Abstract
Eukaryotic cells are equipped with surveillance mechanisms called checkpoints to ensure proper execution of cell cycle events. Among these are the checkpoints that detect DNA damage or replication perturbations and coordinate cellular activities to maintain genome stability. At the forefront of damage sensing is an evolutionarily conserved molecule, known respectively in budding yeast and humans as Mec1 (Mitosis entry checkpoint 1) and ATR (Ataxia telangiectasia and Rad3-related protein). Through phosphorylation, Mec1/ATR activates downstream components of a signaling cascade to maintain nucleotide pool balance, protect replication fork integrity, regulate activation of origins of replication, coordinate DNA repair, and implement cell cycle delay. This list of functions continues to expand as studies have revealed that Mec1/ATR modularly interacts with various protein molecules in response to different cellular cues. Among these newly assigned functions is the regulation of RNA metabolism during checkpoint activation and the coordination of replication–transcription conflicts. In this review, I will highlight some of these new functions of Mec1/ATR with a focus on the yeast model organism.
1. Introduction
Mec1/ATR (Mitosis entry checkpoint 1 and Ataxia telangiectasia and Rad3-related protein)—an evolutionarily conserved protein in Saccharomyces cerevisiae and Homo sapiens, respectively—is virtually ubiquitous in all cellular compartments, dispatching work forces to perform a wide range of tasks. The biochemical function of Mec1/ATR is a kinase acting in a complex with Ddc2 (ATRIP, ATR interacting protein in humans) that controls a signaling cascade to ensure the maintenance of genome integrity. Mec1/ATR’s responsibilities are rooted in DNA metabolisms including replication, repair, and chromosome segregation. Growing evidence also extends Mec1/ATR’s function to RNA metabolisms and, more importantly, implicates Mec1/ATR in resolving conflicts between DNA replication and gene transcription. Mec1/ATR’s function is thus akin to that of a Program Manager in an organization, who serves a strategic role by coordinating teams working on related projects. For these reasons, it is appropriate to bestow such a title upon Mec1/ATR in this coined enterprise (and a feeble attempt at a double entendre)—Nucleic Acids Incorporated.
MEC1 and RAD53 (radiation sensitive mutant 53, human CHEK2) were initially identified in Saccharomyces cerevisiae as genes required for the S and G2 checkpoints that are induced by DNA replication inhibition and DNA damage, respectively [1]. When replication inhibition is induced through treatment with hydroxyurea (HU)—a ribonucleotide reductase (RNR) inhibitor—Mec1 (acting as a sensor) phosphorylates a cluster of residues in the N-terminus of Rad53 (a transducer), which in turn activates downstream effector molecules to play multiple cellular functions [2]. A defective checkpoint via mutations in the Mec1 or Rad53 kinase causes cells to lose control over replication initiation, prevent stalled replication forks from resuming progression, prematurely enter mitosis, and ultimately, lose viability [2]. Along with these two kinases, the Tel1 kinase (human ATM, ataxia telangiectasia mutated) partially substitutes Mec1 during its absence, albeit the two kinases show distinct substrate specificities [3]. A recent study provided structural insights into how the Mec1•Ddc2 dimer and Tel1 dimer function differentially towards substrate [4]. The kinase domains within the Mec1•Ddc2 dimer are quite close to each other, thus potentially requiring a dimer-to-monomer structural change to activate the kinase. In contrast, the kinase domains within the Tel1 dimer are sufficiently far from each other, thus permitting the kinase activity even in the dimer configuration. This structural difference may help explain the substrate specificity of the ATR and ATM protein in specific pathways.
Though premature mitosis is a prominent phenotype associated with the checkpoint mutants upon replication stress [1,5], restraining mitosis apparently does not ameliorate the loss of cell viability and, instead, the essential function of the checkpoint appears to be promoting recovery from stress [6]. Moreover, cell lethality associated with mec1 and rad53 deletions can be rescued by the removal of a protein inhibitor of RNR, Sml1, suggesting that nucleotide pool maintenance is the underpinning for Mec1/Rad53 functions during an unperturbed cell cycle [7]. Thus, it seems that the critical function(s) of the checkpoint pathway vary based on the situation at hand, be it a normal cell cycle or during induced stress. In other words, certain functions of these enzymes only become essential upon induced stress, including nutrient deprivation, external DNA damage, and replication blockage. Incidentally, it is these stress-induced functions of the checkpoint that are better characterized so far. However, recent studies have identified new substrates of the Mec1/ATR in a normal S phase, which promise to further our understanding of the essential function of the checkpoint without external stress. As well, the stress-induced checkpoint functions of Mec1/ATR are an ever-expanding list. Here, I will focus on reviewing the roles that Mec1/ATR play in nucleic acid metabolisms, including DNA replication, gene transcription, and the interface between these processes. I will also highlight recent studies presenting Mec1/ATR’s new functions in various cellular pathways, with a focus on the model organism Saccharomyces cerevisiae.
2. The To-Do List of Mec1/ATR in DNA and RNA Metabolisms
2.1. DNA Metabolism
2.1.1. Nucleotide Pool Maintenance
Following DNA damage or replication blockage, the Mec1/Rad53 checkpoint induces the Dun1 kinase, which in turn down-regulates Sml1 (an inhibitor of RNR), thus allowing a boosted production of deoxyribonucleotides (dNTPs) [8]. Later, it was demonstrated that this signaling pathway is also crucial during unchallenged cell growth. As mentioned above, the lethality associated with mec1 or rad53 deletion can be suppressed by increasing RNR activity through removal of its protein inhibitors, including sml1, crt1, hug1, and dif1, or overexpressing RNR1 or RNR3 [6,7,9,10,11,12]. Therefore, it appears that the essential function of the Mec1/Rad53/Dun1 checkpoint cascade during normal growth is to maintain a proper level of dNTP pools. Indeed, the dNTP levels are depleted—with a broad range of depletion levels—in various mec1, rad53, and dun1 mutants compared to wild-type yeast [9,13,14].
The mechanisms by which Mec1/Rad53 signaling ensures an adequate and balanced pool of dNTPs largely centered on RNR regulation. RNR activity can be turned on/off by its binding to ATP or dATP (deoxyadenosine triphosphate), respectively, at an allosteric activity site [15,16]. It is also subject to direct transcriptional induction and indirect upregulation by the destruction of the aforementioned protein inhibitors, both through the Mec1/Rad53 pathway, as reviewed in [17]. However, what is the molecular consequence of the failure to maintain dNTP levels during normal growth? Using constructs containing regulated expression of Sml1, it was shown that prolonged inhibition of RNR results in a terminal phenotype of incomplete DNA replication in mec1 and rad53 deletion mutants [18]. It was also shown that a mec1-21 mutant is hyper-recombinogenic in an Sml1-dependent manner [14]. In addition, a rad53-4AQ mutant that lacks the N-terminal cluster of phosphorylation sites by Mec1 is unable to activate Dun1, and is synthetically lethal with rad9 without external damage [19]. Together, these data argue that there is a minimal requirement of Dun1 activation by the Mec1/Rad53 checkpoint during a normal S phase to maintain an adequate level of dNTPs, protect cells from DNA damage, prevent hyper-recombination, and ensure complete DNA replication.
The broad range of dNTP pool level reduction in checkpoint mutants is intriguing. In some of the mutants examined, the reduction of dNTP levels was rather modest. Notably, in a mec1 temperature-sensitive (mec1ts) lethal mutant, there was only a 17% drop in dNTP pools at the restrictive temperature [13]. It was argued that the mec1 mutant is exquisitely sensitive to even minute levels of dNTP reduction due to the wide range of DNA metabolic pathways for which dNTPs are required [13]. Therefore, it appears that the next challenge in understanding the essential function of the Mec1/Rad53 checkpoint for normal cell growth is the identification of those checkpoint substrates in a normal cell cycle. Conceivably, although both mec1 and rad53 deletion mutants can be suppressed by up-regulating RNR, the respective essential functions of these kinases might differ. Consistent with this notion, a recent phosphor-proteomic screen has revealed >200 peptide substrates represented by genes of the Mec1/Tel1 kinases during normal S phase. Approximately 50% of these substrates are Rad53-independent, and their phosphorylation is not further induced by HU or DNA damage by methylmethane sulfonate (MMS) [20]. Therefore, it stands to reason that these protein substrates of Mec1/Tel1 might define the essential function of the Mec1 kinase during normal growth (more on this later).
2.1.2. Regulation of Origins of Replication and Replication Forks
As alluded to above, the essential function of the Mec1/Rad53 checkpoint may vary depending on the growth conditions. During replication stress, it is thought that the checkpoint is essential for the preservation of the integrity of the replication fork, facilitated by maintaining a critical level of dNTP pools. It was also demonstrated that HU- or MMS-treated mec1 or rad53 cells fail to inhibit late origin activation, which is considered as another underlying cause of cell death [21,22]. Consistently, checkpoint-deficient cells that sustained irreparable UV damage also activate late origins prematurely during DNA synthesis and lose viability [23]. Together, these studies cemented the notion that premature origin activation during replication stress is detrimental to genome stability. Subsequent studies have revealed mechanisms through which the checkpoint imparts an inhibitory signal to late origins: via two initiation factors—Sld3 and Dbf4 proteins—that are subject to phosphorylation by Mec1/Rad53 and the Cdc7 kinase, respectively [24,25]. It was shown that regulatory domains of the Mcm4 helicase subunit also play a role in the control of late origin activation [26]. A recent study that combined mutations in all three substrates (Sld3, Dbf4, and Mcm4)—rendering them refractory to checkpoint control—showed a global activation of late origins at a similar level as that in the mec1 or rad53 checkpoint mutant [27]. However, the late origin activation phenotype in the triple mutant is only elicited by HU treatment, suggesting that the level of premature late origin activation is not critical enough to jeopardize cell viability during normal growth.
The unrestrained late origin firing in checkpoint mutants during replication stress is accompanied by defective replication fork progression. The function of Mec1/ATR at stalled replication forks during replication stress has been the subject of several comprehensive reviews [28,29,30,31,32]. It is now generally accepted that the absence of checkpoint functions leads to a global level of replication fork collapse, such that the forks are not capable of resumption following the removal of replication stress. The molecular insight into the anatomy of a “collapsed replication fork” was first provided by a seminal study demonstrating extensive single-stranded DNA (ssDNA) accumulation at the replication fork in a rad53-K227A kinase-deficient mutant following replication stress by HU [33]. Concurrently, it was shown by another influential study that conditionally lethal mec1 mutants exhibit breakage at specific regions of the chromosome, akin to the formation of mammalian chromosome fragile sites [34]. Subsequent studies confirmed the formation of fork-associated ssDNA in both rad53 and mec1 mutants upon replication stress, and provided genomic views of the ssDNA at origins of replication [35,36]. It was also shown that the ssDNA, when bound by the eukaryotic ssDNA-binding protein RPA (Replication protein A), constitutes the signal to recruit Mec1/ATR to the replication fork and trigger the signaling cascade [37]. Moreover, it was demonstrated that ssDNA formation at a replication fork destines the fork to DNA double strand breaks and fragile site formation, as previously seen in the mec1 conditional mutant [38].
However, the exact nature of the protein composition and possible transformation at the collapsed fork is still not clear. Previous studies have suggested that replisome stability is compromised in checkpoint mutants when the replication fork is impeded [39,40]. In contrast, recent evidence argues that the replisome components are largely intact in both mec1 and rad53 mutants [41]. Interestingly, in HU-treated human cells, ATR inhibition resulted in genome instability without destabilizing the replisome, but instead involved altered recruitment of other fork-associated proteins [42]. Consistent with this notion, a recent yeast study demonstrated that two DNA helicases involved in replication fork restart—Rrm3 or Pif1—are differentially clustered at replication forks, with a higher retention of Pif1 than Rrm3 [43]. Moreover, removal of either Pif1 or Rrm3 rescues cell lethality in rad53 cells treated with HU [43]. These observations demonstrated an altered architecture of the replication fork during replication stress, and suggested that the restoration to the normal architecture is key to the maintenance of a stalled fork. Rrm3 and Pif1 are both regulated by Mec1/Rad53–mediated phosphorylation, and a phosphor-mimic rrm3-6SD mutant can rescue the phenotypes of the rad53 mutant during replication stress [43]. It would be interesting to test if the altered recruitment of fork-associated proteins is also recapitulated in yeast. It would also be important to determine what dynamic changes might occur behind a stressed replication fork (Figure 1). Future studies could be directed towards comparative analysis of the full architecture of a stalled replication fork vs. a normal one in yeast by capitalizing on a mini-chromosome purification system previously described [44] or similar methods.
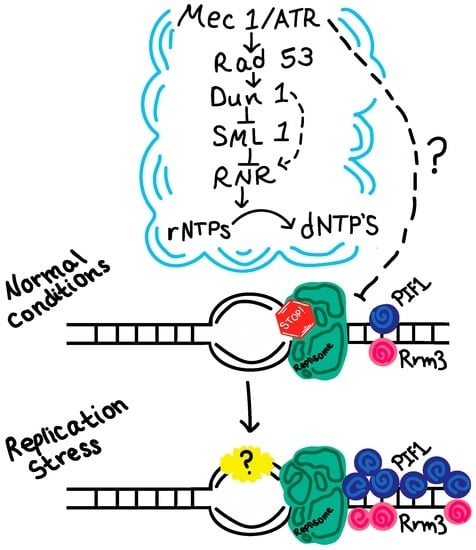
Figure 1.
Schematic representation of replication fork protein dynamics during replication stress. A recent study showing the differential retention of two stressed fork-associated helicases, Pif1 and Rrm3, is featured here. The question mark denotes what future investigation should aim to reveal—potential dynamic changes occurring behind the fork during replication stress. rNTP: ribonucleotide; dNTP: deoxyribonucleotide; Mec1/ATR: Mitosis entry checkpoint 1/ataxia telangiectasia and Rad3-related protein; Rad53: radiation sensitive mutant 53; Dun1: DNA damage uninducible 1, transcriptional inhibitor of SML1; SML1: suppressor of MEC1 lethality 1, inhibitor of RNR; RNR: ribonucleotide reductase.
2.2. RNA Metabolism
2.2.1. RNA Processing during Damage-Sensing in the Checkpoint Pathway
Activation of the Mec1/ATR kinase involves recruitment of the protein to the chromatin, at DNA double strand break (DSB) sites [45,46], at stalled replication forks [37], or at shortened telomeres [47,48]. In the cases of DSBs and stalled forks, RPA-coated ssDNA activates Mec1/ATR, making DNA intermediates the key molecule at the center of checkpoint signaling. However, increasing evidence also places RNA molecules in this pathway. Indeed, there is a clear interplay between pre-mRNA processing and the checkpoint response in metazoans, as reviewed [49].
How does RNA processing play a role in damage sensing of a checkpoint response? It was recently shown that the RNA decay factors in yeast—Xrn1, Rrp6, and Trf4—are important for DSB-sensing as they promote the formation of RPA-ssDNA [50]. The precise function of these RNA processing factors at the damaged DNA site is unknown. Clearing the DNA template for possible DNA:RNA hybrid molecules—also known as the co-transcriptional R loops—does not appear to be the reason, because increased production of RNase H1 (which degrades DNA:RNA hybrids) does not promote RPA-ssDNA formation in the absence of Rrp6 or Trf4 [50]. However, this result does not exclude the possibility that RNA molecules in contexts other than DNA:RNA hybrid might be responsible for the blockage of RPA-ssDNA formation. For instance, an aberrant mRNP (messenger ribonucleoprotein) particle may be obstructing the damaged site. This hypothesis is substantiated by recent findings that Rrp6 plays an important role in the quality control of specific mRNPs [51,52]. Moreover, a previous proteomic analysis revealed a multitude of interactions between RPA and the chromatin remodeling proteins, including Ino80, Isw1, Isw2, Swic, Rsc2, and SWI/SNF [53]. The question then becomes “do these proteins play a role in promoting RPA-ssDNA formation at the damaged fork?” Indeed, there has been some evidence suggesting that in mammalian cells, chromatin remodeling factors such as INO80 facilitate RPA-ssDNA formation during DSB processing [54,55]. Finally, it would also be interesting to determine if RNA processing plays a role in the detection of RPA-ssDNA in the context of a stalled fork.
2.2.2. Transcription Regulation in cis of DNA Damage or Stalled Forks
It has been documented that in mammalian cells, both RNA Pol I- and Pol II-mediated transcriptional silencing/inhibition occurs in the vicinity of damaged DNA (e.g., at induced DSBs), in an ATM-dependent manner [56,57,58]. Similarly, ATR is responsible for transcription repression at clusters of stalled replication forks induced by doxorubicin [59]. However, this checkpoint-dependent transcriptional inhibition response is contentious in yeast, at least at an induced DSB [60]. Yet, in both mammalian cells and yeast, Mec1/ATR and Tel1/ATM phosphorylate histone H2AX at a C-terminal serine to generate gamma-H2AX, thereby causing changes in the chromatin environment at the damaged DNA site [61,62,63,64]. Whether this chromatin remodeling is the cause of transcription inhibition or merely the reflection of the latter is not clear. Therefore, the exact role of the checkpoint in transcription silencing at a DSB site still warrants further investigation.
Proteomic studies in mammalian and yeast systems both identified components of the hnRNP (heterogeneous nuclear ribonucleoprotein) complex as substrates of the Mec1/ATR kinase during replication stress [65,66]. A recent study identified 115 peptides, represented by 71 genes, as Mec1/Tel1- and Rad53-dependent substrates during replication stress [20]. The molecular functions of these genes are enriched in DNA replication and response to DNA damage, as expected [20]. In addition, this gene group is also enriched for those in regulation of transcription [GO:6355], chromatin silencing [GO:6348, 30466], and mRNA transport [GO:51028] (p = 9.44 × 10−7, 2.39 × 10−6, 3.15 × 10−5, and 3.25 × 10−5, respectively). Moreover, as mentioned earlier, 117 peptides represented by 81 genes were identified as Mec1/Tel1-dependent and Rad53-independent substrates in normal S phase, and they are highly enriched for genes involved in transcription, chromatin remodeling, and RNA processing [20]. These findings therefore invite the hypothesis that Mec1/Tel1/Rad53 checkpoint proteins play a role in regulating gene transcription and related activities both during normal DNA synthesis and upon replication stress (see more below).
2.3. The Interface between DNA and RNA Metabolism—Resolving Replication and Transcription Conflicts
The functions of Mec1/ATR in the pathways described above naturally necessitate the checkpoint function at the junction between DNA replication and gene transcription, which share the same chromosome template. Indeed, these two processes can be in physical conflict when the DNA and RNA polymerase complexes are stalled for various reasons [67]. Notably, a progressing replication fork can encounter a Pol II complex blocked by stable R-loop formation. As alluded to before, R-loops are co-transcriptional structures defined by a hybrid between the nascent RNA transcript and one of the DNA template strands, leaving the other DNA strand exposed as single-stranded [68,69,70,71]. It is thought that stable R-loop formation could impede the replication machinery, triggering both homologous recombination and non-homologous end-joining, suggesting that these sites have undergone DSB formation [72,73,74,75,76,77,78]. Thus, stable R-loop formation impedes replication forks and is a detriment to genome stability.
Replication–transcription conflicts can also originate from a defective replication fork encountering unscheduled transcription activities, particularly during induced replication stress. My laboratory recently showed that the replication inhibitor HU can simultaneously stall replication forks and induce unscheduled gene expression, leading to chromosome breakage [79]. Our study provides an explanation for why different replication inhibitors can produce distinct chromosome breakage patterns—it is the result of differential sites of replication–transcription conflicts dependent on the drug-specific gene expression profiles. Consistent with this notion, it was recently shown that estrogen-induced DSBs occur where replication encounters estrogen-responsive genes [80]. These studies thus highlighted the importance of understanding the gene expression profiles of replication inhibitors, which are widely present in the environment and are commonly used in medical practices (e.g., anti-cancer drugs).
What is the role of Mec1/ATR in preventing and/or resolving conflicts between replication and transcription? This topic has recently been extensively reviewed [81]. Here I summarize the two broad aspects of Mec1/ATR’s function in this process reported so far: maintaining fork stability as discussed earlier, and eviction of the transcription complex. In the absence of Mec1 (in a mec1Δ sml1Δ mutant) the replication fork produces extensive ssDNA at replication forks, and ultimately leads to DSBs [38]. However, does Mec1/ATR also exert any function on the transcription complex? A recent study illuminated the other side of the coin, so to speak, by presenting a novel function of Mec1 in removing RNA Pol II from the template to preserve replication fork integrity when replication and transcription are in conflict [82]. In this specific capacity, Mec1 forms a complex with the chromatin remodeling factors Ino80 and Paf1, where Ino80 serves as a substrate—possibly at Ser51 and Thr568—for Mec1 [82]. It stands to reason that Mec1 can also complex with other proteins to modulate the replication fork proteins during replication–transcription conflict. This study also underscores the importance of proteomic studies in identifying novel Mec1-interacting proteins and checkpoint functions. These findings are depicted in a juxtaposition of DNA replication and transcription approaching each other in a head-on configuration (Figure 2).
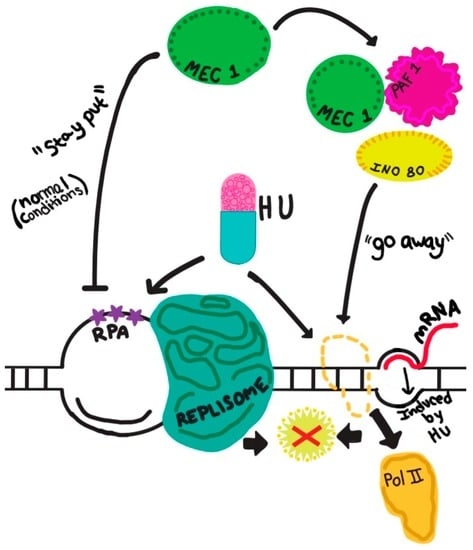
Figure 2.
Schematic representation of converging replication and transcription, endangering the chromosome template for DNA double strand breaks (depicted by a red cross). The dual effects of a replication inhibitor, (e.g., hydroxyurea, HU) simultaneously impacting replication and transcription (shown by two arrows descending from “HU”) are described in the main text. The precise function of Mec1 in the protection of a stressed (e.g., by HU) fork is yet to be defined, and is depicted as a “stay put” signal, which likely also operates during a normal S phase. The inhibitory nature of the signal is sheer speculation at present. The active removal of RNA Pol II by the Mec1-Ino80-Paf1 complex during replication–transcription conflict is featured here. Other novel protein complexes involving Mec1 await future discoveries. RPA: replication protein A, the ssDNA-binding protein.
One of the hurdles arising from replication–transcription conflict is the torsional stress (positive and negative supercoiling) generated in the chromosomal DNA. Many pathways are involved in the prevention of accumulated DNA torsional stress, including (but not limited to) mRNP biogenesis (THO/TREX complex), template unwinding (Rrm3 helicase), chromatin remodeling (FACT complexes), R-loop prevention (RNase H, topoisomerase I, MCM helicase, etc.), and gene gating (nuclear pore complexes) [83,84,85,86,87,88]. Of note, Mec1/ATR is important for severing the actively transcribed genes from the tethered nuclear pore complex during replication–transcription conflicts, representing yet another solution to protect replication forks [87]. Many proteins in these pathways are substrates of Mec1/ATR [20]. For instance, seven nuclear pore complex proteins (Nup60, Gle1, Yrb2, Mlp1, Nup2, Nup188, and Nup1) and ten chromatin silencing factors (Fun30, Mrc1, Sum1, Ino80, Nup2, Spt21, Esc1, Rlf2, Net1, and Top1) were identified as Mec1/Tel1/Rad53-dependent substrates during replication stress from the study by Bastos de Oliveira et al. In addition, four proteins in the mRNA export pathway (Hpr1, Yra1, Sgf73, and Thp2) are Mec1/Tel1-dependent and Rad53-independent substrates in normal S phase. Mutation studies designed to probe the molecular functions of the checkpoint-dependent phosphorylation of these proteins will shed new light on this pathway.
2.4. Other Specialized Tasks
2.4.1. Dealing with Mechanical Stress
The Mec1/ATR-mediated function at the nuclear periphery described above apparently goes beyond the capacity of resolving replication–transcription conflicts. It has been shown that Mec1/ATR also regulates a pathway that senses general stress to the nuclear envelope, produced either by torsional stress of the chromosomal DNA in the processes discussed above, or through osmotic pressure and mechanical force [89]. All these stimuli can increase the location of Mec1/ATR at the nuclear envelope, where it regulates chromatin condensation and nuclear envelope breakdown. It is an exciting property of the Mec1/ATR kinase, and the players involved in this signaling pathway will prove interesting, as mechanical stress was induced without incurring DNA damage. It will also be interesting to determine to what extent the Mec1/ATR substrates in the mechanical stress pathway overlap with those in other stress–response pathways.
2.4.2. Dealing with Nucleolar Stress
Recent studies have demonstrated that ATR/ATM-mediated DNA damage response results in transcriptional regulation and organized DSB repair in the nucleolus, as reviewed in [90]. Indeed, ATM was observed to be localized specifically to the nuclear caps following DNA damage [91]. As a response to nuclear envelope stress described above, ATR was also seen localized in the nucleolus [89]. It has been shown that in response to chromosome breaks, the ATM pathway inhibits Pol I transcription at the ribosomal DNA (rDNA) loci in mouse embryonic fibroblasts [56]. Analogous to the eviction of RNA Pol II by Mec1-Ino80-Paf1, ATM activity was shown to be important for displacing the RNA Pol I elongating complex [56]. Does ATM mediate Pol I transcription directly or through an intermediary? Recent studies showed that ATM signaling following DNA damage triggers Pol I silencing through the interaction between the Nijmegen breakage syndrome (NBS) protein and a nucleolar factor TCOF1-Treacle [92,93]. These studies suggested that the ATM-mediated DNA damage signaling is capable of propagating in-trans through nuclear compartments. The precise mechanism through which nucleolar NBS in conjunction with Treacle causes Pol I inhibition remains to be determined. It is also important to understand the significance of Pol I transcription inhibition in the presence of genomic DNA damage.
2.4.3. Nutrient Sensing
The Mec1 signaling pathway can also cooperate with the nutrient response pathways in yeast. For instance, when cells grown on a non-fermentable carbon source receive glucose, it triggers a transient peak of cyclic adenosine monophosphate (cAMP) production through the Ras pathway, which in turn stimulates the cAMP-dependent protein kinase A (PKA) activity and drives S phase progression [94]. It is thought that this PKA response is also important for restraining mitosis if the daughter cell has not reached a critical size [95]. This negative regulation of cell cycle progression by PKA is apparently exploited by the Mec1-mediated checkpoint pathway in response to DNA damage [96]. Subsequent investigation revealed that Mec1 directly phosphorylates the regulatory subunits of PKA, thereby activating the catalytic subunit of the kinase [97]. Similar to this partnership with the PKA pathway, Mec1/Tel1 can also draw on other substrates in the glucose-sensing pathway, such as Snf1 (the AMP-dependent kinase), and by down-regulating Snf1 steer cells towards aerobic fermentation instead of respiration [98]. It was proposed that the Mec1-mediated DNA damage response produces a cellular decision analogous to the Warburg effect in cancer cells [98].
3. Concluding Remarks
The vast range of the Mec1/ATR-mediated signaling pathways precludes a thorough coverage in this review. Here I highlighted some of the recent studies describing Mec1/ATR’s roles in nucleic acid metabolisms. From these studies, we can glean several key features of the Mec1/ATR protein function. First, it appears that Mec1/ATR functions modularly and complexes with different proteins in specific cellular contexts to exploit existent pathways, or to acquire new functions. Second, RNA molecules play increasingly more complex roles in chromosomal DNA transactions. With the discovery of new substrates of the Mec1/ATR during a normal cell cycle, or when cells are under stress, we will continue to discover new genes and molecules in the crossroads of DNA replication and gene expression. Finally, there are certain topics in the Mec1/ATR-associated biology that are not covered in detail here. For instance, the list of activators of Mec1/ATR continues to expand, one of the latest examples being that the SWI/SNF chromatin remodeling complex specifically regulates Mec1 kinase activity during S phase, independent of the known regulators of Mec1 such as Dpb11 [99]. Though the precise function of this regulation is not clear, one can envisage yet another layer of complexity in the Mec1-mediated pathways in response to the SWI/SNF-regulated processes.
Acknowledgments
I thank Madeline Clark for providing artistic rendition of the Mec1-mediated pathways in the figures and the Feng lab members for general support. I am also grateful to the anonymous reviewers who provided critical insights during revision of this manuscript. This work was supported by the National Institutes of Health (5R01 GM118799-01A1) to W.F.
Conflicts of Interest
The author declare no conflict of interest.
References
- Weinert, T.A.; Kiser, G.L.; Hartwell, L.H. Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes Dev. 1994, 8, 652–665. [Google Scholar] [CrossRef] [PubMed]
- Nyberg, K.A.; Michelson, R.J.; Putnam, C.W.; Weinert, T.A. Toward maintaining the genome: DNA damage and replication checkpoints. Annu. Rev. Genet. 2002, 36, 617–656. [Google Scholar] [CrossRef] [PubMed]
- Morrow, D.M.; Tagle, D.A.; Shiloh, Y.; Collins, F.S.; Hieter, P. TEL1, an S. cerevisiae homolog of the human gene mutated in ataxia telangiectasia, is functionally related to the yeast checkpoint gene MEC1. Cell 1995, 82, 831–840. [Google Scholar] [CrossRef]
- Sawicka, M.; Wanrooij, P.H.; Darbari, V.C.; Tannous, E.; Hailemariam, S.; Bose, D.; Makarova, A.V.; Burgers, P.M.; Zhang, X. The dimeric architecture of checkpoint kinases Mec1ATR and Tel1ATM reveal a common structural organization. J. Biol. Chem. 2016, 291, 13436–13447. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.B.; Zhou, Z.; Siede, W.; Friedberg, E.C.; Elledge, S.J. The SAD1/RAD53 protein kinase controls multiple checkpoints and DNA damage-induced transcription in yeast. Genes Dev. 1994, 8, 2401–2415. [Google Scholar] [CrossRef] [PubMed]
- Desany, B.A.; Alcasabas, A.A.; Bachant, J.B.; Elledge, S.J. Recovery from DNA replicational stress is the essential function of the S-phase checkpoint pathway. Genes Dev. 1998, 12, 2956–2970. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Georgieva, B.; Chabes, A.; Domkin, V.; Ippel, J.H.; Schleucher, J.; Wijmenga, S.; Thelander, L.; Rothstein, R. Mutational and structural analyses of the ribonucleotide reductase inhibitor Sml1 define its Rnr1 interaction domain whose inactivation allows suppression of mec1 and rad53 lethality. Mol. Cell. Biol. 2000, 20, 9076–9083. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, Y.; Desany, B.A.; Jones, W.J.; Liu, Q.H.; Wang, B.; Elledge, S.J. Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science 1996, 271, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Rothstein, R. The Dun1 checkpoint kinase phosphorylates and regulates the ribonucleotide reductase inhibitor Sml1. Proc. Natl. Acad. Sci. USA 2002, 99, 3746–3751. [Google Scholar] [CrossRef] [PubMed]
- Basrai, M.A.; Velculescu, V.E.; Kinzler, K.W.; Hieter, P. NORF5/HUG1 is a component of the MEC1-mediated checkpoint response to DNA damage and replication arrest in Saccharomyces cerevisiae. Mol. Cell. Biol. 1999, 19, 7041–7049. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Zhou, Z.; Elledge, S.J. The DNA replication and damage checkpoint pathways induce transcription by inhibition of the Crt1 repressor. Cell 1998, 94, 595–605. [Google Scholar] [CrossRef]
- Wu, X.; Huang, M. Dif1 controls subcellular localization of ribonucleotide reductase by mediating nuclear import of the R2 subunit. Mol. Cell. Biol. 2008, 28, 7156–7167. [Google Scholar] [CrossRef] [PubMed]
- Earp, C.; Rowbotham, S.; Merényi, G.; Chabes, A.; Cha, R.S. S phase block following MEC1ATR inactivation occurs without severe dNTP depletion. Biol. Open 2015, 4, 1739–1743. [Google Scholar] [CrossRef] [PubMed]
- Fasullo, M.; Tsaponina, O.; Sun, M.; Chabes, A. Elevated dNTP levels suppress hyper-recombination in Saccharomyces cerevisiae S-phase checkpoint mutants. Nucleic Acids Res. 2010, 38, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Chabes, A.; Georgieva, B.; Domkin, V.; Zhao, X.; Rothstein, R.; Thelander, L. Survival of DNA damage in yeast directly depends on increased dNTP levels allowed by relaxed feedback inhibition of ribonucleotide reductase. Cell 2003, 112, 391–401. [Google Scholar] [CrossRef]
- Weinberg, G.; Ullman, B.; Martin, D.W., Jr. Mutator phenotypes in mammalian cell mutants with distinct biochemical defects and abnormal deoxyribonucleoside triphosphate pools. Proc. Natl. Acad. Sci. USA 1981, 78, 2447–2451. [Google Scholar] [CrossRef] [PubMed]
- Guarino, E.; Salguero, I.; Kearsey, S.E. Cellular regulation of ribonucleotide reductase in eukaryotes. Semin. Cell Dev. Biol. 2014, 30, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Chabes, A.; Domkin, V.; Thelander, L.; Rothstein, R. The ribonucleotide reductase inhibitor Sml1 is a new target of the Mec1/Rad53 kinase cascade during growth and in response to DNA damage. EMBO J. 2001, 20, 3544–3553. [Google Scholar] [CrossRef] [PubMed]
- Hoch, N.C.; Chen, E.S.-W.; Buckland, R.; Wang, S.-C.; Fazio, A.; Hammet, A.; Pellicioli, A.; Chabes, A.; Tsai, M.-D.; Heierhorst, J. Molecular basis of the essential s phase function of the rad53 checkpoint kinase. Mol. Cell. Biol. 2013, 33, 3202–3213. [Google Scholar] [CrossRef] [PubMed]
- Bastos de Oliveira, F.M.; Kim, D.; Cussiol, J.R.; Das, J.; Jeong, M.C.; Doerfler, L.; Schmidt, K.H.; Yu, H.; Smolka, M.B. Phosphoproteomics reveals distinct modes of Mec1/ATR signaling during DNA replication. Mol. Cell 2015, 57, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- Santocanale, C.; Diffley, J.F. A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature 1998, 395, 615–618. [Google Scholar] [PubMed]
- Shirahige, K.; Hori, Y.; Shiraishi, K.; Yamashita, M.; Takahashi, K.; Obuse, C.; Tsurimoto, T.; Yoshikawa, H. Regulation of DNA-replication origins during cell-cycle progression. Nature 1998, 395, 618–621. [Google Scholar] [PubMed]
- Neecke, H.; Lucchini, G.; Longhese, M.P. Cell cycle progression in the presence of irreparable DNA damage is controlled by a Mec1- and Rad53-dependent checkpoint in budding yeast. EMBO J. 1999, 18, 4485–4497. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Mosqueda, J.; Maas, N.L.; Jonsson, Z.O.; DeFazio-Eli, L.G.; Wohlschlegel, J.; Toczyski, D.P. Damage-induced phosphorylation of Sld3 is important to block late origin firing. Nature 2010, 467, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Zegerman, P.; Diffley, J.F. Checkpoint-dependent inhibition of DNA replication initiation by Sld3 and Dbf4 phosphorylation. Nature 2010, 467, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Sheu, Y.J.; Kinney, J.B.; Lengronne, A.; Pasero, P.; Stillman, B. Domain within the helicase subunit Mcm4 integrates multiple kinase signals to control DNA replication initiation and fork progression. Proc. Natl. Acad. Sci. USA 2014, 111, E1899–E1908. [Google Scholar] [CrossRef] [PubMed]
- Sheu, Y.J.; Kinney, J.B.; Stillman, B. Concerted activities of Mcm4, Sld3, and Dbf4 in control of origin activation and DNA replication fork progression. Genome Res. 2016, 26, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Foiani, M. Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell. Biol. 2010, 11, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Friedel, A.M.; Pike, B.L.; Gasser, S.M. ATR/Mec1: Coordinating fork stability and repair. Curr. Opin. Cell Biol. 2009, 21, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Jossen, R.; Bermejo, R. The DNA damage checkpoint response to replication stress: A Game of Forks. Front. Genet. 2013, 4, 26. [Google Scholar] [CrossRef] [PubMed]
- Nam, E.A.; Cortez, D. ATR signalling: More than meeting at the fork. Biochem. J. 2011, 436, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Yazinski, S.A.; Zou, L. Functions, Regulation, and Therapeutic Implications of the ATR Checkpoint Pathway. Annu. Rev. Genet. 2016, 50, 155–173. [Google Scholar] [CrossRef] [PubMed]
- Sogo, J.M.; Lopes, M.; Foiani, M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 2002, 297, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Cha, R.S.; Kleckner, N. ATR homolog Mec1 promotes fork progression, thus averting breaks in replication slow zones. Science 2002, 297, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Bachant, J.; Collingwood, D.; Raghuraman, M.K.; Brewer, B.J. Centromere replication timing determines different forms of genomic instability in Saccharomyces cerevisiae checkpoint mutants during replication stress. Genetics 2009, 183, 1249–1260. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Collingwood, D.; Boeck, M.E.; Fox, L.A.; Alvino, G.M.; Fangman, W.L.; Raghuraman, M.K.; Brewer, B.J. Genomic mapping of single-stranded DNA in hydroxyurea-challenged yeasts identifies origins of replication. Nat. Cell Biol. 2006, 8, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Di Rienzi, S.C.; Raghuraman, M.K.; Brewer, B.J. Replication stress-induced chromosome breakage is correlated with replication fork progression and is preceded by single-stranded DNA formation. G3 2011, 1, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Cobb, J.A.; Schleker, T.; Rojas, V.; Bjergbaek, L.; Tercero, J.A.; Gasser, S.M. Replisome instability, fork collapse, and gross chromosomal rearrangements arise synergistically from Mec1 kinase and RecQ helicase mutations. Genes Dev. 2005, 19, 3055–3069. [Google Scholar] [CrossRef] [PubMed]
- Lucca, C.; Vanoli, F.; Cotta-Ramusino, C.; Pellicioli, A.; Liberi, G.; Haber, J.; Foiani, M. Checkpoint-mediated control of replisome-fork association and signalling in response to replication pausing. Oncogene 2004, 23, 1206–1213. [Google Scholar] [CrossRef] [PubMed]
- De Piccoli, G.; De Piccoli, G.; Katou, Y.; Itoh, T.; Nakato, R.; Shirahige, K.; Labib, K. Replisome stability at defective DNA replication forks is independent of S phase checkpoint kinases. Mol. Cell 2012, 45, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Dungrawala, H.; Rose, K.L.; Bhat, K.P.; Mohni, K.N.; Glick, G.G.; Couch, F.B.; Cortez, D. The Replication Checkpoint Prevents Two Types of Fork Collapse without Regulating Replisome Stability. Mol. Cell 2015, 59, 998–1010. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.E.; Ajazi, A.; Carotenuto, W.; Foiani, M.; Giannattasio, M. Rad53-Mediated Regulation of Rrm3 and Pif1 DNA Helicases Contributes to Prevention of Aberrant Fork Transitions under Replication Stress. Cell Rep. 2015, 13, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Unnikrishnan, A.; Akiyoshi, B.; Biggins, S.; Tsukiyama, T. An efficient purification system for native minichromosome from Saccharomyces cerevisiae. Methods Mol. Biol. 2012, 833, 115–123. [Google Scholar] [PubMed]
- Nakada, D.; Hirano, Y.; Tanaka, Y.; Sugimoto, K. Role of the C terminus of Mec1 checkpoint kinase in its localization to sites of DNA damage. Mol. Biol. Cell 2005, 16, 5227–5235. [Google Scholar] [CrossRef] [PubMed]
- Dubrana, K.; van Attikum, H.; Hediger, F.; Gasser, S.M. The processing of double-strand breaks and binding of single-strand-binding proteins RPA and Rad51 modulate the formation of ATR-kinase foci in yeast. J. Cell Sci. 2007, 120, 4209–4220. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhao, R.; Glick, G.G.; Cortez, D. Function of the ATR N-terminal domain revealed by an ATM/ATR chimera. Exp. Cell Res. 2007, 313, 1667–1674. [Google Scholar] [CrossRef] [PubMed]
- Takata, H.; Kanoh, Y.; Gunge, N.; Shirahige, K. Akira Matsuura Reciprocal association of the budding yeast ATM-related proteins Tel1 and Mec1 with telomeres in vivo. Mol. Cell 2004, 14, 515–522. [Google Scholar] [CrossRef]
- Montecucco, A.; Biamonti, G. Pre-mRNA processing factors meet the DNA damage response. Front. Genet. 2013, 4, 102. [Google Scholar] [CrossRef] [PubMed]
- Manfrini, N.; Trovesi, C.; Wery, M.; Martina, M.; Cesena, D.; Descrimes, M.; Morillon, A.; d’Adda di Fagagna, F.; Longhese, M.P. RNA-processing proteins regulate Mec1/ATR activation by promoting generation of RPA-coated ssDNA. EMBO Rep. 2015, 16, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Mosrin-Huaman, C.; Hervouet-Coste, N.; Rahmouni, A.R. Co-transcriptional degradation by the 5′-3′ exonuclease Rat1p mediates quality control of HXK1 mRNP biogenesis in S. cerevisiae. RNA Biol. 2016, 13, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Stuparevic, I.; Mosrin-Huaman, C.; Hervouet-Coste, N.; Remenaric, M.; Rahmouni, A.R. Cotranscriptional recruitment of RNA exosome cofactors Rrp47p and Mpp6p and two distinct Trf-Air-Mtr4 polyadenylation (TRAMP) complexes assists the exonuclease Rrp6p in the targeting and degradation of an aberrant messenger ribonucleoprotein particle (mRNP) in yeast. J. Biol. Chem. 2013, 288, 31816–31829. [Google Scholar] [PubMed]
- Chen, S.H.; Albuquerque, C.P.; Liang, J.; Suhandynata, R.T.; Zhou, H. A proteome-wide analysis of kinase-substrate network in the DNA damage response. J. Biol. Chem. 2010, 285, 12803–12812. [Google Scholar] [CrossRef] [PubMed]
- Gospodinov, A.; Vaissiere, T.; Krastev, D.B.; Legube, G.; Anachkova, B.; Herceg, Z. Mammalian Ino80 mediates double-strand break repair through its role in DNA end strand resection. Mol. Cell. Biol. 2011, 31, 4735–4745. [Google Scholar] [CrossRef] [PubMed]
- Vassileva, I.; Yanakieva, I.; Peycheva, M.; Gospodinov, A.; Anachkova, B. The mammalian INO80 chromatin remodeling complex is required for replication stress recovery. Nucleic Acids Res. 2014, 42, 9074–9086. [Google Scholar] [CrossRef] [PubMed]
- Kruhlak, M.; Crouch, E.E.; Orlov, M.; Montaño, C.; Gorski, S.A.; Nussenzweig, A.; Misteli, T.; Phair, R.D.; Casellas, R. The ATM repair pathway inhibits RNA polymerase I transcription in response to chromosome breaks. Nature 2007, 447, 730–734. [Google Scholar] [CrossRef] [PubMed]
- Shanbhag, N.M.; Rafalska-Metcalf, I.U.; Balane-Bolivar, C.; Janicki, S.M.; Greenberg, R.A. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 2010, 141, 970–981. [Google Scholar] [CrossRef] [PubMed]
- Harding, S.M.; Boiarsky, J.A.; Greenberg, R.A. ATM Dependent Silencing Links Nucleolar Chromatin Reorganization to DNA Damage Recognition. Cell Rep. 2015, 13, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Im, J.S.; Keaton, M.; Lee, K.Y.; Kumar, P.; Park, J.; Dutta, A. ATR checkpoint kinase and CRL1betaTRCP collaborate to degrade ASF1a and thus repress genes overlapping with clusters of stalled replication forks. Genes Dev. 2014, 28, 875–887. [Google Scholar] [CrossRef] [PubMed]
- Manfrini, N.; Clerici, M.; Wery, M.; Colombo, C.V.; Descrimes, M.; Morillon, A.; d’Adda di Fagagna, F.; Longhese, M.P. Resection is responsible for loss of transcription around a double-strand break in Saccharomyces cerevisiae. Elife 2015, 4, e08942. [Google Scholar] [CrossRef] [PubMed]
- Downs, J.A.; Lowndes, N.F.; Jackson, S.P. A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature 2000, 408, 1001–1004. [Google Scholar] [PubMed]
- Burma, S.; Chen, B.P.; Murphy, M.; Kurimasa, A.; Chen, D.J. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 2001, 276, 42462–42467. [Google Scholar] [CrossRef] [PubMed]
- Ward, I.M.; Chen, J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 2001, 276, 47759–47762. [Google Scholar] [PubMed]
- Shroff, R.; Arbel-Eden, A.; Pilch, D.; Ira, G.; Bonner, W.M.; Petrini, J.H.; Haber, J.E.; Lichten, M. Distribution and dynamics of chromatin modification induced by a defined DNA double-strand break. Curr. Biol. 2004, 14, 1703–1711. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., III; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Smolka, M.B.; Albuquerque, C.P.; Chen, S.; Zhou, H. Proteome-wide identification of in vivo targets of DNA damage checkpoint kinases. Proc. Natl. Acad. Sci. USA 2007, 104, 10364–10369. [Google Scholar] [CrossRef] [PubMed]
- Brambati, A.; Colosio, A.; Zardoni, L.; Galanti, L.; Liberi, G. Replication and transcription on a collision course: Eukaryotic regulation mechanisms and implications for DNA stability. Front. Genet. 2015, 6, 166. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; White, R.L.; Davis, R.W. Hybridization of RNA to double-stranded DNA: Formation of R-loops. Proc. Natl. Acad. Sci. USA 1976, 73, 2294–2298. [Google Scholar] [CrossRef] [PubMed]
- Drolet, M.; Phoenix, P.; Menzel, R.; Massé, E.; Liu, L.F.; Crouch, R.J. Overexpression of RNase H partially complements the growth defect of an Escherichia coli delta topA mutant: R-loop formation is a major problem in the absence of DNA topoisomerase I. Proc. Natl. Acad. Sci. USA 1995, 92, 3526–3530. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.; Yu, K.; Lieber, M.R. Mechanism of R-loop formation at immunoglobulin class switch sequences. Mol. Cell. Biol. 2008, 28, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Huertas, P.; Aguilera, A. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol. Cell 2003, 12, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.; Gomez-Gonzalez, B. Genome instability: A mechanistic view of its causes and consequences. Nat. Rev. Genet. 2008, 9, 204–217. [Google Scholar] [CrossRef] [PubMed]
- Gottipati, P.; Cassel, T.N.; Savolainen, L.; Helleday, T. Transcription-associated recombination is dependent on replication in Mammalian cells. Mol. Cell. Biol. 2008, 28, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T. Pathways for mitotic homologous recombination in mammalian cells. Mutat. Res. 2003, 532, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Lo, J.; van Gent, D.C.; Engelward, B.P. DNA double-strand break repair: From mechanistic understanding to cancer treatment. DNA Repair 2007, 6, 923–935. [Google Scholar] [CrossRef] [PubMed]
- Soulas-Sprauel, P.; Rivera-Munoz, P.; Malivert, L.; Le Guyader, G.; Abramowski, V.; Revy, P.; de Villartay, J.-P. V(D)J and immunoglobulin class switch recombinations: A paradigm to study the regulation of DNA end-joining. Oncogene 2007, 26, 7780–7791. [Google Scholar] [CrossRef] [PubMed]
- Gan, W.; Guan, Z.; Liu, J.; Gui, T.; Shen, K.; Manley, J.L.; Li, X. R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev. 2011, 25, 2041–2056. [Google Scholar] [CrossRef] [PubMed]
- Houlard, M.; Artus, J.; Léguillier, T.; Vandormael-Pournin, S.; Cohen-Tannoudji, M. DNA-RNA hybrids contribute to the replication dependent genomic instability induced by Omcg1 deficiency. Cell Cycle 2011, 10, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.A.; McCulley, A.; Haarer, B. Remigiusz Arnak and Wenyi Feng Break-seq reveals hydroxyurea-induced chromosome fragility as a result of unscheduled conflict between DNA replication and transcription. Genome Res. 2015, 25, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Stork, C.T.; Bocek, M.; Crossley, M.P.; Sollier, J.; Sanz, L.A.; Chédin, F.; Swigut, T.; Cimprich, K.A. Co-transcriptional R-loops are the main cause of estrogen-induced DNA damage. Elife 2016, 5, e17548. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Muse, T.; Aguilera, A. Transcription-replication conflicts: How they occur and how they are resolved. Nat. Rev. Mol. Cell. Biol. 2016, 17, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Poli, J.; Gerhold, C.B.; Tosi, A.; Hustedt, N.; Seeber, A.; Sack, R.; Herzog, F.; Pasero, P.; Shimada, K.; Hopfner, K.P.; et al. Mec1, INO80, and the PAF1 complex cooperate to limit transcription replication conflicts through RNAPII removal during replication stress. Genes Dev. 2016, 30, 337–354. [Google Scholar] [CrossRef] [PubMed]
- Luna, R.; Rondon, A.G.; Aguilera, A. New clues to understand the role of THO and other functionally related factors in mRNP biogenesis. Biochim. Biophys. Acta 2012, 1819, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Tuduri, S.; Crabbé, L.; Conti, C.; Tourrière, H.; Holtgreve-Grez, H.; Jauch, A.; Pantesco, V.; De Vos, J.; Thomas, A.; Theillet, C.; et al. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat. Cell Biol. 2009, 11, 1315–1324. [Google Scholar] [CrossRef] [PubMed]
- Azvolinsky, A.; Giresi, P.G.; Lieb, J.D.; Zakian, V.A. Highly transcribed RNA polymerase II genes are impediments to replication fork progression in Saccharomyces cerevisiae. Mol. Cell 2009, 34, 722–734. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Moyano, E.; Mergui, X.; García-Rubio, M.L.; Barroso, S.; Aguilera, A. The yeast and human FACT chromatin-reorganizing complexes solve R-loop-mediated transcription-replication conflicts. Genes Dev. 2014, 28, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Bermejo, R.; Capra, T.; Jossen, R.; Colosio, A.; Frattini, C.; Carotenuto, W.; Cocito, A.; Doksani, Y.; Klein, H.; Gómez-González, B.; et al. The replication checkpoint protects fork stability by releasing transcribed genes from nuclear pores. Cell 2011, 146, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Vijayraghavan, S.; Tsai, F.L.; Schwacha, A. A Checkpoint-Related Function of the MCM Replicative Helicase Is Required to Avert Accumulation of RNA:DNA Hybrids during S-phase and Ensuing DSBs during G2/M. PLoS Genet 2016, 12, e1006277. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Mazzanti, M.; Mistrik, M.; Kosar, M.; Beznoussenko, G.V.; Mironov, A.A.; Garrè, M.; Parazzoli, D.; Shivashankar, G.V.; Scita, G.; et al. ATR mediates a checkpoint at the nuclear envelope in response to mechanical stress. Cell 2014, 158, 633–646. [Google Scholar] [CrossRef] [PubMed]
- Larsen, D.H.; Stucki, M. Nucleolar responses to DNA double-strand breaks. Nucleic Acids Res. 2016, 44, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Van Sluis, M.; McStay, B. A localized nucleolar DNA damage response facilitates recruitment of the homology-directed repair machinery independent of cell cycle stage. Genes Dev. 2015, 29, 1151–1163. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Huang, J.-W.; Izhar, L.; Sowa, M.E.; Harper, J.W.; Elledge, S.J. Treacher Collins syndrome TCOF1 protein cooperates with NBS1 in the DNA damage response. Proc. Natl. Acad. Sci. USA 2014, 111, 18631–18636. [Google Scholar] [CrossRef] [PubMed]
- Larsen, D.H.; Hari, F.; Clapperton, J.A.; Gwerder, M.; Gutsche, K.; Altmeyer, M.; Jungmichel, S.; Toledo, L.I.; Fink, D.; Rask, M.-B.; et al. The NBS1-Treacle complex controls ribosomal RNA transcription in response to DNA damage. Nat. Cell Biol. 2014, 16, 792–803. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.H.; Gasch, A.P. Exploiting the yeast stress-activated signaling network to inform on stress biology and disease signaling. Curr. Genet. 2015, 61, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Anghileri, P.; Branduardi, P.; Sternieri, F.; Monti, P.; Visintin, R.; Bevilacqua, A.; Alberghina, L.; Martegani, E.; Baroni, M.D. Chromosome separation and exit from mitosis in budding yeast: Dependence on growth revealed by cAMP-mediated inhibition. Exp. Cell Res. 1999, 250, 510–523. [Google Scholar] [CrossRef] [PubMed]
- Searle, J.S.; Schollaert, K.L.; Wilkins, B.J.; Sanchez, Y. The DNA damage checkpoint and PKA pathways converge on APC substrates and Cdc20 to regulate mitotic progression. Nat. Cell Biol. 2004, 6, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Searle, J.S.; Wood, M.D.; Kaur, M.; Tobin, D.V.; Sanchez, Y. Proteins in the nutrient-sensing and DNA damage checkpoint pathways cooperate to restrain mitotic progression following DNA damage. PLoS Genet 2011, 7, e1002176. [Google Scholar] [CrossRef] [PubMed]
- Simpson-Lavy, K.J.; Bronstein, A.; Kupiec, M.; Johnston, M. Cross-Talk between Carbon Metabolism and the DNA Damage Response in S. cerevisiae. Cell Rep. 2015, 12, 1865–1875. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, P.; Bao, Y.; Xiao, J.; Luo, J.; Shen, J.; Persinger, J.; Peng, G.; Ranish, J.; Bartholomew, B.; Shen, X. Regulation of Mec1 kinase activity by the SWI/SNF chromatin remodeling complex. Genes Dev. 2015, 29, 591–602. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).