
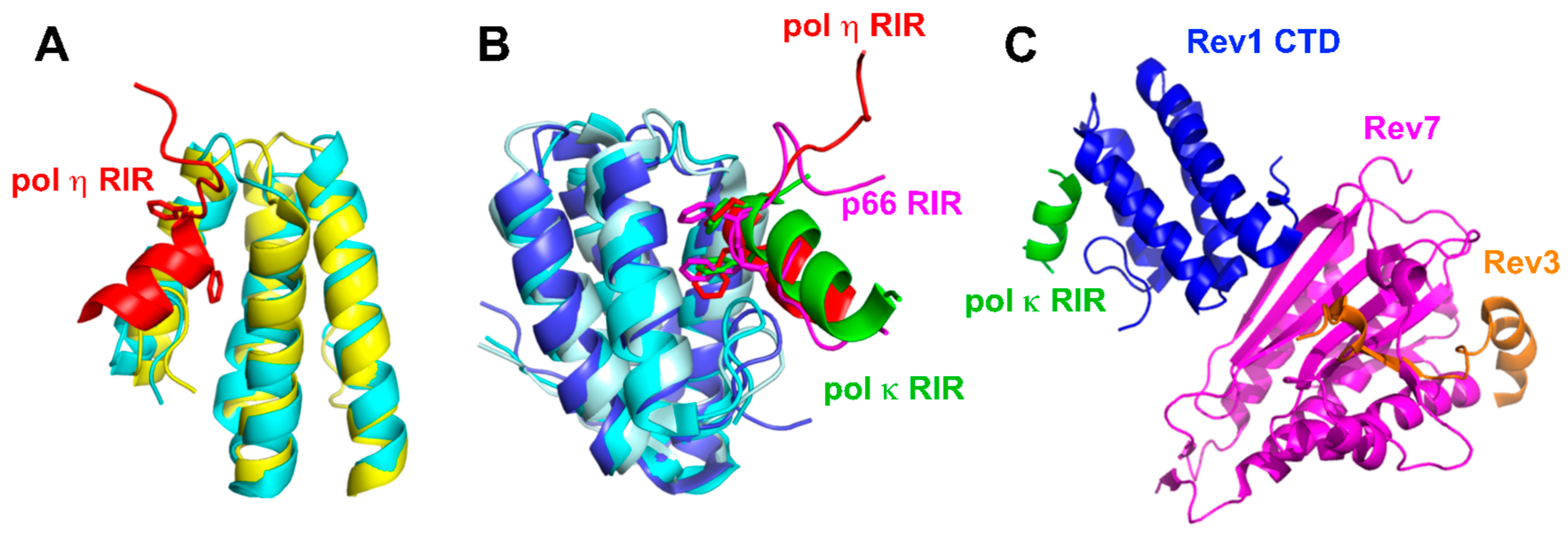
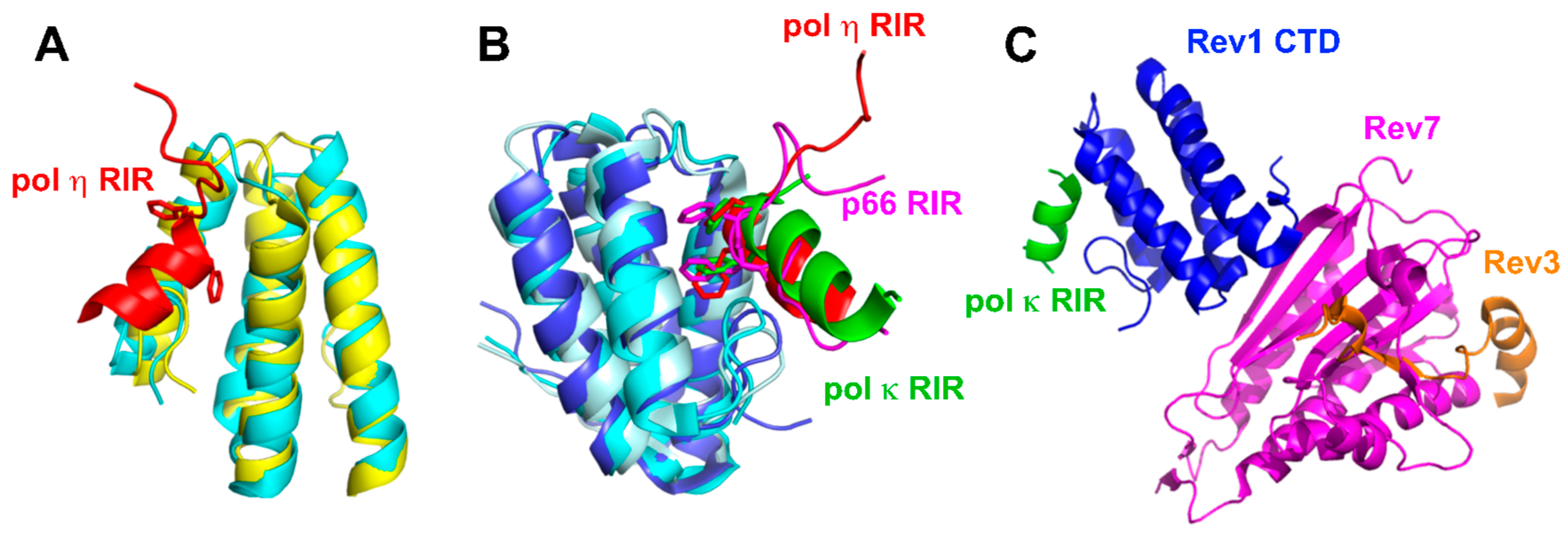
2.3.1. Interactions between Rev1 and Other Pols
2.3.2. Interactions between Rev1 and Pol ζ
2.3.3. Coordination of Multiple Binding Partners by Rev1
2.3.4. PCNA Tool Belts and Rev1 Bridges
2.3.5. Physiological Functions of Rev1-Mediated Protein Interactions
2.4. Subunits Sharing between Pol δ and Pol ζ
2.4.1. The Subunit Organization of Pol ζ
2.4.2. Switching between Pol δ and Pol ζ
2.5. Proteasomal Degradation of DNA Polymerases
2.5.1. Regulation of the Steady-State Levels of TLS Pols
2.5.2. Proteasomal Degradation of TLS Pols
2.5.3. Protein Degradation Creates Binding Sites for TLS Pols
2.5.4. Proteasomal Degradation of Pol δ
3. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hoeijmakers, J.H.J. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Shinoura, Y. Isolation and characterization of mutants of Escherichia coli deficient in induction of mutations by ultraviolet light. Mol. Gen. Genet. 1977, 156, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Steinborn, G. Uvm mutants of Escherichia coli K 12 deficient in UV mutagenesis. Mol. Gen. Genet. 1978, 165, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Lemontt, J.F. Mutants of yeast defective in mutation induced by ultraviolet light. Genetics 1971, 68, 21–33. [Google Scholar] [PubMed]
- Lawrence, C.W.; Christensen, R. UV mutagenesis in radiation-sensitive strains of yeast. Genetics 1976, 82, 207–232. [Google Scholar] [PubMed]
- Lehmann, A.R.; Kirk-Bell, S.; Arlett, C.F.; Paterson, M.C.; Lohman, P.H.; de Weerd-Kastelein, E.A.; Bootsma, D. Xeroderma pigmentosum cells with normal levels of excision repair have a defect in DNA synthesis after UV-irradiation. Proc. Natl. Acad. Sci. USA 1975, 72, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.R.; Lawrence, C.W.; Hinkle, D.C. Deoxycytidyl transferase activity of yeast REV1 protein. Nature 1996, 382, 729–731. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.R.; Lawrence, C.W.; Hinkle, D.C. Thymine-thymine dimer bypass by yeast DNA polymerase zeta. Science 1996, 272, 1646–1649. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.E.; Prakash, S.; Prakash, L. Efficient bypass of a thymine-thymine dimer by yeast DNA polymerase, Polη. Science 1999, 283, 1001–1004. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.E.; Kondratick, C.M.; Prakash, S.; Prakash, L. hRAD30 mutations in the variant form of xeroderma pigmentosum. Science 1999, 285, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Masutani, C.; Kusumoto, R.; Yamada, A.; Dohmae, N.; Yokoi, M.; Yuasa, M.; Araki, M.; Iwai, S.; Takio, K.; Hanaoka, F. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase η. Nature 1999, 399, 700–704. [Google Scholar] [PubMed]
- Wagner, J.; Gruz, P.; Kim, S.; Yamada, M.; Matsui, K.; Fuchs, R.P.P.; Nohmi, T. The dinB gene encodes a novel E. coli DNA polymerase, DNA pol IV, involved in mutagenesis. Mol. Cell 1999, 4, 281–286. [Google Scholar] [CrossRef]
- Reuven, N.B.; Arad, G.; Maor-Shoshani, A.; Livneh, Z. The mutagenesis protein UmuC is a DNA polymerase activated by UmuD′, RecA, and SSB and is specialized for translesion replication. J. Biol. Chem. 1999, 274, 31763–31766. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Shen, X.; Frank, E.G.; O’Donnell, M.; Woodgate, R.; Goodman, M.F. UmuD′ 2C is an error-prone DNA polymerase, Escherichia coli pol V. Proc. Natl. Acad. Sci. USA 1999, 96, 8919–8924. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.P.; Levine, A.S.; Woodgate, R. The Saccharomyces cerevisiae RAD30 gene, a homologue of Escherichia coli dinB and umuC, is DNA damage inducible and functions in a novel error-free postreplication repair mechanism. Genetics 1997, 147, 1557–1568. [Google Scholar] [PubMed]
- Tissier, A.; McDonald, J.P.; Frank, E.G.; Woodgate, R. Pol ι, a remarkably error-prone human DNA polymerase. Genes Dev. 2000, 14, 1642–1650. [Google Scholar] [PubMed]
- Johnson, R.E.; Washington, M.T.; Haracska, L.; Prakash, S.; Prakash, L. Eukaryotic polymerases ι and ζ act sequentially to bypass DNA lesions. Nature 2000, 406, 1015–1019. [Google Scholar] [PubMed]
- Zhang, Y.; Yuan, F.; Wu, X.; Wang, Z. Preferential incorporation of G opposite template T by the low-fidelity human DNA polymerase ι. Mol. Cell. Biol. 2000, 20, 7099–7108. [Google Scholar] [CrossRef] [PubMed]
- Ogi, T.; Kato, T.; Kato, T.; Ohmori, H. Mutation enhancement by DINB1, a mammalian homologue of the Escherichia coli mutagenesis protein DinB. Genes Cells 1999, 4, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, V.L.; Aravind, L.; Gotway, G.; Schultz, R.A.; Koonin, E.V.; Friedberg, E.C. Human and mouse homologs of Escherichia coli DinB (DNA polymerase IV), members of the UmuC/DinB superfamily. Proc. Natl. Acad. Sci. USA 1999, 96, 11922–11927. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, E.; Ogi, T.; Kusumoto, R.; Iwai, S.; Masutani, C.; Hanaoka, F.; Ohmori, H. Error-prone bypass of certain DNA lesions by the human DNA polymerase κ. Genes Dev. 2000, 14, 1589–1594. [Google Scholar] [PubMed]
- Zhang, Y.; Yuan, F.; Wu, X.; Wang, M.; Rechkoblit, O.; Taylor, J.-S.; Geacintov, N.E.; Wang, Z. Error-free and error-prone lesion bypass by human DNA polymerase κ in vitro. Nucleic Acids Res. 2000, 28, 4138–4146. [Google Scholar] [CrossRef] [PubMed]
- Woodgate, R. A plethora of lesion-replicating DNA polymerases. Genes Dev. 1999, 13, 2191–2195. [Google Scholar] [CrossRef] [PubMed]
- Hübscher, U.; Maga, G.; Spadari, S. Eukaryotic DNA polymerases. Annu. Rev. Biochem. 2002, 71, 133–163. [Google Scholar] [CrossRef] [PubMed]
- Lange, S.S.; Takata, K.; Wood, R.D. DNA polymerases and cancer. Nat. Rev. Cancer 2011, 11, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Rudd, S.G.; Bianchi, J.; Doherty, A.J. PrimPol—A new polymerase on the block. Mol. Cell Oncol. 2014, 1, e960754. [Google Scholar] [CrossRef] [PubMed]
- Sale, J.E. Translesion DNA synthesis and mutagenesis in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012708. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, J.; Rudd, S.G.; Jozwiakowski, S.K.; Bailey, L.J.; Soura, V.; Taylor, E.; Stevanovic, I.; Green, A.J.; Stracker, T.H.; Lindsay, H.D. PrimPol bypasses UV photoproducts during eukaryotic chromosomal DNA replication. Mol. Cell 2013, 52, 566–573. [Google Scholar] [CrossRef] [PubMed]
- García-Gómez, S.; Reyes, A.; Martínez-Jiménez, M.I.; Chocrón, E.S.; Mourón, S.; Terrados, G.; Powell, C.; Salido, E.; Méndez, J.; Holt, I.J. PrimPol, an archaic primase/polymerase operating in human cells. Mol. Cell 2013, 52, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Lou, J.; Xia, Y.; Su, B.; Liu, T.; Cui, J.; Sun, Y.; Lou, H.; Huang, J. hPrimpol1/CCDC111 is a human DNA primase-polymerase required for the maintenance of genome integrity. EMBO Rep. 2013, 14, 1104–1112. [Google Scholar] [CrossRef] [PubMed]
- Zafar, M.K.; Ketkar, A.; Lodeiro, M.F.; Cameron, C.E.; Eoff, R.L. Kinetic analysis of human PrimPol DNA polymerase activity reveals a generally error-prone enzyme capable of accurately bypassing 7,8-dihydro-8-oxo-2′-deoxyguanosine. Biochemistry 2014, 53, 6584–6594. [Google Scholar] [CrossRef] [PubMed]
- Mourón, S.; Rodriguez-Acebes, S.; Martínez-Jiménez, M.I.; García-Gómez, S.; Chocrón, S.; Blanco, L.; Méndez, J. Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nat. Struct. Mol. Biol. 2013, 20, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Jiménez, M.I.; García-Gómez, S.; Bebenek, K.; Sastre-Moreno, G.; Calvo, P.A.; Díaz-Talavera, A.; Kunkel, T.A.; Blanco, L. Alternative solutions and new scenarios for translesion DNA synthesis by human PrimPol. DNA Repair 2015, 29, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Schiavone, D.; Jozwiakowski, S.K.; Romanello, M.; Guilbaud, G.; Guilliam, T.A.; Bailey, L.J.; Sale, J.E.; Doherty, A.J. PrimPol is required for replicative tolerance of G quadruplexes in vertebrate cells. Mol. Cell 2016, 61, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Guilliam, T.A.; Tsuda, M.; Yamamoto, J.; Bailey, L.J.; Iwai, S.; Takeda, S.; Doherty, A.J.; Hirota, K. Repriming by PrimPol is critical for DNA replication restart downstream of lesions and chain-terminating nucleosides. Cell Cycle 2016, 15, 1997–2008. [Google Scholar] [CrossRef] [PubMed]
- Jansen, J.G.; Tsaalbi-Shtylik, A.; de Wind, N. Roles of mutagenic translesion synthesis in mammalian genome stability, health and disease. DNA Repair 2015, 29, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Wickramasinghe, C.M.; Arzouk, H.; Frey, A.; Maiter, A.; Sale, J.E. Contributions of the specialised DNA polymerases to replication of structured DNA. DNA Repair 2015, 29, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Sale, J.E.; Lehmann, A.R.; Woodgate, R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol. 2012, 13, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Prakash, S.; Prakash, L. Translesion DNA synthesis in eukaryotes: A one-or two-polymerase affair. Genes Dev. 2002, 16, 1872–1883. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, A.R.; Niimi, A.; Ogi, T.; Brown, S.; Sabbioneda, S.; Wing, J.F.; Kannouche, P.L.; Green, C.M. Translesion synthesis: Y-family polymerases and the polymerase switch. DNA Repair 2007, 6, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, E.C.; Lehmann, A.R.; Fuchs, R.P.P. Trading places: How do DNA polymerases switch during translesion DNA synthesis? Mol. Cell 2005, 18, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Sutton, M.D. Coordinating DNA polymerase traffic during high and low fidelity synthesis. BAA Protein Proteom. 2010, 1804, 1167–1179. [Google Scholar] [CrossRef] [PubMed]
- Korzhnev, D.M.; Hadden, M.K. Targeting the translesion synthesis pathway for the development of anti-cancer chemotherapeutics. J. Med. Chem. 2016, 59, 9321–9336. [Google Scholar] [CrossRef] [PubMed]
- Prakash, S.; Johnson, R.E.; Prakash, L. Eukaryotic translesion synthesis DNA polymerases: Specificity of structure and function. Annu. Rev. Biochem. 2005, 74, 317–353. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Woodgate, R. What a difference a decade makes: Insights into translesion DNA synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 15591–15598. [Google Scholar] [CrossRef] [PubMed]
- Washington, M.T.; Carlson, K.D.; Freudenthal, B.D.; Pryor, J.M. Variations on a theme: Eukaryotic Y-family DNA polymerases. BAA Protein Proteom. 2010, 1804, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Goodman, M.F.; Woodgate, R. Translesion DNA polymerases. Cold Spring Harb. Perspect. Biol. 2013, 5, a010363. [Google Scholar] [CrossRef] [PubMed]
- Hübscher, U.; Maga, G. DNA replication and repair bypass machines. Curr. Opin. Chem. Biol. 2011, 15, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Einolf, H.J.; Guengerich, F.P. Fidelity of nucleotide insertion at 8-oxo-7,8-dihydroguanine by mammalian DNA polymerase δ steady-state and pre-steady-state kinetic analysis. J. Biol. Chem. 2001, 276, 3764–3771. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Tsuda, M.; Mohiuddin; Tsurimoto, T.; Cohen, I.S.; Livneh, Z.; Kobayashi, K.; Narita, T.; Nishihara, K.; Murai, J.; et al. In vivo evidence for translesion synthesis by the replicative DNA polymerase δ. Nucleic Acids Res. 2016, 44, 7242–7250. [Google Scholar] [PubMed]
- Shachar, S.; Ziv, O.; Avkin, S.; Adar, S.; Wittschieben, J.; Reißner, T.; Chaney, S.; Friedberg, E.C.; Wang, Z.; Carell, T.; et al. Two-polymerase mechanisms dictate error-free and error-prone translesion DNA synthesis in mammals. EMBO J. 2009, 28, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Waters, L.S.; Minesinger, B.K.; Wiltrout, M.E.; D’Souza, S.; Woodruff, R.V.; Walker, G.C. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol. Mol. Biol. Rev. 2009, 73, 134–154. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Bebenek, K.; Masutani, C.; Hanaoka, F.; Kunkel, T.A. Low fidelity DNA synthesis by human DNA polymerase η. Nature 2000, 404, 1011–1013. [Google Scholar] [PubMed]
- Ogi, T.; Shinkai, Y.; Tanaka, K.; Ohmori, H. Polκ protects mammalian cells against the lethal and mutagenic effects of benzo[a]pyrene. Proc. Natl. Acad. Sci. USA 2002, 99, 15548–15553. [Google Scholar] [CrossRef]
- Denissenko, M.F.; Pao, A.; Tang, M.-S.; Pfeifer, G.P. Preferential formation of benzo[a]pyrene adducts at lung cancer mutational hotspots in p53. Science 1996, 274, 430–432. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, G.P.; Denissenko, M.F.; Olivier, M.; Tretyakova, N.; Hecht, S.S.; Hainaut, P. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene 2002, 21, 7435–7451. [Google Scholar] [CrossRef] [PubMed]
- Slaga, T.J.; Bracken, W.J.; Gleason, G.; Levin, W.; Yagi, H.; Jerina, D.M.; Conney, A.H. Marked differences in the skin tumor-initiating activities of the optical enantiomers of the diastereomeric benzo[a]pyrene 7,8-diol-9,10-epoxides. Cancer Res. 1979, 39, 67–71. [Google Scholar] [PubMed]
- Thakker, D.R.; Yagi, H.; Lu, A.Y.; Levin, W.; Conney, A.H. Metabolism of benzo[a]pyrene: Conversion of (+/−)-trans-7,8-dihydroxy-7,8-dihydrobenzo[a]pyrene to highly mutagenic 7,8-diol-9,10-epoxides. Proc. Natl. Acad. Sci. USA 1976, 73, 3381–3385. [Google Scholar] [CrossRef] [PubMed]
- Kapitulnik, J.; Wislocki, P.G.; Levin, W.; Yagi, H.; Thakker, D.R.; Akagi, H.; Koreeda, M.; Jerina, D.M.; Conney, A.H. Marked differences in the carcinogenic activity of optically pure (+)-and (−)-trans-7,8-dihydroxy-7,8-dihydrobenzo[a]pyrene in newborn mice. Cancer Res. 1978, 38, 2661–2665. [Google Scholar] [PubMed]
- Straub, K.M.; Meehan, T.; Burlingame, A.L.; Calvin, M. Identification of the major adducts formed by reaction of benzo[a]pyrene diol epoxide with DNA in vitro. Proc. Natl. Acad. Sci. USA 1977, 74, 5285–5289. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.C.; Hilton, B.D.; Roman, J.M.; Dipple, A. DNA adducts from carcinogenic and noncarcinogenic enantiomers of benzo[a]pyrenedihydrodiol epoxide. Chem. Res. Toxicol. 1989, 2, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Devanesan, P.D.; RamaKrishna, N.V.S.; Todorovic, R.; Rogan, E.G.; Cavalieri, E.L.; Jeong, H.; Jankowiak, R.; Small, G.J. Identification and quantitation of benzo[a]pyrene-DNA adducts formed by rat liver microsomes in vitro. Chem. Res. Toxicol. 1992, 5, 302–309. [Google Scholar] [CrossRef] [PubMed]
- McCoull, K.D.; Rindgen, D.; Blair, I.A.; Penning, T.M. Synthesis and characterization of polycyclic aromatic hydrocarbon o-quinone depurinating N7-guanine adducts. Chem. Res. Toxicol. 1999, 12, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Balu, N.; Padgett, W.T.; Lambert, G.R.; Swank, A.E.; Richard, A.M.; Nesnow, S. Identification and characterization of novel stable deoxyguanosine and deoxyadenosine adducts of benzo[a]pyrene-7, 8-quinone from reactions at physiological pH. Chem. Res. Toxicol. 2004, 17, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Shukla, R.; Geacintov, N.E.; Loechler, E.L. The major, N2-dG adduct of (+)-anti-B[a]PDE induces G→A mutations in a 5′-AGA-3′ sequence context. Carcinogenesis 1999, 20, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Seo, K.-Y.; Jelinsky, S.A.; Loechler, E.L. Factors that influence the mutagenic patterns of DNA adducts from chemical carcinogens. Mutat. Res. 2000, 463, 215–246. [Google Scholar] [CrossRef]
- Chary, P.; Stone, M.P.; Lloyd, R.S. Sequence context modulation of polycyclic aromatic hydrocarbon-induced mutagenesis. Environ. Mol. Mutagen. 2013, 54, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Menzies, G.E.; Reed, S.H.; Brancale, A.; Lewis, P.D. Base damage, local sequence context and TP53 mutation hotspots: A molecular dynamics study of benzo[a]pyrene induced DNA distortion and mutability. Nucleic Acids Res. 2015, 43, 9133–9146. [Google Scholar] [CrossRef] [PubMed]
- Geacintov, N.E.; Cosman, M.; Hingerty, B.E.; Amin, S.; Broyde, S.; Patel, D.J. NMR solution structures of stereoisomeric covalent polycyclic aromatic carcinogen-DNA adducts: Principles, patterns, and diversity. Chem. Res. Toxicol. 1997, 10, 111–146. [Google Scholar] [CrossRef] [PubMed]
- Moriya, M.; Spiegel, S.; Fernandes, A.; Amin, S.; Liu, T.; Geacintov, N.; Grollman, A.P. Fidelity of translesional synthesis past benzo[a]pyrene diol epoxide-2′-deoxyguanosine DNA adducts: Marked effects of host cell, sequence context, and chirality. Biochemistry 1996, 35, 16646–16651. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, X.; Guo, D.; Rechkoblit, O.; Wang, Z. Activities of human DNA polymerase κ in response to the major benzo[a]pyrene DNA adduct: Error-free lesion bypass and extension synthesis from opposite the lesion. DNA Repair 2002, 1, 559–569. [Google Scholar] [CrossRef]
- Choi, J.-Y.; Angel, K.C.; Guengerich, F.P. Translesion synthesis across bulky N2-alkyl guanine DNA adducts by human DNA polymerase κ. J. Biol. Chem. 2006, 281, 21062–21072. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Ohashi, E.; Kolbanovskiy, A.; Geacintov, N.E.; Grollman, A.P.; Ohmori, H.; Shibutani, S. Translesion synthesis by human DNA polymerase κ on a DNA template containing a single stereoisomer of dG-(+)-or dG-(−)-anti-N2-BPDE (7,8-dihydroxy-anti-9,10-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene). Biochemistry 2002, 41, 6100–6106. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Kolbanovskiy, A.; Wu, X.; Zhang, Y.; Wang, Z.; Zhuang, P.; Amin, S.; Geacintov, N.E. Effects of base sequence context on translesion synthesis past a bulky (+)-trans-anti-B[a]P-N2-dG lesion catalyzed by the Y-family polymerase pol κ. Biochemistry 2003, 42, 2456–2466. [Google Scholar] [CrossRef] [PubMed]
- Avkin, S.; Goldsmith, M.; Velasco-Miguel, S.; Geacintov, N.; Friedberg, E.C.; Livneh, Z. Quantitative analysis of translesion DNA synthesis across a benzo[a]pyrene-guanine adduct in mammalian cells: The role of DNA polymerase κ. J. Biol. Chem. 2004, 279, 53298–53305. [Google Scholar] [CrossRef] [PubMed]
- Rechkoblit, O.; Zhang, Y.; Guo, D.; Wang, Z.; Amin, S.; Krzeminsky, J.; Louneva, N.; Geacintov, N.E. trans-Lesion synthesis past bulky benzo[a]pyrene diol epoxide N2-dG and N6-dA lesions catalyzed by DNA bypass polymerases. J. Biol. Chem. 2002, 277, 30488–30494. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.J.; Chang, R.L.; Wong, C.-Q.; Bhachech, N.; Cui, X.X.; Hennig, E.; Yagi, H.; Sayer, J.M.; Jerina, D.M.; Preston, B.D. Dose-dependent differences in the profile of mutations induced by an ultimate carcinogen from benzo[a]pyrene. Proc. Natl. Acad. Sci. USA 1991, 88, 11227–11230. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Chang, R.L.; Bhachech, N.; Cui, X.X.; Merkler, K.A.; Wong, C.Q.; Hennig, E.; Yagi, H.; Jerina, D.M.; Conney, A.H. Dose-dependent differences in the profile of mutations induced by (+)-7R,8S-dihydroxy-9S,10R-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene in the coding region of the hypoxanthine (guanine) phosphoribosyltransferase gene in Chinese hamster V-79 cells. Cancer Res. 1993, 53, 3294–3301. [Google Scholar] [PubMed]
- Conney, A.H.; Chang, R.L.; Jerina, D.M.; Caroline Wei, S.J. Studies on the metabolism of benzo[a]pyrene and dose-dependent differences in the mutagenic profile of its ultimate carcinogenic metabolite. Drug Metab. Rev. 1994, 26, 125–163. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Cho, Y.; Yang, I.-Y.; Akagi, J.-I.; Ohashi, E.; Tateishi, S.; De Wind, N.; Hanaoka, F.; Ohmori, H.; Moriya, M. The vital role of polymerase ζ and REV1 in mutagenic, but not correct, DNA synthesis across benzo[a]pyrene-dG and recruitment of polymerase ζ by REV1 to replication-stalled site. J. Biol. Chem. 2012, 287, 9613–9622. [Google Scholar] [CrossRef]
- Frank, E.G.; Woodgate, R. Increased catalytic activity and altered fidelity of human DNA polymerase ι in the presence of manganese. J. Biol. Chem. 2007, 282, 24689–24696. [Google Scholar] [CrossRef] [PubMed]
- Frank, E.G.; Sayer, J.M.; Kroth, H.; Ohashi, E.; Ohmori, H.; Jerina, D.M.; Woodgate, R. Translesion replication of benzo[a]pyrene and benzo[c]phenanthrene diol epoxide adducts of deoxyadenosine and deoxyguanosine by human DNA polymerase ι. Nucleic Acids Res. 2002, 30, 5284–5292. [Google Scholar] [CrossRef] [PubMed]
- Jha, V.; Bian, C.; Xing, G.; Ling, H. Structure and mechanism of error-free replication past the major benzo[a]pyrene adduct by human DNA polymerase κ. Nucleic Acids Res. 2016, 44, 4957–4967. [Google Scholar] [CrossRef] [PubMed]
- Cosman, M.; De Los Santos, C.; Fiala, R.; Hingerty, B.E.; Singh, S.B.; Ibanez, V.; Margulis, L.A.; Live, D.; Geacintov, N.E.; Broyde, S. Solution conformation of the major adduct between the carcinogen (+)-anti-benzo[a]pyrene diol epoxide and DNA. Proc. Natl. Acad. Sci. USA 1992, 89, 1914–1918. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Gorin, A.; Hingerty, B.E.; Geacintov, N.E.; Broyde, S.; Patel, D.J. Structural alignment of the (+)-trans-anti-benzo[a]pyrene-dG adduct positioned opposite dC at a DNA template-primer junction. Biochemistry 1997, 36, 13769–13779. [Google Scholar] [CrossRef] [PubMed]
- Donny-Clark, K.; Broyde, S. Influence of local sequence context on damaged base conformation in human DNA polymerase ι: Molecular dynamics studies of nucleotide incorporation opposite a benzo[a]pyrene-derived adenine lesion. Nucleic Acids Res. 2009, 37, 7095–7109. [Google Scholar] [CrossRef] [PubMed]
- Beland, F.A.; Churchwell, M.I.; Von Tungeln, L.S.; Chen, S.; Fu, P.P.; Culp, S.J.; Schoket, B.; Gyorffy, E.; Minárovits, J.; Poirier, M.C. High-performance liquid chromatography electrospray ionization tandem mass spectrometry for the detection and quantitation of benzo[a]pyrene-DNA adducts. Chem. Res. Toxicol. 2005, 18, 1306–1315. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, X.; Rechkoblit, O.; Geacintov, N.E.; Taylor, J.-S.; Wang, Z. Response of human REV1 to different DNA damage: Preferential dCMP insertion opposite the lesion. Nucleic Acids Res. 2002, 30, 1630–1638. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.R.; Gibbs, P.E.M.; Nowicka, A.M.; Hinkle, D.C.; Lawrence, C.W. Evidence for a second function for Saccharomyces cerevisiae Rev1p. Mol. Microbiol. 2000, 37, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Fischhaber, P.L.; Luk-Paszyc, M.J.; Masuda, Y.; Zhou, J.; Kamiya, K.; Kisker, C.; Friedberg, E.C. Mouse Rev1 protein interacts with multiple DNA polymerases involved in translesion DNA synthesis. EMBO J. 2003, 22, 6621–6630. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, E.; Murakumo, Y.; Kanjo, N.; Akagi, J.i.; Masutani, C.; Hanaoka, F.; Ohmori, H. Interaction of hREV1 with three human Y-family DNA polymerases. Genes Cells 2004, 9, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Tissier, A.; Kannouche, P.; Reck, M.-P.; Lehmann, A.R.; Fuchs, R.P.P.; Cordonnier, A. Co-localization in replication foci and interaction of human Y-family members, DNA polymerase polη and REVl protein. DNA Repair 2004, 3, 1503–1514. [Google Scholar] [CrossRef] [PubMed]
- Murakumo, Y.; Ogura, Y.; Ishii, H.; Numata, S.-I.; Ichihara, M.; Croce, C.M.; Fishel, R.; Takahashi, M. Interactions in the error-prone postreplication repair proteins hREV1, hREV3, and hREV7. J. Biol. Chem. 2001, 276, 35644–35651. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, E.; Hanafusa, T.; Kamei, K.; Song, I.; Tomida, J.; Hashimoto, H.; Vaziri, C.; Ohmori, H. Identification of a novel REV1-interacting motif necessary for DNA polymerase κ function. Genes Cells 2009, 14, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Prelich, G.; Tan, C.-K.; Kostura, M.; Mathews, M.B.; So, A.G.; Downey, K.M.; Stillman, B. Functional identity of proliferating cell nuclear antigen and a DNA polymerase-delta auxiliary protein. Nature 1986, 326, 517–520. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.-K.; Castillo, C.; So, A.G.; Downey, K.M. An auxiliary protein for DNA polymerase-delta from fetal calf thymus. J. Biol. Chem. 1986, 261, 12310–12316. [Google Scholar] [PubMed]
- Maga, G.; Hübscher, U. Proliferating cell nuclear antigen (PCNA): A dancer with many partners. J. Cell Sci. 2003, 116, 3051–3060. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, G.-L.; Pfander, B.; Jentsch, S. PCNA, the maestro of the replication fork. Cell 2007, 129, 665–679. [Google Scholar] [CrossRef] [PubMed]
- Boehm, E.M.; Gildenberg, M.S.; Washington, M.T. The many roles of PCNA in eukaryotic DNA replication. In The Enzymes; Laurie, S.K., Marcos Túlio, O., Eds.; Academic Press: London, UK, 2016; Volume 39, pp. 231–254. [Google Scholar]
- Mailand, N.; Gibbs-Seymour, I.; Bekker-Jensen, S. Regulation of PCNA–protein interactions for genome stability. Nat. Rev. Mol. Cell Biol. 2013, 14, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Haracska, L.; Johnson, R.E.; Unk, I.; Phillips, B.; Hurwitz, J.; Prakash, L.; Prakash, S. Physical and functional interactions of human DNA polymerase η with PCNA. Mol. Cell. Biol. 2001, 21, 7199–7206. [Google Scholar] [CrossRef] [PubMed]
- Haracska, L.; Johnson, R.E.; Unk, I.; Phillips, B.B.; Hurwitz, J.; Prakash, L.; Prakash, S. Targeting of human DNA polymerase ι to the replication machinery via interaction with PCNA. Proc. Natl. Acad. Sci. USA 2001, 98, 14256–14261. [Google Scholar] [CrossRef] [PubMed]
- Haracska, L.; Kondratick, C.M.; Unk, I.; Prakash, S.; Prakash, L. Interaction with PCNA is essential for yeast DNA polymerase η function. Mol. Cell 2001, 8, 407–415. [Google Scholar] [CrossRef]
- Haracska, L.; Unk, I.; Johnson, R.E.; Phillips, B.B.; Hurwitz, J.; Prakash, L.; Prakash, S. Stimulation of DNA synthesis activity of human DNA polymerase κ by PCNA. Mol. Cell. Biol. 2002, 22, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Ohmori, H.; Hanafusa, T.; Ohashi, E.; Vaziri, C. Separate roles of structured and unstructured regions of Y-family DNA polymerases. Adv. Protein Chem. Struct. Biol. 2009, 78, 99–146. [Google Scholar] [PubMed]
- Krishna, T.S.R.; Kong, X.-P.; Gary, S.; Burgers, P.M.; Kuriyan, J. Crystal structure of the eukaryotic DNA polymerase processivity factor PCNA. Cell 1994, 79, 1233–1243. [Google Scholar] [CrossRef]
- Gulbis, J.M.; Kelman, Z.; Hurwitz, J.; O’Donnell, M.; Kuriyan, J. Structure of the C-terminal region of p21 WAF1/CIP1 complexed with human PCNA. Cell 1996, 87, 297–306. [Google Scholar] [CrossRef]
- Kochaniak, A.B.; Habuchi, S.; Loparo, J.J.; Chang, D.J.; Cimprich, K.A.; Walter, J.C.; van Oijen, A.M. Proliferating cell nuclear antigen uses two distinct modes to move along DNA. J. Biol. Chem. 2009, 284, 17700–17710. [Google Scholar] [CrossRef] [PubMed]
- Garg, P.; Stith, C.M.; Majka, J.; Burgers, P.M.J. Proliferating cell nuclear antigen promotes translesion synthesis by DNA polymerase ζ. J. Biol. Chem. 2005, 280, 23446–23450. [Google Scholar] [CrossRef] [PubMed]
- Makarova, A.V.; Stodola, J.L.; Burgers, P.M. A four-subunit DNA polymerase ζ complex containing Pol δ accessory subunits is essential for PCNA-mediated mutagenesis. Nucleic Acids Res. 2012, 40, 11618–11626. [Google Scholar] [CrossRef] [PubMed]
- Garg, P.; Burgers, P.M. Ubiquitinated proliferating cell nuclear antigen activates translesion DNA polymerases η and REV1. Proc. Natl. Acad. Sci. USA 2005, 102, 18361–18366. [Google Scholar] [CrossRef] [PubMed]
- Bruning, J.B.; Shamoo, Y. Structural and thermodynamic analysis of human PCNA with peptides derived from DNA polymerase-δ p66 subunit and flap endonuclease-1. Structure 2004, 12, 2209–2219. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, S.; Kitano, K.; Yamaguchi, H.; Hamada, K.; Okada, K.; Fukuda, K.; Uchida, M.; Ohtsuka, E.; Morioka, H.; Hakoshima, T. Structural basis for recruitment of human flap endonuclease 1 to PCNA. EMBO J. 2005, 24, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Hishiki, A.; Hashimoto, H.; Hanafusa, T.; Kamei, K.; Ohashi, E.; Shimizu, T.; Ohmori, H.; Sato, M. Structural basis for novel interactions between human translesion synthesis polymerases and proliferating cell nuclear antigen. J. Biol. Chem. 2009, 284, 10552–10560. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Sonoda, E.; Tang, T.-S.; Parker, J.L.; Bielen, A.B.; Takeda, S.; Ulrich, H.D.; Friedberg, E.C. REV1 protein interacts with PCNA: Significance of the REV1 BRCT domain in vitro and in vivo. Mol. Cell 2006, 23, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Pustovalova, Y.; Maciejewski, M.W.; Korzhnev, D.M. NMR mapping of PCNA interaction with translesion synthesis DNA polymerase Rev1 mediated by Rev1-BRCT domain. J. Mol. Biol. 2013, 425, 3091–3105. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.M.; Kochenova, O.V.; Shcherbakova, P.V. The non-canonical protein binding site at the monomer-monomer interface of yeast proliferating cell nuclear antigen (PCNA) regulates the Rev1-PCNA interaction and Polζ/Rev1-dependent translesion DNA synthesis. J. Biol. Chem. 2011, 286, 33557–33566. [Google Scholar] [CrossRef] [PubMed]
- Boehm, E.M.; Powers, K.T.; Kondratick, C.M.; Spies, M.; Houtman, J.C.D.; Washington, M.T. The proliferating cell nuclear antigen (PCNA)-interacting protein (PIP) motif of DNA polymerase η mediates its interaction with the C-terminal domain of Rev1. J. Biol. Chem. 2016, 291, 8735–8744. [Google Scholar] [CrossRef] [PubMed]
- Baldeck, N.; Janel-Bintz, R.; Wagner, J.; Tissier, A.; Fuchs, R.P.; Burkovics, P.; Haracska, L.; Despras, E.; Bichara, M.; Chatton, B. FF483-484 motif of human Polη mediates its interaction with the POLD2 subunit of Polδ and contributes to DNA damage tolerance. Nucleic Acids Res. 2015, 2116–2125. [Google Scholar] [CrossRef] [PubMed]
- Boehm, E.M.; Washington, M.T. R.I.P. to the PIP: PCNA-binding motif no longer considered specific. Bioessays 2016, 38, 1117–1122. [Google Scholar] [CrossRef] [PubMed]
- Hedglin, M.; Pandey, B.; Benkovic, S.J. Characterization of human translesion DNA synthesis across a UV-induced DNA lesion. eLife 2016, 5, e19788. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Weinacht, C.P.; Zhuang, Z. Regulatory role of ubiquitin in eukaryotic DNA translesion synthesis. Biochemistry 2013, 52, 3217–3228. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Bennett, E.J.; Huttlin, E.L.; Guo, A.; Li, J.; Possemato, A.; Sowa, M.E.; Rad, R.; Rush, J.; Comb, M.J. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 2011, 44, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.A.; Beli, P.; Weinert, B.T.; Nielsen, M.L.; Cox, J.; Mann, M.; Choudhary, C. A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol. Cell. Proteom. 2011. [Google Scholar] [CrossRef] [PubMed]
- Bienko, M.; Green, C.M.; Crosetto, N.; Rudolf, F.; Zapart, G.; Coull, B.; Kannouche, P.; Wider, G.; Peter, M.; Lehmann, A.R. Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science 2005, 310, 1821–1824. [Google Scholar] [CrossRef] [PubMed]
- Freudenthal, B.D.; Gakhar, L.; Ramaswamy, S.; Washington, M.T. Structure of monoubiquitinated PCNA and implications for translesion synthesis and DNA polymerase exchange. Nat. Struct. Mol. Biol. 2010, 17, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Dieckman, L.M.; Washington, M.T. PCNA trimer instability inhibits translesion synthesis by DNA polymerase η and by DNA polymerase δ. DNA Repair 2013, 12, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Z.; Johnson, R.E.; Haracska, L.; Prakash, L.; Prakash, S.; Benkovic, S.J. Regulation of polymerase exchange between Pol η and Pol δ by monoubiquitination of PCNA and the movement of DNA polymerase holoenzyme. Proc. Natl. Acad. Sci. USA 2008, 105, 5361–5366. [Google Scholar] [CrossRef] [PubMed]
- Hoege, C.; Pfander, B.; Moldovan, G.-L.; Pyrowolakis, G.; Jentsch, S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 2002, 419, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Stelter, P.; Ulrich, H.D. Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature 2003, 425, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Kannouche, P.L.; Wing, J.; Lehmann, A.R. Interaction of human DNA polymerase η with monoubiquitinated PCNA: A possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell 2004, 14, 491–500. [Google Scholar] [CrossRef]
- Watanabe, K.; Tateishi, S.; Kawasuji, M.; Tsurimoto, T.; Inoue, H.; Yamaizumi, M. Rad18 guides polη to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 2004, 23, 3886–3896. [Google Scholar] [CrossRef] [PubMed]
- Despras, E.; Delrieu, N.; Garandeau, C.; Ahmed-Seghir, S.; Kannouche, P.L. Regulation of the specialized DNA polymerase eta: Revisiting the biological relevance of its PCNA-and ubiquitin-binding motifs. Environ. Mol. Mutagen. 2012, 53, 752–765. [Google Scholar] [CrossRef] [PubMed]
- Acharya, N.; Yoon, J.-H.; Gali, H.; Unk, I.; Haracska, L.; Johnson, R.E.; Hurwitz, J.; Prakash, L.; Prakash, S. Roles of PCNA-binding and ubiquitin-binding domains in human DNA polymerase η in translesion DNA synthesis. Proc. Natl. Acad. Sci. USA 2008, 105, 17724–17729. [Google Scholar] [CrossRef] [PubMed]
- Sabbioneda, S.; Green, C.M.; Bienko, M.; Kannouche, P.; Dikic, I.; Lehmann, A.R. Ubiquitin-binding motif of human DNA polymerase η is required for correct localization. Proc. Natl. Acad. Sci. USA 2009, 106. [Google Scholar] [CrossRef] [PubMed]
- Sabbioneda, S.; Gourdin, A.M.; Green, C.M.; Zotter, A.; Giglia-Mari, G.; Houtsmuller, A.; Vermeulen, W.; Lehmann, A.R. Effect of proliferating cell nuclear antigen ubiquitination and chromatin structure on the dynamic properties of the Y-family DNA polymerases. Mol. Biol. Cell 2008, 19, 5193–5202. [Google Scholar] [CrossRef] [PubMed]
- Göhler, T.; Sabbioneda, S.; Green, C.M.; Lehmann, A.R. ATR-mediated phosphorylation of DNA polymerase η is needed for efficient recovery from UV damage. J. Cell Biol. 2011, 192, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Hendel, A.; Krijger, P.H.L.; Diamant, N.; Goren, Z.; Langerak, P.; Kim, J.; Reißner, T.; Lee, K.-y.; Geacintov, N.E.; Carell, T. PCNA ubiquitination is important, but not essential for translesion DNA synthesis in mammalian cells. PLoS Genet. 2011, 7, e1002262. [Google Scholar] [CrossRef] [PubMed]
- Langerak, P.; Nygren, A.O.H.; Krijger, P.H.L.; van den Berk, P.C.M.; Jacobs, H. A/T mutagenesis in hypermutated immunoglobulin genes strongly depends on PCNAK164 modification. J. Exp. Med. 2007, 204, 1989–1998. [Google Scholar] [CrossRef] [PubMed]
- Szüts, D.; Marcus, A.P.; Himoto, M.; Iwai, S.; Sale, J.E. REV1 restrains DNA polymerase ζ to ensure frame fidelity during translesion synthesis of UV photoproducts in vivo. Nucleic Acids Res. 2008, 36, 6767–6780. [Google Scholar] [CrossRef] [PubMed]
- Edmunds, C.E.; Simpson, L.J.; Sale, J.E. PCNA ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. Mol. Cell 2008, 30, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Lau, W.C.; Li, Y.; Zhang, Q.; Huen, M.S. Molecular architecture of the Ub-PCNA/Pol η complex bound to DNA. Sci. Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Prindle, M.J.; Loeb, L.A. DNA polymerase delta in DNA replication and genome maintenance. Environ. Mol. Mutagen. 2012, 53, 666–682. [Google Scholar] [CrossRef] [PubMed]
- Netz, D.J.A.; Stith, C.M.; Stümpfig, M.; Köpf, G.; Vogel, D.; Genau, H.M.; Stodola, J.L.; Lill, R.; Burgers, P.M.J.; Pierik, A.J. Eukaryotic DNA polymerases require an iron-sulfur cluster for the formation of active complexes. Nat. Chem. Biol. 2012, 8, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Nair, D.T.; Johnson, R.E.; Prakash, L.; Prakash, S.; Aggarwal, A.K. Rev1 employs a novel mechanism of DNA synthesis using a protein template. Science 2005, 309, 2219–2222. [Google Scholar] [CrossRef] [PubMed]
- Haracska, L.; Unk, I.; Johnson, R.E.; Johansson, E.; Burgers, P.M.J.; Prakash, S.; Prakash, L. Roles of yeast DNA polymerases δ and ζ and of Rev1 in the bypass of abasic sites. Genes Dev. 2001, 15, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.-L.; Simpson, L.J.; Sale, J.E. Vertebrate DNA damage tolerance requires the C-terminus but not BRCT or transferase domains of REV1. Nucleic Acids Res. 2005, 33, 1280–1289. [Google Scholar] [CrossRef] [PubMed]
- Baranovskiy, A.G.; Lada, A.G.; Siebler, H.M.; Zhang, Y.; Pavlov, Y.I.; Tahirov, T.H. DNA polymerase δ and ζ switch by sharing accessory subunits of DNA polymerase δ. J. Biol. Chem. 2012, 287, 17281–17287. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.E.; Prakash, L.; Prakash, S. Pol31 and Pol32 subunits of yeast DNA polymerase δ are also essential subunits of DNA polymerase ζ. Proc. Natl. Acad. Sci. USA 2012, 109, 12455–12460. [Google Scholar] [CrossRef] [PubMed]
- Pustovalova, Y.; Magalhães, M.T.Q.; D’Souza, S.; Rizzo, A.A.; Korza, G.; Walker, G.C.; Korzhnev, D.M. Interaction between the Rev1 C-terminal domain and the PolD3 subunit of Polζ suggests a mechanism of polymerase exchange upon Rev1/Polζ-dependent translesion synthesis. Biochemistry 2016, 55, 2043–2053. [Google Scholar] [CrossRef] [PubMed]
- Gabel, S.A.; DeRose, E.F.; London, R.E. XRCC1 interaction with the REV1 C-terminal domain suggests a role in post replication repair. DNA Repair 2013, 12, 1105–1113. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Lin, A.; Zhou, C.; Blackwell, S.R.; Zhang, Y.; Wang, Z.; Feng, Q.; Guan, R.; Hanna, M.D.; Chen, Z.; et al. Involvement of budding yeast Rad5 in translesion DNA synthesis through physical interaction with Rev1. Nucleic Acids Res. 2016, 44, 5231–5245. [Google Scholar] [CrossRef] [PubMed]
- Pozhidaeva, A.; Pustovalova, Y.; D’Souza, S.; Bezsonova, I.; Walker, G.C.; Korzhnev, D.M. NMR structure and dynamics of the C-terminal domain from human Rev1 and its complex with Rev1 interacting region of DNA polymerase η. Biochemistry 2012, 51, 5506–5520. [Google Scholar] [CrossRef] [PubMed]
- Wojtaszek, J.; Liu, J.; D’Souza, S.; Wang, S.; Xue, Y.; Walker, G.C.; Zhou, P. Multifaceted recognition of vertebrate Rev1 by translesion polymerases ζ and κ. J. Biol. Chem. 2012, 287, 26400–26408. [Google Scholar] [CrossRef] [PubMed]
- Livneh, Z.; Shachar, S. Multiple two-polymerase mechanisms in mammalian translesion DNA synthesis. Cell Cycle 2010, 9, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.-H.; Prakash, L.; Prakash, S. Error-free replicative bypass of (6–4) photoproducts by DNA polymerase ζ in mouse and human cells. Genes Dev. 2010, 24, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Wojtaszek, J.; Lee, C.-J.; D’Souza, S.; Minesinger, B.; Kim, H.; D’Andrea, A.D.; Walker, G.C.; Zhou, P. Structural basis of Rev1-mediated assembly of a quaternary vertebrate translesion polymerase complex consisting of Rev1, heterodimeric polymerase (Pol) ζ, and Pol κ. J. Biol. Chem. 2012, 287, 33836–33846. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Yang, X.; Xu, M.; Jiang, T. Structural insights into the assembly of human translesion polymerase complexes. Protein Cell 2012, 3, 864–874. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, S.; Hara, K.; Shimizu, T.; Sato, M.; Hashimoto, H. Structural basis of recruitment of DNA polymerase ζ by interaction between REV1 and REV7 proteins. J. Biol. Chem. 2012, 287, 33847–33852. [Google Scholar] [CrossRef] [PubMed]
- Boehm, E.M.; Spies, M.; Washington, M.T. PCNA tool belts and polymerase bridges form during translesion synthesis. Nucleic Acids Res. 2016, 44, 8250–8260. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.L.; Xu, F.; Ziola, B.; McGregor, W.G.; Xiao, W. Sequential assembly of translesion DNA polymerases at UV-induced DNA damage sites. Mol. Biol. Cell 2011, 22, 2373–2383. [Google Scholar] [CrossRef] [PubMed]
- Akagi, J.-I.; Masutani, C.; Kataoka, Y.; Kan, T.; Ohashi, E.; Mori, T.; Ohmori, H.; Hanaoka, F. Interaction with DNA polymerase η is required for nuclear accumulation of REV1 and suppression of spontaneous mutations in human cells. DNA Repair 2009, 8, 585–599. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-S.; Gregory, M.T.; Yang, W. Human Pol ζ purified with accessory subunits is active in translesion DNA synthesis and complements Pol η in cisplatin bypass. Proc. Natl. Acad. Sci. USA 2014, 111, 2954–2959. [Google Scholar] [CrossRef] [PubMed]
- Johansson, E.; Garg, P.; Burgers, P.M.J. The Pol32 subunit of DNA polymerase δ contains separable domains for processive replication and proliferating cell nuclear antigen (PCNA) binding. J. Biol. Chem. 2004, 279, 1907–1915. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Llorente, Y.; Malik, R.; Jain, R.; Choudhury, J.R.; Johnson, R.E.; Prakash, L.; Prakash, S.; Ubarretxena-Belandia, I.; Aggarwal, A.K. The architecture of yeast DNA polymerase ζ. Cell reports 2013, 5, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Makarova, A.V.; Burgers, P.M. Eukaryotic DNA polymerase ζ. DNA Repair 2015, 29, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhou, Y.; Trusa, S.; Meng, X.; Lee, E.Y.C.; Lee, M.Y.W.T. A novel DNA damage response: Rapid degradation of the p12 subunit of DNA polymerase δ. J. Biol. Chem. 2007, 282, 15330–15340. [Google Scholar] [CrossRef] [PubMed]
- Murga, M.; Lecona, E.; Kamileri, I.; Díaz, M.; Lugli, N.; Sotiriou, S.K.; Anton, M.E.; Méndez, J.; Halazonetis, T.D.; Fernandez-Capetillo, O. POLD3 is haploinsufficient for DNA replication in mice. Mol. Cell 2016, 63, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Stepchenkova, E.I.; Tarakhovskaya, E.R.; Siebler, H.M.; Pavlov, Y.I. Defect of Fe-S cluster binding by DNA polymerase δ in yeast suppresses UV-induced mutagenesis, but enhances DNA polymerase ζ—Dependent spontaneous mutagenesis. DNA Repair 2016. [Google Scholar] [CrossRef]
- Little, J.W.; Mount, D.W. The SOS regulatory system of Escherichia coli. Cell 1982, 29, 11–22. [Google Scholar] [CrossRef]
- Goodman, M.F.; McDonald, J.P.; Jaszczur, M.M.; Woodgate, R. Insights into the complex levels of regulation imposed on Escherichia coli DNA polymerase V. DNA Repair 2016, 44, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Wiltrout, M.E.; Walker, G.C. Proteasomal regulation of the mutagenic translesion DNA polymerase, Saccharomyces cerevisiae Rev1. DNA Repair 2011, 10, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Pabla, R.; Rozario, D.; Siede, W. Regulation of Saccharomyces cerevisiae DNA polymerase η transcript and protein. Radiat. Environ. Biophys. 2008, 47, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Waters, L.S.; Walker, G.C. The critical mutagenic translesion DNA polymerase Rev1 is highly expressed during G2/M phase rather than S phase. Proc. Natl. Acad. Sci. USA 2006, 103, 8971–8976. [Google Scholar] [CrossRef] [PubMed]
- Plachta, M.; Halas, A.; McIntyre, J.; Sledziewska-Gojska, E. The steady-state level and stability of TLS polymerase eta are cell cycle dependent in the yeast S. cerevisiae. DNA Repair 2015, 29, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, M.; Terunuma, J.; Hanaoka, F. The protein level of Rev1, a TLS polymerase in fission yeast, is strictly regulated during the cell cycle and after DNA damage. PLoS ONE 2015, 10, e0130000. [Google Scholar] [CrossRef] [PubMed]
- King, N.M.; Nikolaishvili-Feinberg, N.; Bryant, M.F.; Luche, D.D.; Heffernan, T.P.; Simpson, D.A.; Hanaoka, F.; Kaufmann, W.K.; Cordeiro-Stone, M. Overproduction of DNA polymerase eta does not raise the spontaneous mutation rate in diploid human fibroblasts. DNA Repair 2005, 4, 714–724. [Google Scholar] [CrossRef] [PubMed]
- Thakur, M.; Wernick, M.; Collins, C.; Limoli, C.L.; Crowley, E.; Cleaver, J.E. DNA polymerase η undergoes alternative splicing, protects against UV sensitivity and apoptosis, and suppresses Mre11-dependent recombination. Genes Chromosomes Cancer 2001, 32, 222–235. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Zhu, H.; Lou, M.; Fan, Y.; Liu, H.; Shen, J.; Li, Z.; Lv, X.; Shan, J.; Zhu, L. Interferon regulatory factor 1 transactivates expression of human DNA polymerase η in response to carcinogen N-methyl-N′-nitro-N-nitrosoguanidine. J. Biol. Chem. 2012, 287, 12622–12633. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.-L.; Johnson, R.E.; Prakash, S.; Prakash, L. Requirement of DNA polymerase η for error-free bypass of UV-induced CC and TC photoproducts. Mol. Cell. Biol. 2001, 21, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Haruta, N.; Kubota, Y.; Hishida, T. Chronic low-dose ultraviolet-induced mutagenesis in nucleotide excision repair-deficient cells. Nucleic Acids Res. 2012, 40, 8406–8415. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, Y.I.; Nguyen, D.; Kunkel, T.A. Mutator effects of overproducing DNA polymerase η (Rad30) and its catalytically inactive variant in yeast. Mutat. Res. Fundam. Mol. Mech. Mutag. 2001, 478, 129–139. [Google Scholar] [CrossRef]
- McIntyre, J.; Woodgate, R. Regulation of translesion DNA synthesis: Posttranslational modification of lysine residues in key proteins. DNA Repair 2015, 29, 166–179. [Google Scholar] [CrossRef] [PubMed]
- García-Rodríguez, N.; Wong, R.P.; Ulrich, H.D. Functions of ubiquitin and SUMO in DNA replication and replication stress. Front. Genet. 2016. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.-S.; Liu, G.; Chen, X. Pirh2 E3 ubiquitin ligase targets DNA polymerase eta for 20S proteasomal degradation. Mol. Cell. Biol. 2010, 30, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.-S.; Hakem, A.; Hakem, R.; Chen, X. Pirh2 E3 ubiquitin ligase monoubiquitinates DNA polymerase eta to suppress translesion DNA synthesis. Mol. Cell. Biol. 2011, 31, 3997–4006. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.-S.; Qian, Y.; Chen, X. DNA polymerase eta is targeted by Mdm2 for polyubiquitination and proteasomal degradation in response to ultraviolet irradiation. DNA Repair 2012, 11, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Wallace, H.A.; Merkle, J.A.; Yu, M.C.; Berg, T.G.; Lee, E.; Bosco, G.; Lee, L.A. TRIP/NOPO E3 ubiquitin ligase promotes ubiquitylation of DNA polymerase η. Development 2014, 141, 1332–1341. [Google Scholar] [CrossRef] [PubMed]
- Havens, C.G.; Walter, J.C. Mechanism of CRL4Cdt2, a PCNA-dependent E3 ubiquitin ligase. Genes Dev. 2011, 25, 1568–1582. [Google Scholar] [CrossRef] [PubMed]
- Higa, L.A.A.; Mihaylov, I.S.; Banks, D.P.; Zheng, J.; Zhang, H. Radiation-mediated proteolysis of CDT1 by CUL4–ROC1 and CSN complexes constitutes a new checkpoint. Nat. Cell Biol. 2003, 5, 1008–1015. [Google Scholar] [CrossRef] [PubMed]
- Shiomi, Y.; Hayashi, A.; Ishii, T.; Shinmyozu, K.; Nakayama, J.-I.; Sugasawa, K.; Nishitani, H. Two different replication factor C proteins, Ctf18 and RFC1, separately control PCNA-CRL4Cdt2-mediated Cdt1 proteolysis during S phase and following UV irradiation. Mol. Cell. Biol. 2012, 32, 2279–2288. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Michael, W.M. Regulated proteolysis of DNA polymerase η during the DNA-damage response in C. elegans. Mol. Cell 2008, 32, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Havens, C.G.; Walter, J.C. Docking of a specialized PIP box onto chromatin-bound PCNA creates a degron for the ubiquitin ligase CRL4Cdt2. Mol. Cell 2009, 35, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Michishita, M.; Morimoto, A.; Ishii, T.; Komori, H.; Shiomi, Y.; Higuchi, Y.; Nishitani, H. Positively charged residues located downstream of PIP box, together with TD amino acids within PIP box, are important for CRL4Cdt2-mediated proteolysis. Genes Cells 2011, 16, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Tsanov, N.; Kermi, C.; Coulombe, P.; van der Laan, S.; Hodroj, D.; Maiorano, D. PIP degron proteins, substrates of CRL4Cdt2, and not PIP boxes, interfere with DNA polymerase η and κ focus formation on UV damage. Nucleic Acids Res. 2014, 42, 3692–3706. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xie, B.; Zhou, Y.; Rahmeh, A.; Trusa, S.; Zhang, S.; Gao, Y.; Lee, E.Y.; Lee, M.Y. Functional roles of p12, the fourth subunit of human DNA polymerase δ. J. Biol. Chem. 2006, 281, 14748–14755. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Zhou, Y.; Lee, E.Y.C.; Lee, M.Y.W.T.; Frick, D.N. The p12 subunit of human polymerase δ modulates the rate and fidelity of DNA synthesis. Biochemistry 2010, 49, 3545–3554. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Zhou, Y.; Zhang, S.; Lee, E.Y.C.; Frick, D.N.; Lee, M.Y.W.T. DNA damage alters DNA polymerase δ to a form that exhibits increased discrimination against modified template bases and mismatched primers. Nucleic Acids Res. 2009, 37, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Zhang, S.; Xu, D.; Lee, M.Y.W.T.; Zhang, Z.; Lee, E.Y.C.; Darzynkiewicz, Z. Expression of the p12 subunit of human DNA polymerase δ (Pol δ), CDK inhibitor p21WAF1, Cdt1, cyclin A, PCNA and Ki-67 in relation to DNA replication in individual cells. Cell Cycle 2014, 13, 3529–3540. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.W.T.; Zhang, S.; Hua Lin, S.; Wang, X.; Darzynkiewicz, Z.; Zhang, Z.; Lee, E. The tail that wags the dog: P12, the smallest subunit of DNA polymerase δ, is degraded by ubiquitin ligases in response to DNA damage and during cell cycle progression. Cell Cycle 2014, 13, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Darzynkiewicz, Z.; Zhao, H.; Zhang, S.; Marietta, Y.W.T.L.; Ernest, Y.C.L.; Zhang, Z. Initiation and termination of DNA replication during S phase in relation to cyclins D1, E and A, p21WAF1, Cdt1 and the p12 subunit of DNA polymerase δ revealed in individual cells by cytometry. Oncotarget 2015, 6, 11735–11750. [Google Scholar] [CrossRef] [PubMed]
- Hedglin, M.; Pandey, B.; Benkovic, S.J. Stability of the human polymerase δ holoenzyme and its implications in lagging strand DNA synthesis. Proc. Natl. Acad. Sci. USA 2016, 113, E1777–E1786. [Google Scholar] [CrossRef] [PubMed]
- Tsurimoto, T.; Stillman, B. Replication factors required for SV40 DNA replication in vitro. II. Switching of DNA polymerase alpha and delta during initiation of leading and lagging strand synthesis. J. Biol. Chem. 1991, 266, 1961–1968. [Google Scholar] [PubMed]
- Yurieva, O.; O’Donnell, M. Reconstitution of a eukaryotic replisome reveals the mechanism of asymmetric distribution of DNA polymerases. Nucleus 2016, 7, 360–368. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DNA Polymerase | Sequence | Kd (μM) |
|---|---|---|
| pol δ PIP | 451GKANRQVSITGFFQRK | 16 1 |
| pol δ holoenzyme | <0.010 2 | |
| pol η PIP2 | C694KRPRPEGMQTLESFFKPLTH | 0.40 3 |
| pol η holoenzyme | 0.12 4 | |
| pol κ PIP + PLTH | C856IKPNNPKHTLDIFFKPLTH | 4.9 3 |
| pol ι PIP | C419AKKGLIDYYLMPSLST | 0.39 3 |
| DNA Polymerase | Sequence | Kd (μM) SPR | Fluorescence 3 |
|---|---|---|---|
| p66 | 231KGNMMSNFFGKAAMNK | 2.3 1 | |
| pol η | 524QSTGTEPFFKQKSLLL | 13 2 | 4.4 |
| pol κ | 560EMSHKKSFFDKKRSER | 7.6 2 | |
| pol κ | 560EMSHKKSFFDKKRSER | 1.7 1 | 0.28 |
| pol ι PIP | 539ASRGVLSFFSKKQMQD | 69 2 | 5.5 |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, L.; Washington, M.T. Translesion Synthesis: Insights into the Selection and Switching of DNA Polymerases. Genes 2017, 8, 24. https://doi.org/10.3390/genes8010024
Zhao L, Washington MT. Translesion Synthesis: Insights into the Selection and Switching of DNA Polymerases. Genes. 2017; 8(1):24. https://doi.org/10.3390/genes8010024
Chicago/Turabian StyleZhao, Linlin, and M. Todd Washington. 2017. "Translesion Synthesis: Insights into the Selection and Switching of DNA Polymerases" Genes 8, no. 1: 24. https://doi.org/10.3390/genes8010024
APA StyleZhao, L., & Washington, M. T. (2017). Translesion Synthesis: Insights into the Selection and Switching of DNA Polymerases. Genes, 8(1), 24. https://doi.org/10.3390/genes8010024