
Functions of the RNA Editing Enzyme ADAR1 and Their Relevance to Human Diseases


Abstract
:1. Introduction
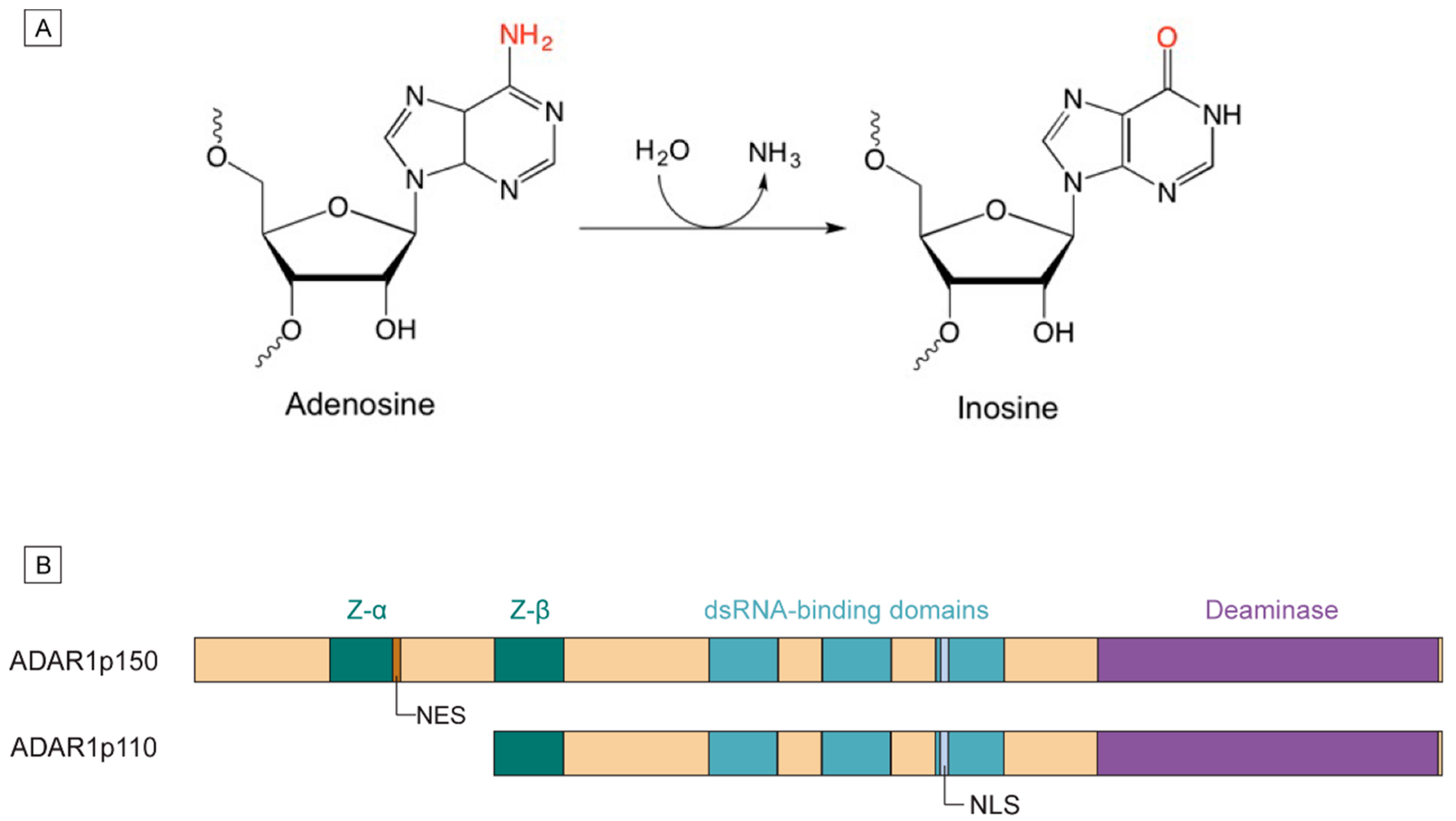
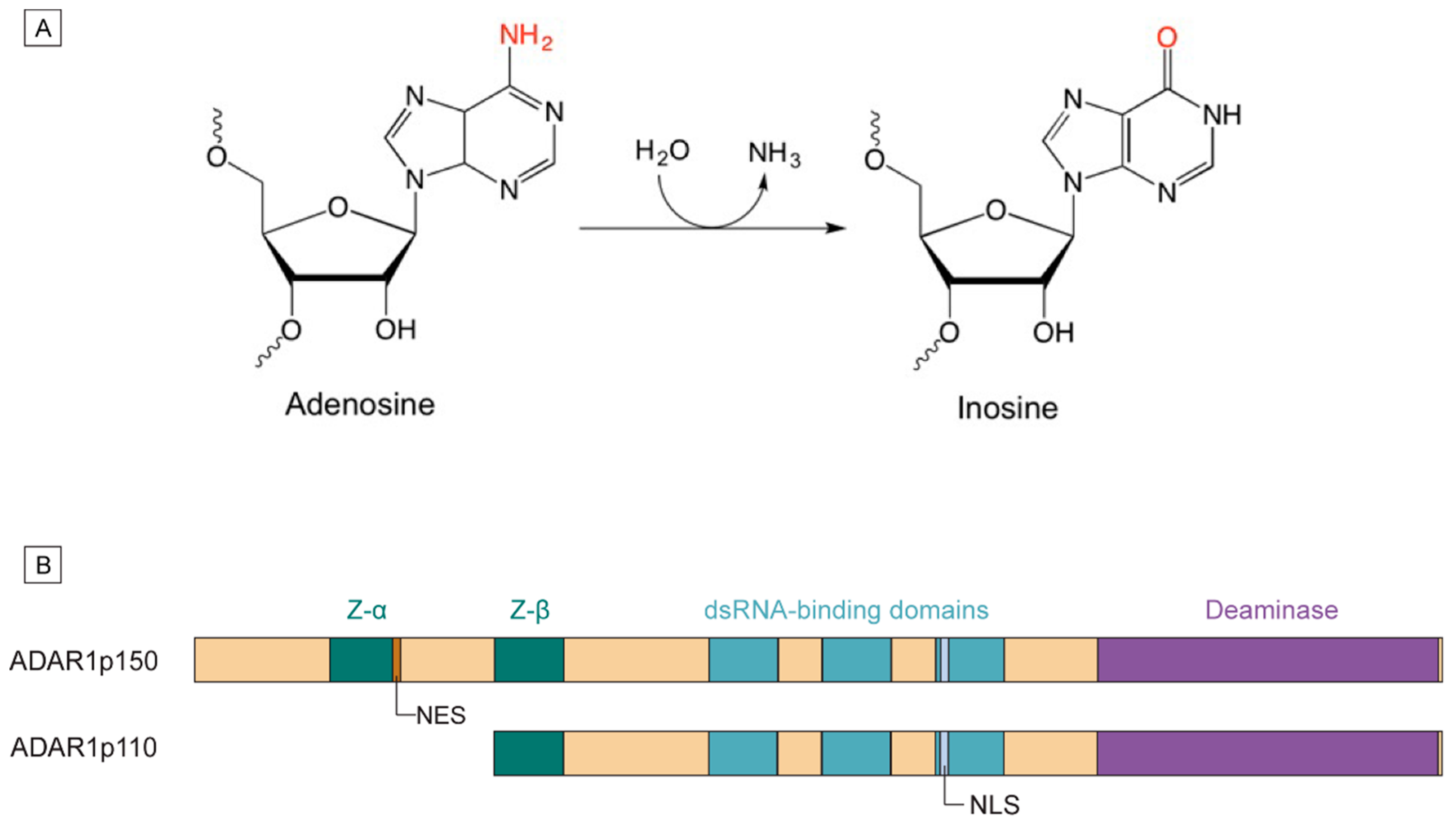
1.1. Mechanism of A-to-I Editing
1.2. ADAR1 Isoforms and Domain Structures
2. ADAR1′s Role in Immunity
2.1. Hematopoiesis
2.2. Type I Interferonopathies
3. Editing of Exogenous Viral dsRNA
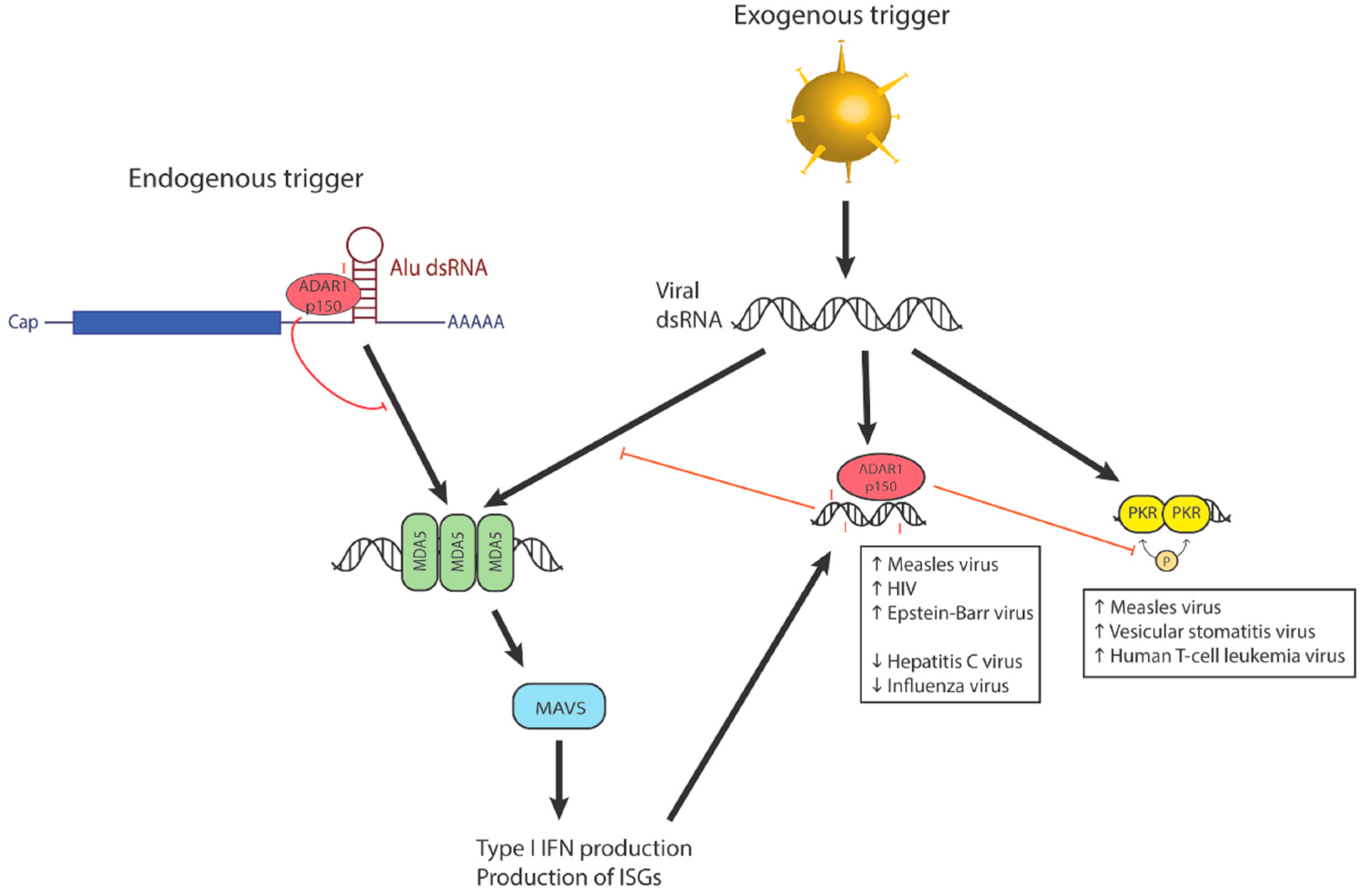
3.1. Editing Affects Viral Recognition by RLRs
3.2. Editing on Viral Coding Sequences Affects Viral Life Cycles
3.3. Editing-Independent Interactions of ADAR1 with Other Interferon-Stimulated Proteins
4. Editing of Coding Endogenous dsRNA
4.1. AZIN1
4.2. FLNB
4.3. NEIL1
4.4. GLI1
4.5. PTPN6
4.6. GABRA3
4.7. RHOQ
4.8. CAPS1
4.9. HTR2C
4.10. GRIA2
4.11. Comparing ADAR1′s and ADAR2′s Substrates
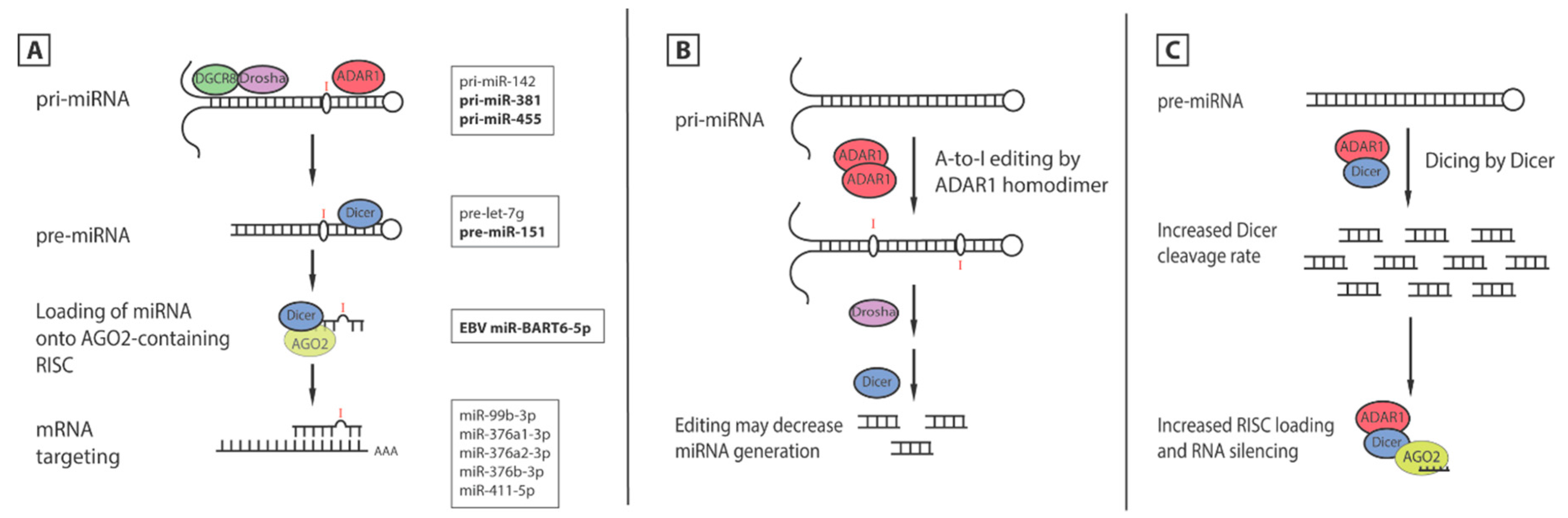
5. ADAR1′s Effects on MicroRNA
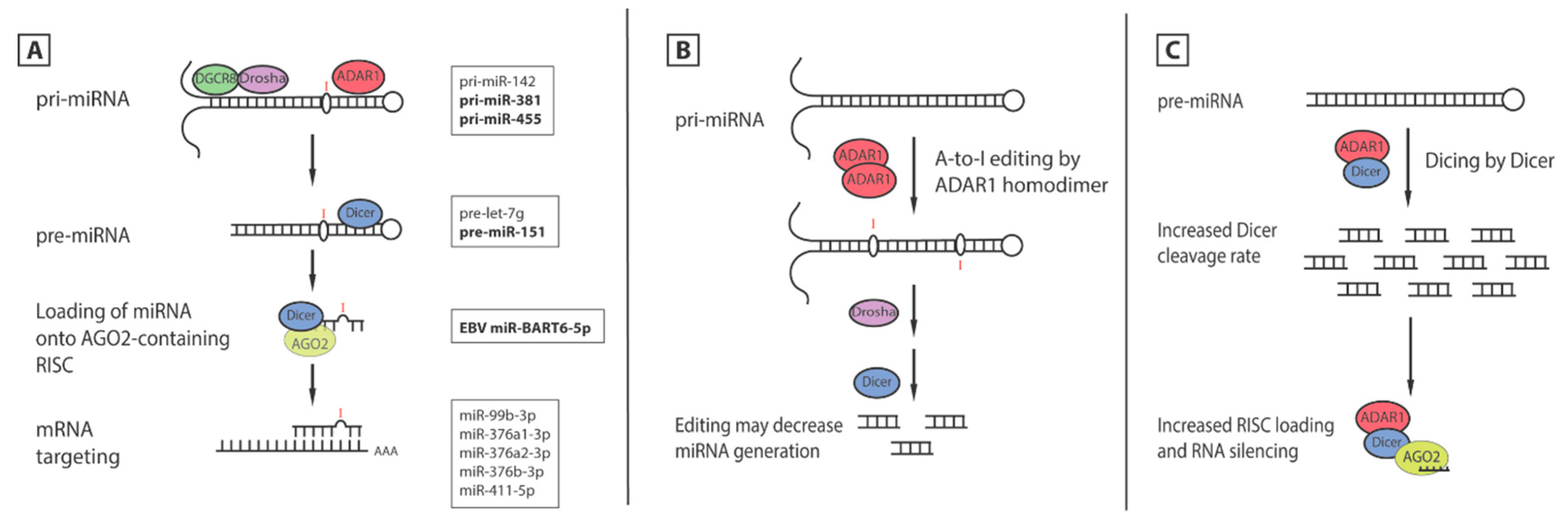
5.1. Editing of miRNA
5.2. Editing of Seed Sequence Changes mRNA Targets
5.3. Editing-Independent Effects of ADAR1 on miRNA Processing
6. ADAR′s Effects on Other Noncoding RNA
6.1. Editing of Alu dsRNA
6.2. Editing Affects Interactions between Intronic lncRNA and Coding mRNA
6.3. Editing of LINE1 Retrotransposon
7. Regulation of ADAR1 Expression
8. Conclusions and Future Outlook
Acknowledgments
Conflicts of Interest
References
- Bass, B.L.; Weintraub, H. A developmentally regulated activity that unwinds RNA duplexes. Cell 1987, 48, 607–613. [Google Scholar] [CrossRef]
- Rebagliati, M.R.; Melton, D.A. Antisense RNA injections in fertilized frog eggs reveal an RNA duplex unwinding activity. Cell 1987, 48, 599–605. [Google Scholar] [CrossRef]
- Bass, B.L.; Weintraub, H. An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell 1988, 55, 1089–1098. [Google Scholar] [CrossRef]
- Wagner, R.W.; Nishikura, K. Cell cycle expression of RNA duplex unwindase activity in mammalian cells. Mol. Cell. Biol. 1988, 8, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Bass, B.L. RNA editing and hypermutation by adenosine deamination. Trends Biochem. Sci. 1997, 22, 157–162. [Google Scholar] [CrossRef]
- Polson, A.G.; Bass, B.L. Preferential selection of adenosines for modification by double-stranded RNA adenosine deaminase. EMBO J. 1994, 13, 5701–5711. [Google Scholar] [PubMed]
- Lehmann, K.A.; Bass, B.L. Double-stranded RNA adenosine deaminases ADAR1 and ADAR2 have overlapping specificities. Biochemistry 2000, 39, 12875–12884. [Google Scholar] [CrossRef] [PubMed]
- Stefl, R.; Oberstrass, F.C.; Hood, J.L.; Jourdan, M.; Zimmermann, M.; Skrisovska, L.; Maris, C.; Peng, L.; Hofr, C.; Emeson, R.B.; et al. The solution structure of the ADAR2 dsrbm-RNA complex reveals a sequence-specific readout of the minor groove. Cell 2010, 143, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Barraud, P.; Allain, F.H. Adar proteins: Double-stranded RNA and z-DNA binding domains. Curr. Top. Microbiol. Immunol. 2012, 353, 35–60. [Google Scholar] [PubMed]
- Kim, U.; Wang, Y.; Sanford, T.; Zeng, Y.; Nishikura, K. Molecular cloning of cdna for double-stranded RNA adenosine deaminase, a candidate enzyme for nuclear RNA editing. Proc. Natl. Acad. Sci. USA 1994, 91, 11457–11461. [Google Scholar] [CrossRef] [PubMed]
- Melcher, T.; Maas, S.; Herb, A.; Sprengel, R.; Seeburg, P.H.; Higuchi, M. A mammalian RNA editing enzyme. Nature 1996, 379, 460–464. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.X.; Cho, D.S.; Wang, Q.; Lai, F.; Carter, K.C.; Nishikura, K. A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. RNA 2000, 6, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Melcher, T.; Maas, S.; Herb, A.; Sprengel, R.; Higuchi, M.; Seeburg, P.H. Red2, a brain-specific member of the RNA-specific adenosine deaminase family. J. Biol. Chem. 1996, 271, 31795–31798. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, H.; Nilsson, J.; Damgaard, C.K.; Egebjerg, J.; Kjems, J. CRM1 mediates the export of ADAR1 through a nuclear export signal within the Z-DNA binding domain. Mol. Cell. Biol 2001, 21, 7862–7871. [Google Scholar] [CrossRef] [PubMed]
- George, C.X.; Samuel, C.E. Human RNA-specific adenosine deaminase ADAR1 transcripts possess alternative exon 1 structures that initiate from different promoters, one constitutively active and the other interferon inducible. Proc. Natl. Acad. Sci. USA 1999, 96, 4621–4626. [Google Scholar] [CrossRef] [PubMed]
- Strehblow, A.; Hallegger, M.; Jantsch, M.F. Nucleocytoplasmic distribution of human RNA-editing enzyme ADAR1 is modulated by double-stranded RNA-binding domains, a leucine-rich export signal, and a putative dimerization domain. Mol. Biol. Cell 2002, 13, 3822–3835. [Google Scholar] [CrossRef] [PubMed]
- Desterro, J.M.; Keegan, L.P.; Lafarga, M.; Berciano, M.T.; O′Connell, M.; Carmo-Fonseca, M. Dynamic association of RNA-editing enzymes with the nucleolus. J. Cell Sci. 2003, 116, 1805–1818. [Google Scholar] [CrossRef] [PubMed]
- Nishikura, K. A-to-I editing of coding and non-coding RNAs by ADARS. Nat. Rev. Mol. Cell Biol. 2016, 17, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Nishikura, K. Functions and regulation of RNA editing by ADAR deaminases. Annu. Rev. Biochem. 2010, 79, 321–349. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Khillan, J.; Gadue, P.; Nishikura, K. Requirement of the RNA editing deaminase ADAR1 gene for embryonic erythropoiesis. Science 2000, 290, 1765–1768. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Luo, X.; Nie, Y.; Su, Y.; Zhao, Q.; Kabir, K.; Zhang, D.; Rabinovici, R. Widespread inosine-containing mRNA in lymphocytes regulated by ADAR1 in response to inflammation. Immunology 2003, 109, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Liddicoat, B.J.; Chalk, A.M.; Walkley, C.R. ADAR1, inosine and the immune sensing system: Distinguishing self from non-self. Wiley Interdiscip. Rev. RNA 2016, 7, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Deng, P.; Zhu, Z.; Zhu, J.; Wang, G.; Zhang, L.; Chen, A.F.; Wang, T.; Sarkar, S.N.; Billiar, T.R.; et al. Adenosine deaminase acting on RNA 1 limits RIG-I RNA detection and suppresses IFN production responding to viral and endogenous RNAs. J. Immunol. 2014, 193, 3436–3445. [Google Scholar] [CrossRef] [PubMed]
- Pestal, K.; Funk, C.C.; Snyder, J.M.; Price, N.D.; Treuting, P.M.; Stetson, D.B. Isoforms of RNA-editing enzyme ADAR1 independently control nucleic acid sensor MDA5-driven autoimmunity and multi-organ development. Immunity 2015, 43, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Liddicoat, B.J.; Piskol, R.; Chalk, A.M.; Ramaswami, G.; Higuchi, M.; Hartner, J.C.; Li, J.B.; Seeburg, P.H.; Walkley, C.R. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 2015, 349, 1115–1120. [Google Scholar] [CrossRef] [PubMed]
- George, C.X.; Ramaswami, G.; Li, J.B.; Samuel, C.E. Editing of cellular self-RNAs by adenosine deaminase ADAR1 suppresses innate immune stress responses. J. Biol. Chem. 2016, 291, 6158–6168. [Google Scholar] [CrossRef] [PubMed]
- Patterson, J.B.; Samuel, C.E. Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: Evidence for two forms of the deaminase. Mol. Cell. Biol. 1995, 15, 5376–5388. [Google Scholar] [CrossRef] [PubMed]
- Hartner, J.C.; Schmittwolf, C.; Kispert, A.; Muller, A.M.; Higuchi, M.; Seeburg, P.H. Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J. Biol. Chem. 2004, 279, 4894–4902. [Google Scholar] [CrossRef] [PubMed]
- Hartner, J.C.; Walkley, C.R.; Lu, J.; Orkin, S.H. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat. Immunol. 2009, 10, 109–115. [Google Scholar] [CrossRef] [PubMed]
- XuFeng, R.; Boyer, M.J.; Shen, H.; Li, Y.; Yu, H.; Gao, Y.; Yang, Q.; Wang, Q.; Cheng, T. ADAR1 is required for hematopoietic progenitor cell survival via RNA editing. Proc. Natl. Acad. Sci. USA 2009, 106, 17763–17768. [Google Scholar] [CrossRef] [PubMed]
- Mannion, N.M.; Greenwood, S.M.; Young, R.; Cox, S.; Brindle, J.; Read, D.; Nellaker, C.; Vesely, C.; Ponting, C.P.; McLaughlin, P.J.; et al. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep. 2014, 9, 1482–1494. [Google Scholar] [CrossRef] [PubMed]
- Crow, Y.J.; Chase, D.S.; Lowenstein Schmidt, J.; Szynkiewicz, M.; Forte, G.M.; Gornall, H.L.; Oojageer, A.; Anderson, B.; Pizzino, A.; Helman, G.; et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am. J. Med. Genet. A 2015, 167A, 296–312. [Google Scholar] [CrossRef] [PubMed]
- Lebon, P.; Badoual, J.; Ponsot, G.; Goutieres, F.; Hemeury-Cukier, F.; Aicardi, J. Intrathecal synthesis of interferon-alpha in infants with progressive familial encephalopathy. J. Neurol. Sci. 1988, 84, 201–208. [Google Scholar] [CrossRef]
- Li, M.; Yang, L.; Li, C.; Jin, C.; Lai, M.; Zhang, G.; Hu, Y.; Ji, J.; Yao, Z. Mutational spectrum of the ADAR1 gene in dyschromatosis symmetrica hereditaria. Arch. Dermatol. Res. 2010, 302, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Suzuki, T.; Mitsuhashi, Y.; Ito, S.; Kono, M.; Komine, M.; Akita, H.; Tomita, Y. Six novel mutations of the ADAR1 gene in patients with dyschromatosis symmetrica hereditaria: Histological observation and comparison of genotypes and clinical phenotypes. J. Dermatol. 2008, 35, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Livingston, J.H.; Lin, J.P.; Dale, R.C.; Gill, D.; Brogan, P.; Munnich, A.; Kurian, M.A.; Gonzalez-Martinez, V.; De Goede, C.G.; Falconer, A.; et al. A type I interferon signature identifies bilateral striatal necrosis due to mutations in adar1. J. Med. Genet. 2014, 51, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Crow, Y.J.; Zaki, M.S.; Abdel-Hamid, M.S.; Abdel-Salam, G.; Boespflug-Tanguy, O.; Cordeiro, N.J.; Gleeson, J.G.; Gowrinathan, N.R.; Laugel, V.; Renaldo, F.; et al. Mutations in ADAR1, IFIH1, and RNASEH2B presenting as spastic paraplegia. Neuropediatrics 2014, 45, 386–393. [Google Scholar] [PubMed]
- Rice, G.I.; Kasher, P.R.; Forte, G.M.; Mannion, N.M.; Greenwood, S.M.; Szynkiewicz, M.; Dickerson, J.E.; Bhaskar, S.S.; Zampini, M.; Briggs, T.A.; et al. Mutations in ADAR1 cause aicardi-goutieres syndrome associated with a type I interferon signature. Nat. Genet. 2012, 44, 1243–1248. [Google Scholar] [PubMed]
- Hayashi, M.; Suzuki, T. Dyschromatosis symmetrica hereditaria. J. Dermatol. 2013, 40, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Jiang, M.; Zhao, J. Two novel mutations in the DSRAD gene in two chinese pedigrees with dyschromatosis symmetrica hereditaria. Eur. J. Dermatol. 2013, 23, 782–785. [Google Scholar] [PubMed]
- Zhu, C.Y.; Zhu, K.J.; Zhou, Y.; Fan, Y.M. A novel insertion mutation in the ADAR1 gene of a Chinese family with dyschromatosis symmetrica hereditaria. Genet. Mol. Res. 2013, 12, 2858–2862. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wang, Z.; Wu, Y.; Cao, L.; Tang, Q.; Xing, X.; Ma, H.; Zhang, S.; Luo, Y. Five novel mutations in the ADAR1 gene associated with dyschromatosis symmetrica hereditaria. BMC Med. Genet. 2014. [Google Scholar] [CrossRef] [PubMed]
- Kono, M.; Matsumoto, F.; Suzuki, Y.; Suganuma, M.; Saitsu, H.; Ito, Y.; Fujiwara, S.; Moriwaki, S.; Matsumoto, K.; Matsumoto, N.; et al. Dyschromatosis symmetrica hereditaria and aicardi-goutieres syndrome 6 are phenotypic variants caused by ADAR1 mutations. J. Invest. Dermatol. 2016, 136, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, Z.; Mu, Y.; Xiong, F.; Chen, X.; Yang, H.; Yang, P.; Liu, L. Two novel mutations of the ADAR1 gene associated with dyschromatosis symmetrica hereditaria. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2016, 33, 173–176. (in Chinese). [Google Scholar] [PubMed]
- Zhang, G.; Shao, M.; Li, Z.; Gu, Y.; Du, X.; Wang, X.; Li, M. Genetic spectrum of dyschromatosis symmetrica hereditaria in Chinese patients including a novel nonstop mutation in ADAR1 gene. BMC Med. Genet. 2016. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.S.; Yang, W.; Lee, J.T.; Shiekhattar, R.; Murray, J.M.; Nishikura, K. Requirement of dimerization for RNA editing activity of adenosine deaminases acting on RNA. J. Biol. Chem. 2003, 278, 17093–17102. [Google Scholar] [CrossRef] [PubMed]
- Valente, L.; Nishikura, K. RNA binding-independent dimerization of adenosine deaminases acting on RNA and dominant negative effects of nonfunctional subunits on dimer functions. J. Biol. Chem. 2007, 282, 16054–16061. [Google Scholar] [CrossRef] [PubMed]
- Vitali, P.; Scadden, A.D. Double-stranded RNAs containing multiple IU pairs are sufficient to suppress interferon induction and apoptosis. Nat. Struct. Mol. Biol. 2010, 17, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Pfaller, C.K.; Mastorakos, G.M.; Matchett, W.E.; Ma, X.; Samuel, C.E.; Cattaneo, R. Measles virus defective interfering RNAs are generated frequently and early in the absence of C protein and can be destabilized by adenosine deaminase acting on RNA-1-like hypermutations. J. Virol. 2015, 89, 7735–7747. [Google Scholar] [CrossRef] [PubMed]
- Ward, S.V.; George, C.X.; Welch, M.J.; Liou, L.Y.; Hahm, B.; Lewicki, H.; de la Torre, J.C.; Samuel, C.E.; Oldstone, M.B. RNA editing enzyme adenosine deaminase is a restriction factor for controlling measles virus replication that also is required for embryogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Sarvestani, S.T.; Tate, M.D.; Moffat, J.M.; Jacobi, A.M.; Behlke, M.A.; Miller, A.R.; Beckham, S.A.; McCoy, C.E.; Chen, W.; Mintern, J.D.; et al. Inosine-mediated modulation of rna sensing by toll-like receptor 7 (TLR7) and TLR8. J. Virol. 2014, 88, 799–810. [Google Scholar] [CrossRef] [PubMed]
- Macnaughton, T.B.; Li, Y.I.; Doughty, A.L.; Lai, M.M. Hepatitis delta virus RNA encoding the large delta antigen cannot sustain replication due to rapid accumulation of mutations associated with RNA editing. J. Virol. 2003, 77, 12048–12056. [Google Scholar] [CrossRef] [PubMed]
- Casey, J.L. Control of ADAR1 editing of hepatitis delta virus RNAs. Curr. Top. Microbiol. Immunol. 2012, 353, 123–143. [Google Scholar] [PubMed]
- Taylor, D.R.; Puig, M.; Darnell, M.E.; Mihalik, K.; Feinstone, S.M. New antiviral pathway that mediates hepatitis C virus replicon interferon sensitivity through ADAR1. J. Virol. 2005, 79, 6291–6298. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Moss, W.; O’Grady, T.; Concha, M.; Strong, M.J.; Wang, X.; Yu, Y.; Baddoo, M.; Zhang, K.; Fewell, C.; et al. New noncoding lytic transcripts derived from the epstein-barr virus latency origin of replication, orip, are hyperedited, bind the paraspeckle protein, NONO/p54nrb, and support viral lytic transcription. J. Virol. 2015, 89, 7120–7132. [Google Scholar] [CrossRef] [PubMed]
- Weiden, M.D.; Hoshino, S.; Levy, D.N.; Li, Y.; Kumar, R.; Burke, S.A.; Dawson, R.; Hioe, C.E.; Borkowsky, W.; Rom, W.N.; et al. Adenosine deaminase acting on RNA-1 (ADAR1) inhibits HIV-1 replication in human alveolar macrophages. PLoS ONE 2014, 9, e108476. [Google Scholar] [CrossRef] [PubMed]
- Cachat, A.; Alais, S.; Chevalier, S.A.; Journo, C.; Fusil, F.; Dutartre, H.; Boniface, A.; Ko, N.L.; Gessain, A.; Cosset, F.L.; et al. ADAR1 enhances HTLV-1 and HTLV-2 replication through inhibition of PKR activity. Retrovirology 2014. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, E.; Booiman, T.; van Hamme, J.L.; Jansen, M.H.; van Dort, K.A.; Vanderver, A.; Rice, G.I.; Crow, Y.J.; Kootstra, N.A.; Kuijpers, T.W. ADAR1 facilitates HIV-1 replication in primary CD4+ T cells. PLoS ONE 2015, 10, e0143613. [Google Scholar] [CrossRef] [PubMed]
- Phuphuakrat, A.; Kraiwong, R.; Boonarkart, C.; Lauhakirti, D.; Lee, T.H.; Auewarakul, P. Double-stranded RNA adenosine deaminases enhance expression of human immunodeficiency virus type 1 proteins. J. Virol. 2008, 82, 10864–10872. [Google Scholar] [CrossRef] [PubMed]
- Doria, M.; Neri, F.; Gallo, A.; Farace, M.G.; Michienzi, A. Editing of HIV-1 RNA by the double-stranded RNA deaminase ADAR1 stimulates viral infection. Nucleic Acids Res. 2009, 37, 5848–5858. [Google Scholar] [CrossRef] [PubMed]
- Orecchini, E.; Federico, M.; Doria, M.; Arenaccio, C.; Giuliani, E.; Ciafre, S.A.; Michienzi, A. The ADAR1 editing enzyme is encapsidated into HIV-1 virions. Virology 2015, 485, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Munir, M.; Berg, M. The multiple faces of proteinkinase R in antiviral defense. Virulence 2013, 4, 85–89. [Google Scholar] [CrossRef] [PubMed]
- George, C.X.; John, L.; Samuel, C.E. An RNA editor, adenosine deaminase acting on double-stranded RNA (ADAR1). J. Interferon Cytokine Res. 2014, 34, 437–446. [Google Scholar] [CrossRef] [PubMed]
- John, L.; Samuel, C.E. Induction of stress granules by interferon and down-regulation by the cellular RNA adenosine deaminase ADAR1. Virology 2014, 454, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Ding, L.; Kao, P.N.; Braun, R.; Yang, J.H. ADAR1 interacts with NF90 through double-stranded RNA and regulates NF90-mediated gene expression independently of Rna editing. Mol. Cell Biol. 2005, 25, 6956–6963. [Google Scholar] [CrossRef] [PubMed]
- Reichman, T.W.; Muniz, L.C.; Mathews, M.B. The RNA binding protein nuclear factor 90 functions as both a positive and negative regulator of gene expression in mammalian cells. Mol. Cell Biol. 2002, 22, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.R.; Qiao, J.J.; Chan, T.H.; Zhu, Y.H.; Li, F.F.; Liu, H.; Fei, J.; Li, Y.; Guan, X.Y.; Chen, L. Adenosine-to-inosine RNA editing mediated by ADARs in esophageal squamous cell carcinoma. Cancer Res. 2014, 74, 840–851. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.H.; Lin, C.H.; Qi, L.; Fei, J.; Li, Y.; Yong, K.J.; Liu, M.; Song, Y.; Chow, R.K.; Ng, V.H.; et al. A disrupted RNA editing balance mediated by ADARs (adenosine deaminases that act on RNA) in human hepatocellular carcinoma. Gut 2014, 63, 832–843. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Diao, L.; Yu, S.; Xu, X.; Li, J.; Zhang, R.; Yang, Y.; Werner, H.M.; Eterovic, A.K.; Yuan, Y.; et al. The genomic landscape and clinical relevance of A-to-I RNA editing in human cancers. Cancer Cell 2015, 28, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zeng, Y.; Murray, J.M.; Nishikura, K. Genomic organization and chromosomal location of the human dsRNA adenosine deaminase gene: The enzyme for glutamate-activated ion channel RNA editing. J. Mol. Biol. 1995, 254, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Anadon, C.; Guil, S.; Simo-Riudalbas, L.; Moutinho, C.; Setien, F.; Martinez-Cardus, A.; Moran, S.; Villanueva, A.; Calaf, M.; Vidal, A.; et al. Gene amplification-associated overexpression of the RNA editing enzyme ADAR1 enhances human lung tumorigenesis. Oncogene 2015. [Google Scholar] [CrossRef]
- Yeo, J.; Goodman, R.A.; Schirle, N.T.; David, S.S.; Beal, P.A. RNA editing changes the lesion specificity for the DNA repair enzyme neil1. Proc. Natl. Acad. Sci. USA 2010, 107, 20715–20719. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, T.; Rahman, M.F.; Tostar, U.; Sonkoly, E.; Stahle, M.; Pivarcsi, A.; Palaniswamy, R.; Zaphiropoulos, P.G. RNA editing of the GLI1 transcription factor modulates the output of hedgehog signaling. RNA Biol. 2013, 10, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Zipeto, M.A.; Jiang, Q.; Melese, E.; Jamieson, C.H. RNA rewriting, recoding, and rewiring in human disease. Trends Mol. Med. 2015, 21, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Quelen, C.; Eloit, Y.; Noirot, C.; Bousquet, M.; Brousset, P. RNA editing in acute myeloid leukaemia with normal karyotype. Br. J. Haematol. 2016, 173, 788–790. [Google Scholar] [CrossRef] [PubMed]
- Beghini, A.; Ripamonti, C.B.; Peterlongo, P.; Roversi, G.; Cairoli, R.; Morra, E.; Larizza, L. RNA hyperediting and alternative splicing of hematopoietic cell phosphatase (PTPN6) gene in acute myeloid leukemia. Hum. Mol. Genet. 2000, 9, 2297–2304. [Google Scholar] [CrossRef] [PubMed]
- Gumireddy, K.; Li, A.; Kossenkov, A.V.; Sakurai, M.; Yan, J.; Li, Y.; Xu, H.; Wang, J.; Zhang, P.J.; Zhang, L.; et al. The mRNA-edited form of GABRA3 suppresses GABRA3-mediated AKT activation and breast cancer metastasis. Nat. Commun. 2016. [Google Scholar] [CrossRef] [PubMed]
- Ohlson, J.; Pedersen, J.S.; Haussler, D.; Ohman, M. Editing modifies the GABA(A) receptor subunit α3. RNA 2007, 13, 698–703. [Google Scholar] [CrossRef] [PubMed]
- Daniel, C.; Veno, M.T.; Ekdahl, Y.; Kjems, J.; Ohman, M. A distant cis acting intronic element induces site-selective RNA editing. Nucleic Acids Res. 2012, 40, 9876–9886. [Google Scholar] [CrossRef] [PubMed]
- Han, S.W.; Kim, H.P.; Shin, J.Y.; Jeong, E.G.; Lee, W.C.; Kim, K.Y.; Park, S.Y.; Lee, D.W.; Won, J.K.; Jeong, S.Y.; et al. RNA editing in RHOQ promotes invasion potential in colorectal cancer. J. Exp. Med. 2014, 211, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Miyake, K.; Ohta, T.; Nakayama, H.; Doe, N.; Terao, Y.; Oiki, E.; Nagatomo, I.; Yamashita, Y.; Abe, T.; Nishikura, K.; et al. CAPS1 RNA editing promotes dense core vesicle exocytosis. Cell Rep. 2016, 17, 2004–2014. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, Y.; Grimberg, A.; Teegarden, S.; Mombereau, C.; Liu, S.; Bale, T.L.; Blendy, J.A.; Nishikura, K. Dysregulated editing of serotonin 2C receptor mRNAs results in energy dissipation and loss of fat mass. J. Neurosci. 2008, 28, 12834–12844. [Google Scholar] [CrossRef] [PubMed]
- Niswender, C.M.; Herrick-Davis, K.; Dilley, G.E.; Meltzer, H.Y.; Overholser, J.C.; Stockmeier, C.A.; Emeson, R.B.; Sanders-Bush, E. RNA editing of the human serotonin 5-HT2C receptor. Alterations in suicide and implications for serotonergic pharmacotherapy. Neuropsychopharmacology 2001, 24, 478–491. [Google Scholar] [CrossRef]
- Di Narzo, A.F.; Kozlenkov, A.; Roussos, P.; Hao, K.; Hurd, Y.; Lewis, D.A.; Sibille, E.; Siever, L.J.; Koonin, E.; Dracheva, S. A unique gene expression signature associated with serotonin 2C receptor RNA editing in the prefrontal cortex and altered in suicide. Hum. Mol. Genet. 2014, 23, 4801–4813. [Google Scholar] [CrossRef] [PubMed]
- Anastasio, N.C.; Stutz, S.J.; Fox, R.G.; Sears, R.M.; Emeson, R.B.; DiLeone, R.J.; O′Neil, R.T.; Fink, L.H.; Li, D.; Green, T.A.; et al. Functional status of the serotonin 5-HT2C receptor (5-HT2CR) drives interlocked phenotypes that precipitate relapse-like behaviors in cocaine dependence. Neuropsychopharmacology 2014, 39, 370–382. [Google Scholar] [CrossRef] [PubMed]
- Lomeli, H.; Mosbacher, J.; Melcher, T.; Hoger, T.; Geiger, J.R.; Kuner, T.; Monyer, H.; Higuchi, M.; Bach, A.; Seeburg, P.H. Control of kinetic properties of AMPA receptor channels by nuclear RNA editing. Science 1994, 266, 1709–1713. [Google Scholar] [CrossRef] [PubMed]
- Krampfl, K.; Schlesinger, F.; Zorner, A.; Kappler, M.; Dengler, R.; Bufler, J. Control of kinetic properties of GluR2 flop AMPA-type channels: Impact of R/G nuclear editing. Eur. J. Neurosci. 2002, 15, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Vollmar, W.; Gloger, J.; Berger, E.; Kortenbruck, G.; Kohling, R.; Speckmann, E.J.; Musshoff, U. RNA editing (R/G site) and flip-flop splicing of the AMPA receptor subunit GluR2 in nervous tissue of epilepsy patients. Neurobiol. Dis. 2004, 15, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Barbon, A.; Fumagalli, F.; Caracciolo, L.; Madaschi, L.; Lesma, E.; Mora, C.; Carelli, S.; Slotkin, T.A.; Racagni, G.; Di Giulio, A.M.; et al. Acute spinal cord injury persistently reduces R/G RNA editing of AMPA receptors. J. Neurochem. 2010, 114, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Penn, A.C.; Balik, A.; Greger, I.H. Steric antisense inhibition of AMPA receptor Q/R editing reveals tight coupling to intronic editing sites and splicing. Nucleic Acids Res. 2013, 41, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Bonini, D.; Filippini, A.; La Via, L.; Fiorentini, C.; Fumagalli, F.; Colombi, M.; Barbon, A. Chronic glutamate treatment selectively modulates AMPA RNA editing and ADAR expression and activity in primary cortical neurons. RNA Biol. 2015, 12, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, S.S.; Thai, K.H.; Chen, K.; Ziff, E. Exposure of neurons to excitotoxic levels of glutamate induces cleavage of the RNA editing enzyme, adenosine deaminase acting on RNA 2, and loss of GluR2 editing. Neuroscience 2011, 189, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Sailer, A.; Swanson, G.T.; Perez-Otano, I.; O’Leary, L.; Malkmus, S.A.; Dyck, R.H.; Dickinson-Anson, H.; Schiffer, H.H.; Maron, C.; Yaksh, T.L.; et al. Generation and analysis of GluR5(Q636R) kainate receptor mutant mice. J. Neurosci. 1999, 19, 8757–8764. [Google Scholar] [PubMed]
- Egebjerg, J.; Heinemann, S.F. Ca2+ permeability of unedited and edited versions of the kainate selective glutamate receptor GluR6. Proc. Natl. Acad. Sci. USA 1993, 90, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Kohler, M.; Burnashev, N.; Sakmann, B.; Seeburg, P.H. Determinants of Ca2+ permeability in both TM1 and TM2 of high affinity kainate receptor channels: Diversity by RNA editing. Neuron 1993, 10, 491–500. [Google Scholar] [CrossRef]
- Riedmann, E.M.; Schopoff, S.; Hartner, J.C.; Jantsch, M.F. Specificity of ADAR-mediated RNA editing in newly identified targets. RNA 2008, 14, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Matthews, M.M.; Thomas, J.M.; Zheng, Y.; Tran, K.; Phelps, K.J.; Scott, A.I.; Havel, J.; Fisher, A.J.; Beal, P.A. Structures of human ADAR2 bound to dsRNA reveal base-flipping mechanism and basis for site selectivity. Nat. Struct. Mol. Biol. 2016, 23, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.K.; Sato, S.; Lazinski, D.W. Substrate recognition by ADAR1 and ADAR2. RNA 2001, 7, 846–858. [Google Scholar] [CrossRef] [PubMed]
- Nishikura, K.; Yoo, C.; Kim, U.; Murray, J.M.; Estes, P.A.; Cash, F.E.; Liebhaber, S.A. Substrate specificity of the dsRNA unwinding/modifying activity. EMBO J. 1991, 10, 3523–3532. [Google Scholar] [PubMed]
- Macbeth, M.R.; Lingam, A.T.; Bass, B.L. Evidence for auto-inhibition by the N terminus of hADAR2 and activation by dsRNA binding. RNA 2004, 10, 1563–1571. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Nemlich, Y.; Greenberg, E.; Ortenberg, R.; Besser, M.J.; Barshack, I.; Jacob-Hirsch, J.; Jacoby, E.; Eyal, E.; Rivkin, L.; Prieto, V.G.; et al. Microrna-mediated loss of ADAR1 in metastatic melanoma promotes tumor growth. J. Clin. Invest. 2013, 123, 2703–2718. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, C.S.; Varelas, X.; Monti, S. Altered RNA editing in 3′ UTR perturbs microRNA-mediated regulation of oncogenes and tumor-suppressors. Sci. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Shoshan, E.; Mobley, A.K.; Braeuer, R.R.; Kamiya, T.; Huang, L.; Vasquez, M.E.; Salameh, A.; Lee, H.J.; Kim, S.J.; Ivan, C.; et al. Reduced adenosine-to-inosine miR-455–5p editing promotes melanoma growth and metastasis. Nat. Cell Biol. 2015, 17, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Li, N.F.; Gemenetzidis, E.; Marshall, F.J.; Davies, D.; Yu, Y.; Frese, K.; Froeling, F.E.; Woolf, A.K.; Feakins, R.M.; Naito, Y.; et al. RhoC interacts with integrin α5β1 and enhances its trafficking in migrating pancreatic carcinoma cells. PLoS ONE 2013, 8, e81575. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.; Sharma, S.; Teknos, T.N. RhoC regulates cancer stem cells in head and neck squamous cell carcinoma by overexpressing IL-6 and phosphorylation of STAT3. PLoS ONE 2014, 9, e88527. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, D.T.; Zhang, J.; Bao, L.; Zhu, L.; Wu, Z.; Toy, K.; Kleer, C.G.; Merajver, S.D. Rhoc impacts the metastatic potential and abundance of breast cancer stem cells. PLoS ONE 2012, 7, e40979. [Google Scholar] [CrossRef] [PubMed]
- Gembarska, A.; Luciani, F.; Fedele, C.; Russell, E.A.; Dewaele, M.; Villar, S.; Zwolinska, A.; Haupt, S.; de Lange, J.; Yip, D.; et al. MDM4 is a key therapeutic target in cutaneous melanoma. Nat. Med. 2012, 18, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- McEvoy, J.; Ulyanov, A.; Brennan, R.; Wu, G.; Pounds, S.; Zhang, J.; Dyer, M.A. Analysis of MDM2 and MDM4 single nucleotide polymorphisms, mRNA splicing and protein expression in retinoblastoma. PLoS ONE 2012, 7, e42739. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.Q.; Wang, R.J.; Diao, C.F.; Li, J.W.; Su, J.L.; Zhang, S. The PTTG1-targeting miRNAs mir-329, miR-300, miR-381, and miR-655 inhibit pituitary tumor cell tumorigenesis and are involved in a p53/PTTG1 regulation feedback loop. Oncotarget 2015, 6, 29413–29427. [Google Scholar] [PubMed]
- Formosa, A.; Markert, E.K.; Lena, A.M.; Italiano, D.; Finazzi-Agro, E.; Levine, A.J.; Bernardini, S.; Garabadgiu, A.V.; Melino, G.; Candi, E. MicroRNAs, mir-154, mir-299–5p, mir-376a, mir-376c, mir-377, mir-381, mir-487b, mir-485–3p, mir-495 and mir-654–3p, mapped to the 14q32.31 locus, regulate proliferation, apoptosis, migration and invasion in metastatic prostate cancer cells. Oncogene 2014, 33, 5173–5182. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Lopez, J.; Hourcade Jde, D.; Del Mazo, J. Reprogramming of microRNAs by adenosine-to-inosine editing and the selective elimination of edited microRNA precursors in mouse oocytes and preimplantation embryos. Nucleic Acids Res. 2013, 41, 5483–5493. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, Y.; Zinshteyn, B.; Chendrimada, T.P.; Shiekhattar, R.; Nishikura, K. RNA editing of the microRNA-151 precursor blocks cleavage by the Dicer-TRBP complex. EMBO Rep. 2007, 8, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Iizasa, H.; Wulff, B.E.; Alla, N.R.; Maragkakis, M.; Megraw, M.; Hatzigeorgiou, A.; Iwakiri, D.; Takada, K.; Wiedmer, A.; Showe, L.; et al. Editing of Epstein-Barr virus-encoded BART6 micrornas controls their dicer targeting and consequently affects viral latency. J. Biol. Chem. 2010, 285, 33358–33370. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, Y.; Zinshteyn, B.; Sethupathy, P.; Iizasa, H.; Hatzigeorgiou, A.G.; Nishikura, K. Redirection of silencing targets by adenosine-to-inosine editing of mirnas. Science 2007, 315, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Heale, B.S.; Keegan, L.P.; McGurk, L.; Michlewski, G.; Brindle, J.; Stanton, C.M.; Caceres, J.F.; O′Connell, M.A. Editing independent effects of ADARs on the miRNA/siRNA pathways. EMBO J. 2009, 28, 3145–3156. [Google Scholar] [CrossRef] [PubMed]
- Bahn, J.H.; Ahn, J.; Lin, X.; Zhang, Q.; Lee, J.H.; Civelek, M.; Xiao, X. Genomic analysis of ADAR1 binding and its involvement in multiple RNA processing pathways. Nat. Commun. 2015. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Xiang, J.F.; Zhu, S.; Chen, S.; Yin, Q.F.; Zhang, X.O.; Zhang, J.; Feng, H.; Dong, R.; Li, X.J.; et al. Adar1 is required for differentiation and neural induction by regulating microRNA processing in a catalytically independent manner. Cell Res. 2015, 25, 459–476. [Google Scholar] [CrossRef] [PubMed]
- Galore-Haskel, G.; Nemlich, Y.; Greenberg, E.; Ashkenazi, S.; Hakim, M.; Itzhaki, O.; Shoshani, N.; Shapira-Fromer, R.; Ben-Ami, E.; Ofek, E.; et al. A novel immune resistance mechanism of melanoma cells controlled by the ADAR1 enzyme. Oncotarget 2015, 6, 28999–29015. [Google Scholar] [PubMed]
- Ota, H.; Sakurai, M.; Gupta, R.; Valente, L.; Wulff, B.E.; Ariyoshi, K.; Iizasa, H.; Davuluri, R.V.; Nishikura, K. ADAR1 forms a complex with dicer to promote microRNA processing and RNA-induced gene silencing. Cell 2013, 153, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Ramaswami, G.; Zhang, R.; Piskol, R.; Keegan, L.P.; Deng, P.; O′Connell, M.A.; Li, J.B. Identifying RNA editing sites using RNA sequencing data alone. Nat. Methods 2013, 10, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Bazak, L.; Haviv, A.; Barak, M.; Jacob-Hirsch, J.; Deng, P.; Zhang, R.; Isaacs, F.J.; Rechavi, G.; Li, J.B.; Eisenberg, E.; et al. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res. 2014, 24, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Lev-Maor, G.; Sorek, R.; Levanon, E.Y.; Paz, N.; Eisenberg, E.; Ast, G. RNA-editing-mediated exon evolution. Genome Biol. 2007. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, M.; Yano, T.; Kawabata, H.; Ueda, H.; Suzuki, T. Inosine cyanoethylation identifies a-to-i rna editing sites in the human transcriptome. Nat. Chem Biol 2010, 6, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.; Memczak, S.; Wyler, E.; Torti, F.; Porath, H.T.; Orejuela, M.R.; Piechotta, M.; Levanon, E.Y.; Landthaler, M.; Dieterich, C.; et al. Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals. Cell Rep. 2015, 10, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Kjems, J.; Damgaard, C.K. Circular RNA and mir-7 in cancer. Cancer Res. 2013, 73, 5609–5612. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Landweber, L.F. Hypothesis: RNA editing of microRNA target sites in humans? RNA 2007, 13, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Borchert, G.M.; Gilmore, B.L.; Spengler, R.M.; Xing, Y.; Lanier, W.; Bhattacharya, D.; Davidson, B.L. Adenosine deamination in human transcripts generates novel microrna binding sites. Hum. Mol. Genet. 2009, 18, 4801–4807. [Google Scholar] [CrossRef] [PubMed]
- Stellos, K.; Gatsiou, A.; Stamatelopoulos, K.; Perisic Matic, L.; John, D.; Lunella, F.F.; Jae, N.; Rossbach, O.; Amrhein, C.; Sigala, F.; et al. Adenosine-to-inosine RNA editing controls cathepsin S expression in atherosclerosis by enabling hur-mediated post-transcriptional regulation. Nat. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Daniel, C.; Silberberg, G.; Behm, M.; Ohman, M. Alu elements shape the primate transcriptome by cis-regulation of RNA editing. Genome Biol 2014, 15, R28. [Google Scholar] [CrossRef] [PubMed]
- Osenberg, S.; Paz Yaacov, N.; Safran, M.; Moshkovitz, S.; Shtrichman, R.; Sherf, O.; Jacob-Hirsch, J.; Keshet, G.; Amariglio, N.; Itskovitz-Eldor, J.; et al. Alu sequences in undifferentiated human embryonic stem cells display high levels of A-to-I RNA editing. PLoS ONE 2010, 5, e11173. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [PubMed]
- Salameh, A.; Lee, A.K.; Cardo-Vila, M.; Nunes, D.N.; Efstathiou, E.; Staquicini, F.I.; Dobroff, A.S.; Marchio, S.; Navone, N.M.; Hosoya, H.; et al. PRUNE2 is a human prostate cancer suppressor regulated by the intronic long noncoding RNA PCA3. Proc. Natl. Acad. Sci. USA 2015, 112, 8403–8408. [Google Scholar] [CrossRef] [PubMed]
- Orecchini, E.; Doria, M.; Antonioni, A.; Galardi, S.; Ciafre, S.A.; Frassinelli, L.; Mancone, C.; Montaldo, C.; Tripodi, M.; Michienzi, A. ADAR1 restricts LINE-1 retrotransposition. Nucleic Acids Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Nishisho, I.; Horii, A.; Miyoshi, Y.; Utsunomiya, J.; Kinzler, K.W.; Vogelstein, B.; Nakamura, Y. Disruption of the APC gene by a retrotransposal insertion of L1 sequence in a colon cancer. Cancer Res. 1992, 52, 643–645. [Google Scholar] [PubMed]
- Iskow, R.C.; McCabe, M.T.; Mills, R.E.; Torene, S.; Pittard, W.S.; Neuwald, A.F.; Van Meir, E.G.; Vertino, P.M.; Devine, S.E. Natural mutagenesis of human genomes by endogenous retrotransposons. Cell 2010, 141, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Beck, C.R.; Garcia-Perez, J.L.; Badge, R.M.; Moran, J.V. Line-1 elements in structural variation and disease. Annu. Rev. Genomics Hum. Genet. 2011, 12, 187–215. [Google Scholar] [CrossRef] [PubMed]
- Paz-Yaacov, N.; Bazak, L.; Buchumenski, I.; Porath, H.T.; Danan-Gotthold, M.; Knisbacher, B.A.; Eisenberg, E.; Levanon, E.Y. Elevated RNA editing activity is a major contributor to transcriptomic diversity in tumors. Cell Rep. 2015, 13, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Cenci, C.; Barzotti, R.; Galeano, F.; Corbelli, S.; Rota, R.; Massimi, L.; Di Rocco, C.; O′Connell, M.A.; Gallo, A. Down-regulation of RNA editing in pediatric astrocytomas: ADAR2 editing activity inhibits cell migration and proliferation. J. Biol. Chem. 2008, 283, 7251–7260. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.H.; Chong, J.H.; Guo, Y.; Zeng, H.M.; Liu, S.Y.; Xu, L.L.; Wei, J.; Lin, Y.M.; Zhu, X.F.; Zheng, G.G. Abnormal expression of ADAR1 isoforms in Chinese pediatric acute leukemias. Biochem. Biophys. Res. Commun. 2011, 406, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.H.; Qamra, A.; Tan, K.T.; Guo, J.; Yang, H.; Qi, L.; Lin, J.S.; Ng, V.H.; Song, Y.; Hong, H.; et al. Adar-mediated RNA editing predicts progression and prognosis of gastric cancer. Gastroenterology 2016, 151, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Dou, N.; Yu, S.; Ye, X.; Yang, D.; Li, Y.; Gao, Y. Aberrant overexpression of ADAR1 promotes gastric cancer progression by activating mTOR/p70S6K signaling. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Zipeto, M.A.; Court, A.C.; Sadarangani, A.; Delos Santos, N.P.; Balaian, L.; Chun, H.J.; Pineda, G.; Morris, S.R.; Mason, C.N.; Geron, I.; et al. ADAR1 activation drives leukemia stem cell self-renewal by impairing Let-7 biogenesis. Cell Stem Cell 2016. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Crews, L.A.; Barrett, C.L.; Chun, H.J.; Court, A.C.; Isquith, J.M.; Zipeto, M.A.; Goff, D.J.; Minden, M.; Sadarangani, A.; et al. ADAR1 promotes malignant progenitor reprogramming in chronic myeloid leukemia. Proc. Natl. Acad. Sci. USA 2013, 110, 1041–1046. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Hu, P.; Lin, X.; Han, W.; Zhu, L.; Tan, X.; Ye, F.; Wang, G.; Wu, F.; Yin, B.; et al. PTBP1 induces ADAR1 p110 isoform expression through IRES-like dependent translation control and influences cell proliferation in gliomas. Cell Mol. Life Sci. 2015, 72, 4383–4397. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Disease | Disease Classification | ADAR1 Isoform | Editing-Dependent | Target RNA | Regulation | Ref. |
|---|---|---|---|---|---|---|
| Acute myeloid leukemia | Growth receptor proliferation | ? * | Yes | PTPN6 mRNA | Inhibition of post-transcriptional processing of phosphatase | [74] |
| Aicardi-Goutieres syndrome (AGS) | Autoimmune | P150 | Yes | Endogenous dsRNA | Interferon; Inhibition of melanoma differentiation-associated protein 5 – mitochondrial activation signaling complex (MDA5-MAVS) | [33] |
| Atherosclerosis | Inflammation | P150 | Yes | Alu dsRNA in CTSS mRNA | Disruption of dsRNA structure | [130] |
| Bilateral striatal necrosis (BSN) | Autoimmune | P150 | Yes | Endogenous dsRNA | Interferon; Inhibition of MDA5-MAVS | [36] |
| Breast cancer | Metastasis | P110 | Yes | GABRA3 mRNA | Inhibition of Akt activation, disruption of localization | [77] |
| Cerebellar tumor | Reduced tumor formation | ? | Yes | GLI1 mRNA | Change in structure of mRNA of glioma associated oncogene 1 (GLI1) | [73] |
| Dyschromatosis symmetrica hereditaria (DSH) | Autoimmune | P150 | Yes | Endogenous dsRNA | Interferon; Inhibition of MDA5-MAVS | [34] |
| Epilepsy, acute spinal cord injury | Neurological | ? | Yes | GRIA2 mRNA | Enhanced synaptic responses | [88,89,90,91] |
| Epstein-Barr virus | Anti-viral | ? | Yes | Viral miR-BART6-5p | Suppression of RNA-induced silencing complex (RISC) loading, inhibition of Dicer | [114] |
| Epstein-Barr virus | Switch to lytic cycle | ? | Yes | OriP transcripts | Inhibition of miRNA processing; direct interaction between OriPtL and ADAR1 | [55] |
| Esophageal squamous cell cancer (ESCC) | Cancer cell proliferation | P110 | Yes | FLNB mRNA | ADAR1 gene amplification | [67] |
| Gastric cancer, Non-squamous cell lung carcinoma (NSCLC), colorectal cancer | Metastasis | ? | Yes | RHOQ mRNA | ? | [80] |
| Hepatocellular carcinoma (HCC), ESCC | Cancer cell proliferation | P110 | Yes | AZIN1 mRNA | Increase in ADAR1 levels and editing | [63] |
| Hepatitis C virus | Antiviral | P150 | Yes | Viral RNA | Degradation of edited RNA | [54] |
| Hepatitis D virus | Switch from proliferation to packaging | P110 | Yes | Viral HDAg-S gene | Disruption of translation termination | [52,53] |
| HIV-1 | Proviral | P110 and P150 | Yes | Viral p24 Gag, Rev, and Tat mRNA | Increase in ADAR1 expression | [57,58,59,60,135] |
| HIV-1 | Pro-viral | P150 | No | - | Inhibition of protein kinase R (PKR) phosphorylation | [60] |
| HIV-1 | Antiviral | ? | Yes | HIV-1 envelope glycoprotein | Degradation of edited RNA | [56] |
| Human T-cell leukemia virus 1 and -2 (HTLV-1 and -2) | Pro-viral | P150 | No | - | Inhibition of PKR phosphorylation | [57] |
| Influenza A virus | Antiviral | ? | Yes | Viral ssRNA | TLR7 sensing; IFN | [50] |
| Locomotion, learning disorders | Neurological | ? | Yes | CAPS1 mRNA | Increase in catecholamine secretion | [81] |
| Lung cancer | Metastasis | ? | Yes | miR-381 | ? | [71] |
| Measles virus | Proviral | P150 | Yes | Viral DI-RNA | Destabilization of DI-RNA | [49] |
| Measles virus | Pro-viral | P150 | No | - | Inhibition of PKR phosphorylation | [63] |
| Measles virus | Anti-viral | P150 | Yes | ? | ? | [50] |
| Enhanced metabolism Depression | Neurological | P110 | Yes | HTR2C mRNA | Synaptic transmission | [82,83] |
| Metastatic melanoma, gastric, thyroid and lung cancers | Suppress metastasis | ? | Yes | Pri-miR-455-5p | miRNA processing | [103] |
| NSCLC | Cancer cell proliferation | ? | Yes | NEIL1 mRNA | ADAR1 gene amplification | [72] |
| Pancreatic cancer, papillary thyroid carcinoma, prostate carcinoma and metastatic melanoma | Cancer, metastasis | ? | No | miR-221/222 | Decrease in miR-221/222 synthesis | [119] |
| Prostate cancer | Malignancy | ? | Yes | PCA3/PRUNE2 complex | mRNA translation | [133] |
| Spastic paraplegia | Autoimmune | ? | Yes | Endogenous dsRNA | Interferon; Inhibition of MDA5-MAVS | [37] |
| Subacute sclerosing panencephalitis (SSPE) in measles virus | Decreased viral assembly and release | ? | Yes | Viral M gene | Inhibition of M protein production | [50] |
| Vesicular stomatitis virus | Pro-viral | P150 | No | - | Inhibition of PKR phosphorylation | [22] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, C.; Sakurai, M.; Shiromoto, Y.; Nishikura, K. Functions of the RNA Editing Enzyme ADAR1 and Their Relevance to Human Diseases. Genes 2016, 7, 129. https://doi.org/10.3390/genes7120129
Song C, Sakurai M, Shiromoto Y, Nishikura K. Functions of the RNA Editing Enzyme ADAR1 and Their Relevance to Human Diseases. Genes. 2016; 7(12):129. https://doi.org/10.3390/genes7120129
Chicago/Turabian StyleSong, Chunzi, Masayuki Sakurai, Yusuke Shiromoto, and Kazuko Nishikura. 2016. "Functions of the RNA Editing Enzyme ADAR1 and Their Relevance to Human Diseases" Genes 7, no. 12: 129. https://doi.org/10.3390/genes7120129
APA StyleSong, C., Sakurai, M., Shiromoto, Y., & Nishikura, K. (2016). Functions of the RNA Editing Enzyme ADAR1 and Their Relevance to Human Diseases. Genes, 7(12), 129. https://doi.org/10.3390/genes7120129