
Development of Genetic Testing for Fragile X Syndrome and Associated Disorders, and Estimates of the Prevalence of FMR1 Expansion Mutations

Abstract
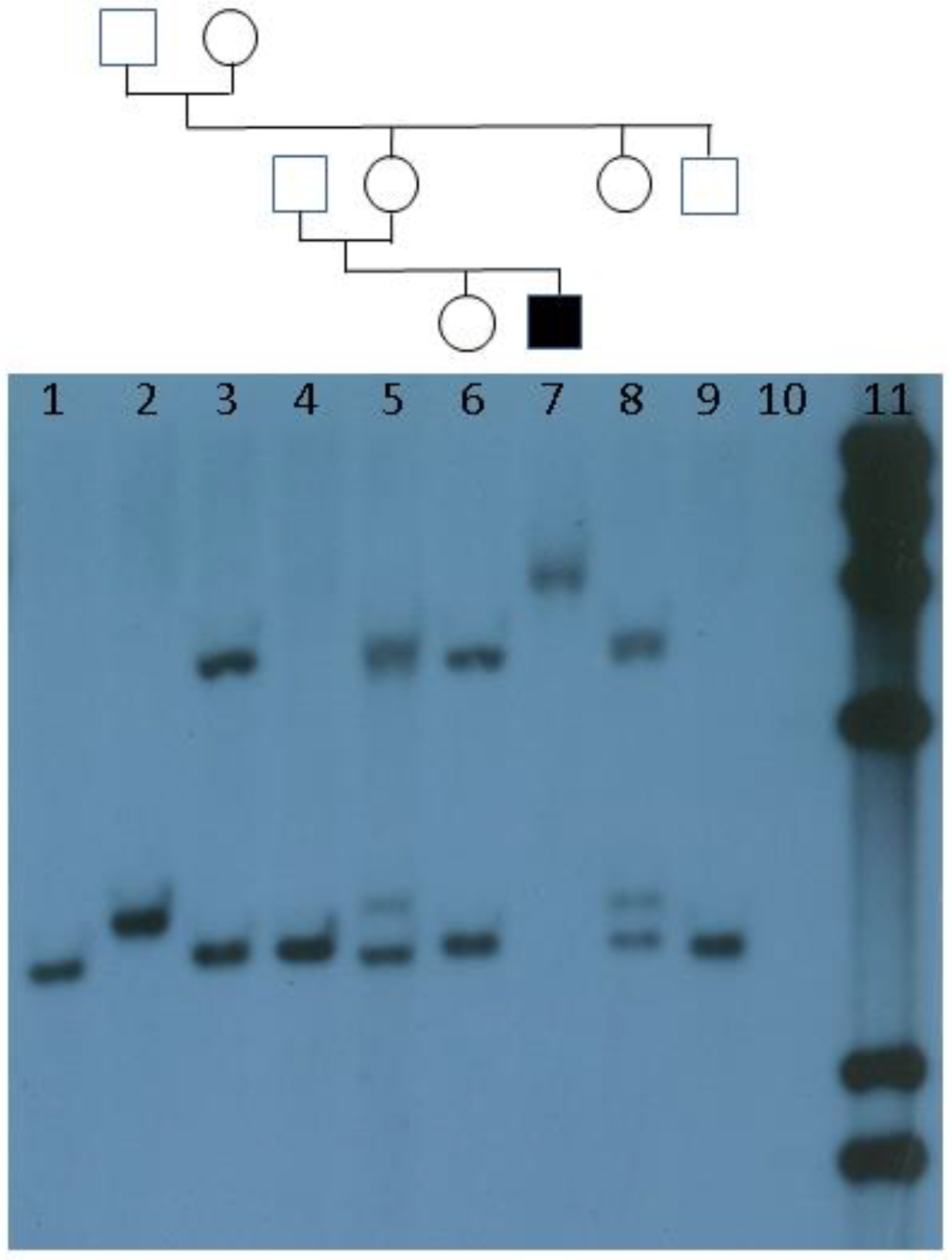
:1. Introduction and History
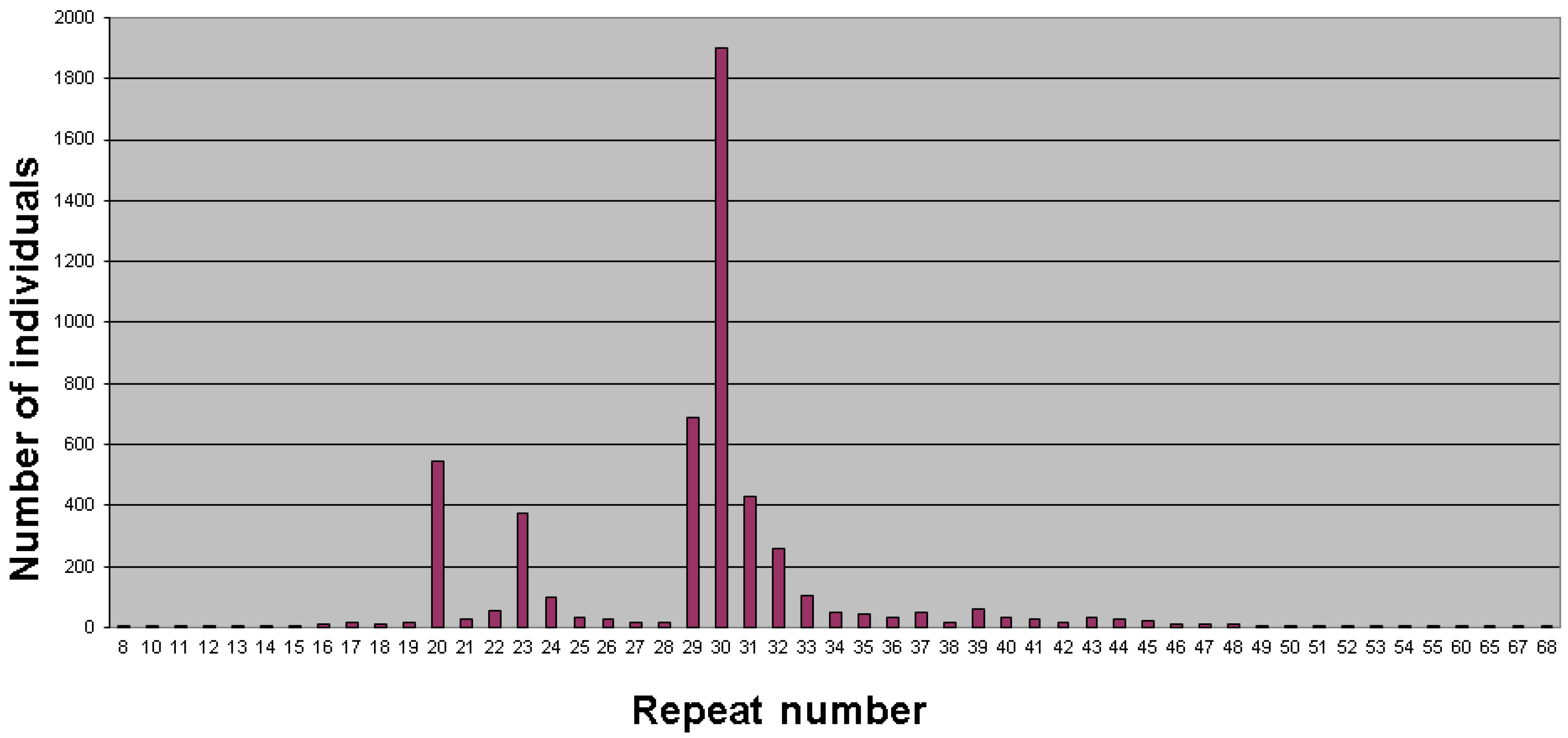
2. Population Studies and Prevalence
3. Factors Affecting Stability and Expansion of the CGG/AGG Repeat Tract
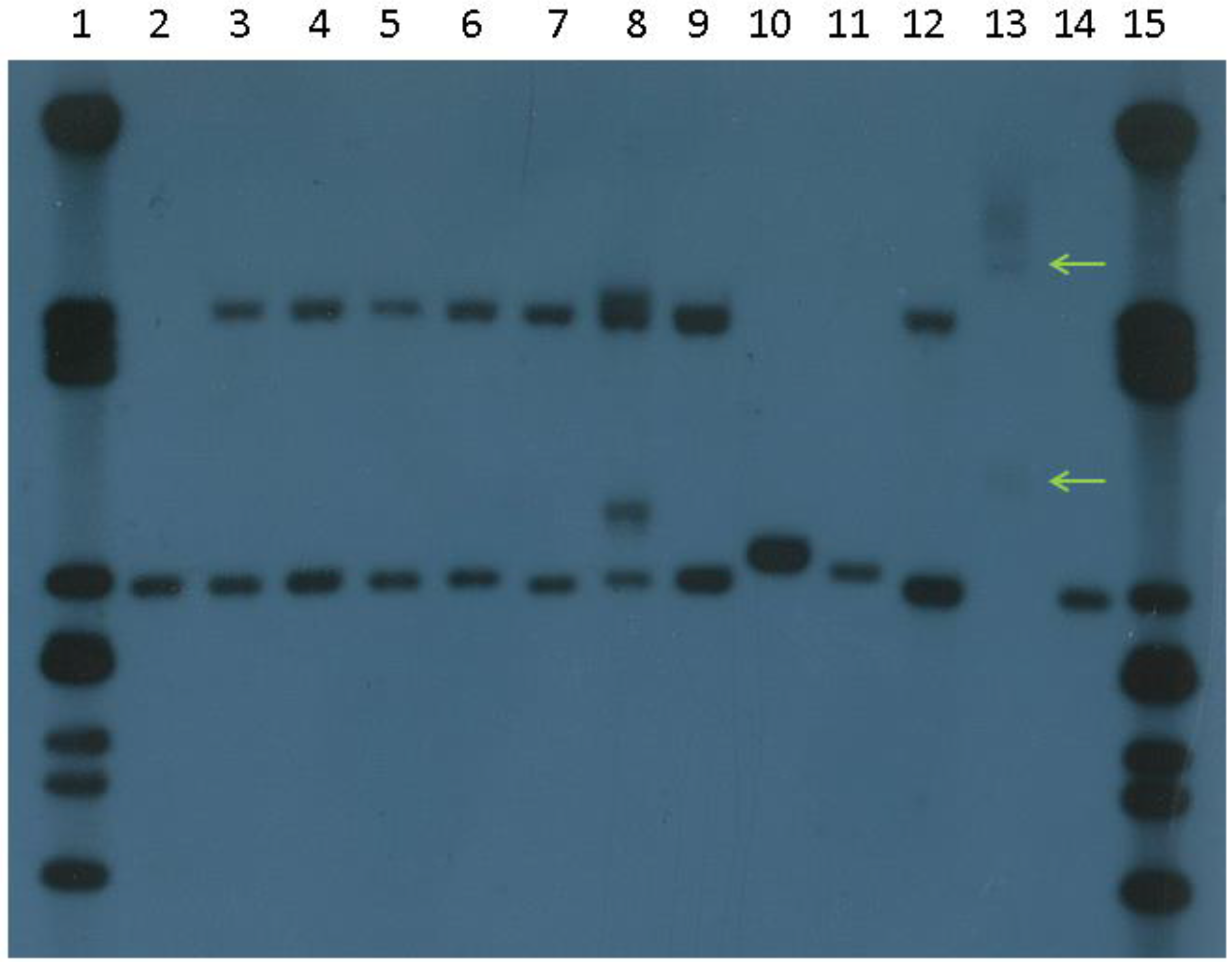
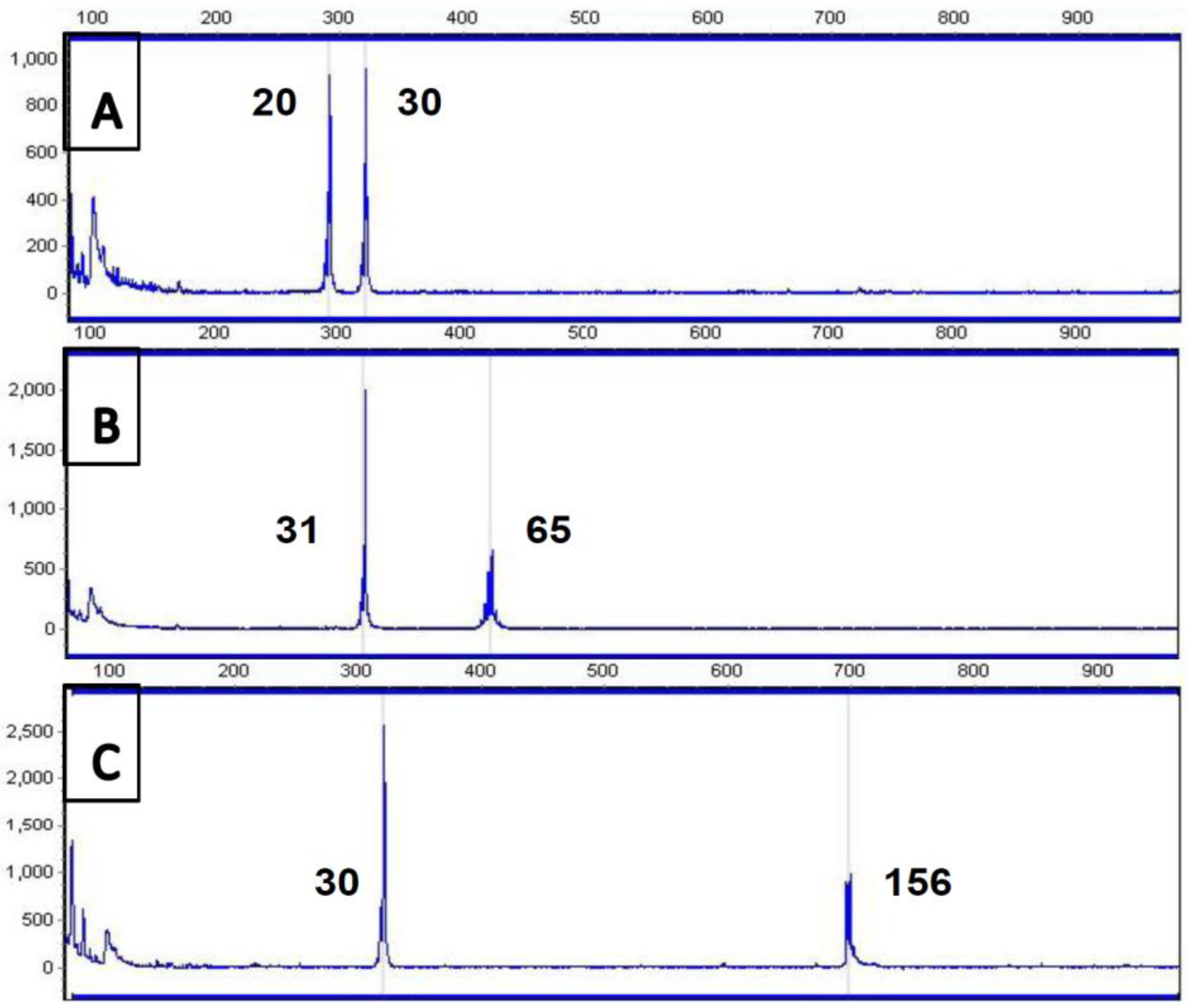
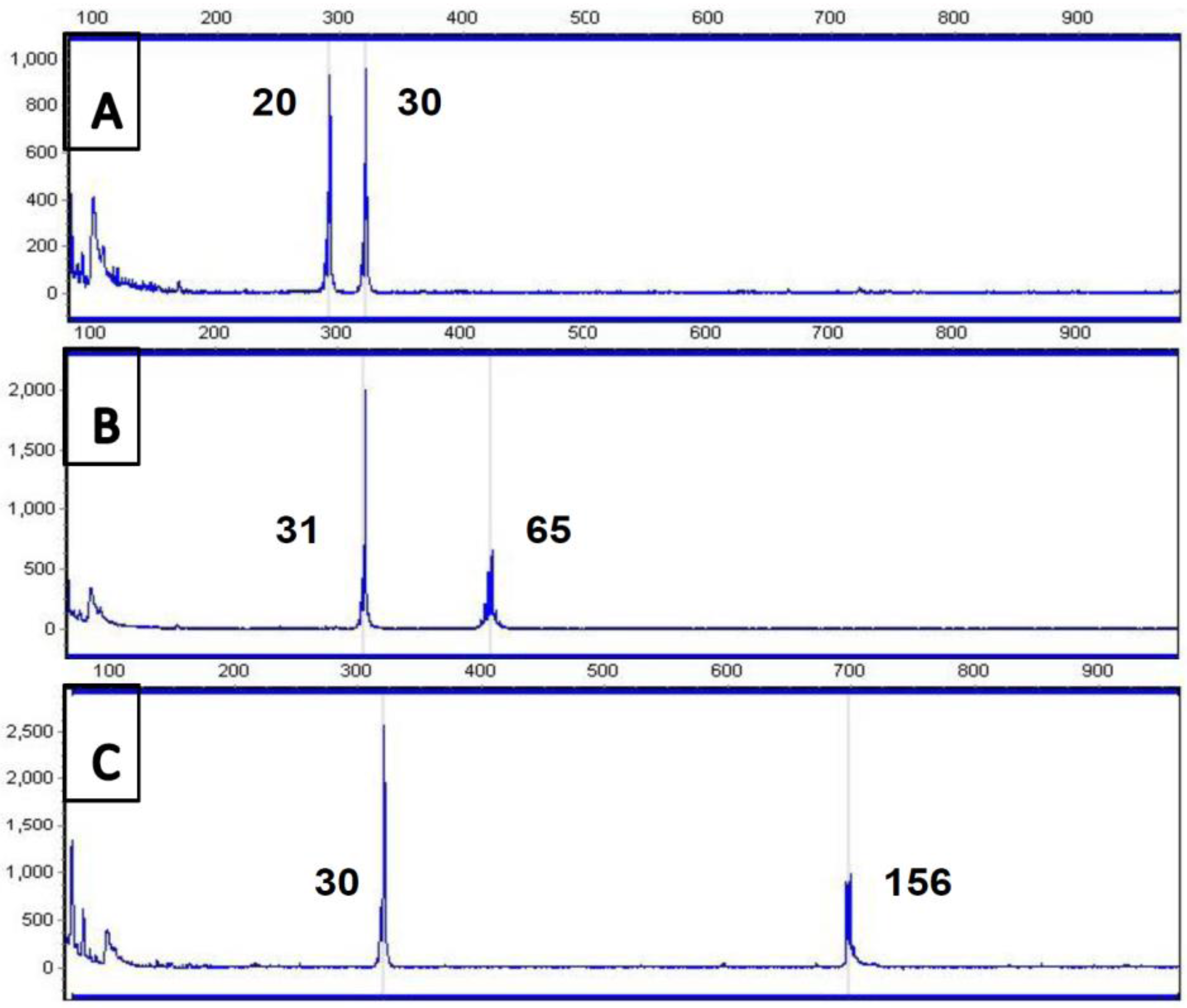
4. Current Diagnostic Procedure
5. Testing Criteria
6. Prenatal Diagnosis
7. Testing for Other FMR1-Related Disorders
8. Interpretation and Reporting
9. Future Directions and Conclusions
9.1. Diagnosis
9.2. Population Screening
9.3. Therapy
9.4. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jacobs, P.A.; Sherman, S.L. The Fragile (X): A marker for the Martin-Bell syndrome. Dis. Markers 1985, 3, 9–25. [Google Scholar]
- Webb, T.P.; Bundey, S.; Thake, A.; Todd, J. The frequency of the fragile X chromosome among schoolchildren in Coventry. J. Med. Genet. 1986, 23, 396–399. [Google Scholar] [CrossRef] [PubMed]
- Turner, G.; Robinson, H.; Laing, S.; van den Berk, M.; Colley, A.; Goddard, A.; Sherman, S.; Partington, M. Population screening for fragile X. Lancet 1992, 339, 1210–1213. [Google Scholar] [CrossRef]
- Chudley, A.E.; Hagerman, R.J. Fragile X syndrome. J. Paediatr. 1987, 110, 821–831. [Google Scholar] [CrossRef]
- Sherman, S.L.; Jacobs, P.A.; Morton, N.E.; Froster-Iskenius, U.; Howard-Peebles, P.N.; Nielsen, K.B.; Partington, M.W.; Sutherland, G.R.; Turner, G.; Watson, M. Further segregation analysis of the fragile X syndrome with special reference to transmitting males. Hum. Genet. 1985, 69, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Pembrey, M.E.; Winter, R.M.; Davies, K.E.; Opitz, J.M.; Reynolds, J.F. A premutation that generates a defect at crossing-over explains the inheritance of fragile X mental retardation. Am. J. Med. Genet. Part A 1985, 21, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Oberlé, I.; Rousseau, F.; Heitz, D.; Kretz, C.; Devys, D.; Hanauer, A.; Boué, J.; Bertheas, M.F.; Mandel, J.L. Instability of a 550-base pair DNA segment and abnormal methylation in Fragile X syndrome. Science 1991, 252, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.J.M.H.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.-H.; Kuhl, D.P.A.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in Fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Kremer, E.J.; Pritchard, M.; Lynch, M.; Yu, S.; Holman, K.; Baker, E.; Warren, S.T.; Schlessinger, D.; Sutherland, G.R.; Richards, R.I. Mapping of DNA instability at the Fragile X to a trinucleotide repeat sequence p(CCG)n. Science 1991, 252, 1711–1714. [Google Scholar] [CrossRef] [PubMed]
- Nakahori, Y.; Knight, S.J.L.; Holland, J.; Schwartz, C.; Roche, A.; Tarleton, J.; Wong, S.; Flint, T.J.; Froster-Iskenius, U.; Bentley, D.; et al. Molecular heterogeneity of the fragile X syndrome. Nucl. Acids Res. 1991, 19, 4355–4359. [Google Scholar] [CrossRef] [PubMed]
- Jin, P.; Warren, S.T. Understanding the molecular basis of fragile X syndrome. Hum. Mol. Genet. 2000, 9, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, G.R.; Richards, R.I. Simple tandem repeats and human genetic disease. Proc. Natl. Acad. Sci. USA 1995, 92, 3636–3641. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.-H.; Kuhl, D.P.A.; Pizzuti, A.; Pieretti, M.; Sutcliffe, J.S.; Richards, S.; Verkerk, A.J.M.H.; Holden, J.J.A.; Fenwick, R.G., Jr.; Warren, S.T.; et al. Variation of the CGG repeat at the Fragile X site results in genetic instability: Resolution of the Sherman paradox. Cell 1991, 67, 1047–1058. [Google Scholar] [CrossRef]
- Rousseau, F.; Heitz, D.; Biancalana, V.; Blumenfeld, S.; Kretz, C.; Boué, J.; Tommerup, N.; van der Hagen, C.; DeLozier-Blanchet, C.; Croquette, M.-F.; et al. Direct diagnosis by DNA analysis of the Fragile X syndrome of mental retardation. New Eng. J. Med. 1991, 325, 1673–1681. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.J.; Hull, C.E.; Safanda, J.F.; Carpenter, I.; Staley, L.W.; O’Connor, R.A.; Seydel, C.; Mazzocco, M.M.M.; Snow, K.; Thibodeau, S.N.; et al. High functioning Fragile X males: Demonstration of an unmethylated fully expanded FMR-1 mutation associated with protein expression. Am. J. Med. Genet. 1994, 51, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Knight, S.J.L.; Flannery, A.V.; Hirst, M.C.; Campbell, L.; Christodoulou, Z.; Phelps, S.R.; Pointon, J.; Middleton-Price, H.R.; Barnicoat, A.; Pembrey, M.E.; et al. Trinucleotide repeat amplification and hypermethylation of a CpG island in FRAXE mental retardation. Cell 1993, 74, 127–134. [Google Scholar] [CrossRef]
- Macpherson, J.N.; Nelson, D.L.; Jacobs, P.A. Frequent small amplifications in the FMR-1 gene in fra(X) families: Limits to the diagnosis of ‘premutations’. J. Med. Genet. 1992, 29, 802–806. [Google Scholar] [CrossRef] [PubMed]
- Turner, G.; Webb, T.; Wake, S.; Robinson, H. Prevalence of Fragile X Syndrome. Am. J. Med. Genet. 1996, 64, 196–197. [Google Scholar] [CrossRef]
- Youings, S.A.; Murray, A.; Dennis, N.; Ennis, S.; Lewis, C.; McKechnie, N.; Pound, M.; Sharrock, A.; Jacobs, P. FRAXA and FRAXE: The results of a five year survey. J. Med. Genet. 2000, 37, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Falik-Zaccai, T.C.; Shachak, E.; Yalon, M.; Lis, Z.; Borochowitz, Z.; Macpherson, J.N.; Nelson, D.L.; Eichler, E.E. Predisposition to the Fragile X Syndrome in Jews of Tunisian descent is due to the absence of AGG interruptions on a rare Mediterranean haplotype. Am. J. Hum. Genet. 1997, 60, 103–112. [Google Scholar] [PubMed]
- Crawford, D. C.; Meadows, K.L.; Newman, J.L.; Taft, L.F.; Scott, E.; Leslie, M.; Shubek, L.; Holmgreen, P.; Yeargin-Allsopp, M.; Boyle, C.; et al. Prevalence of the fragile X syndrome in African-Americans. Am. J. Med. Genet. 2002, 110, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Nolin, S.L.; Brown, W.T.; Glicksman, A.; Houck, G.E.; Gargano, A.D.; Sullivan, A.; Biancalana, V.; Bröndum-Nielsen, K.; Hjalgrim, H.; Holinski-Feder, E.; et al. Expansion of the Fragile X CGG repeat in females with premutation or intermediate alleles. Am. J. Hum. Genet. 2003, 72, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Carvajal, I.; Lopez Posadas, B.; Pan, R.; Raske, C.; Hagerman, P.J.; Tassone, F. Expansion of an FMR1 grey-zone allele to a full mutation in two generations. J. Mol. Diagn. 2009, 11, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Murray, A.; Macpherson, J.N.; Pound, M.C.; Sharrock, A.; Youings, S.A.; Dennis, N.R.; McKechnie, N.; Linehan, P.; Morton, N.E.; Jacobs, P.A. The role of size, sequence and haplotype in the stability of FRAXA and FRAXE alleles during transmission. Hum. Mol. Genet. 1997, 6, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Eichler, E.E.; Holden, J.J.A.; Popovich, B.W.; Reiss, A.L.; Snow, K.; Thibodeau, S.N.; Richards, C.S.; Ward, P.A.; Nelson, D.L. Length of uninterrupted CGG repeats determines instability in the FMR1 gene. Nature Genet. 1994, 8, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Eichler, E.E.; Macpherson, J.N.; Murray, A.; Jacobs, P.A.; Chakravarti, A.; Nelson, D.L. Haplotype and interspersion analysis of the FMR1 CGG repeat identifies two different mutational pathways for the origin of the fragile X syndrome. Hum. Mol. Genet. 1996, 5, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Gunter, C.; Paradee, W.; Crawford, D.C.; Meadows, K.A.; Newman, J.; Kunst, C.B.; Nelson, D.L.; Schwartz, C.; Murray, A.; Macpherson, J.N.; et al. Re-examination of factors associated with expansion of CGG repeats using a single nucleotide polymorphism in FMR1. Hum. Mol. Genet. 1998, 7, 1935–1946. [Google Scholar] [CrossRef] [PubMed]
- Larsen, L.A.; Armstrong, J.S.M.; Grønskov, K.; Hjalgrim, H.; Macpherson, J.N.; Brøndum-Nielsen, K.; Hasholt, L.; Nørgaard-Pedersen, B.; Vuust, J. Haplotype and AGG-interspersion analysis of FMR1 (CGG)n alleles in the Danish population: Implications for multiple mutational pathways towards Fragile X alleles. Am. J. Med. Genet. 2000, 93, 99–106. [Google Scholar] [CrossRef]
- Nolin, S.L.; Glicksman, A.; Ersalesi, N.; Dobkin, C.; Brown, W.T.; Cao, R.; Blatt, E.; Sah, S.; Latham, G.J.; Hadd, A.G. Fragile X full mutation expansions are inhibited by one or more AGG interruptions in premutation carriers. Genet. Med. 2015, 17, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Kunst, C.B.; Warren, S.T. Cryptic and polar variation of the Fragile X repeat could result in predisposing normal alleles. Cell 1994, 77, 853–861. [Google Scholar] [CrossRef]
- Zhong, N.; Yang, W.; Dobkin, C.; Brown, W.T. Fragile X gene instability: Anchoring AGGs and linked microsatellites. Am. J. Hum. Genet. 1995, 57, 351–361. [Google Scholar] [PubMed]
- Macpherson, J.N.; Bullman, H.; Youings, S.A.; Jacobs, P.A. Insert size and flanking haplotype in fragile X and normal populations: Possible multiple origins for the fragile X mutation. Hum. Mol. Genet. 1994, 3, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Snow, K.; Tester, D.J.; Kruckeberg, K.E.; Schaid, D.J.; Thibodeau, S.N. Sequence analysis of the fragile X trinucleotide repeat: Implications for the origin of the fragile X mutation. Hum. Mol. Genet. 1994, 3, 1543–1551. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Santhana Mariappan, S.V.; Catasti, P.; Ratliff, R.; Moyzis, R.K.; Laayoun, A.; Smith, S.S.; Morton Bradbury, E.; Gupta, G. Hairpins are formed by the single DNA strands of the fragile X triplet repeats: Structure and biological implications. Proc. Natl. Acad. Sci. USA 1995, 92, 5199–5203. [Google Scholar] [CrossRef] [PubMed]
- Sinden, R.R.; Potaman, V.N.; Oussatcheva, E.A.; Pearson, C.E.; Lyubchenko, Y.L.; Shlyakhtenko, L.S. Triplet repeat DNA structures and human genetic disease: Dynamic mutations from dynamic DNA. J. Biosci. 2002, 27, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Ennis, S.; Murray, A.; Brightwell, G.; Morton, N.E.; Jacobs, P.A. Closely linked cis-acting modifier of expansion of the CGG repeat in high risk FMR1 haplotypes. Hum. Mutat. 2007, 28, 1216–1224. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, J.; Zaninovic, N.; Zhan, Q.; Madireddy, A.; Nolin, S.L.; Ersalesi, N.; Yan, Z.; Rosenwaks, Z.; Schildkraut, C.L. Cis-acting DNA sequence at a replication origin promotes repeat expansion to a fragile X full mutation. J. Cell Biol. 2014, 206, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Saluto, A.; Brussino, A.; Tassone, F.; Arduino, C.; Cagnoli, C.; Pappi, P.; Hagerman, P.; Migone, N.; Brusco, A. An enhanced polymerase chain reaction assay to detect pre- and full mutation alleles of the Fragile X Mental Retardation 1 gene. J. Mol. Diagn. 2005, 7, 605–612. [Google Scholar] [CrossRef]
- De Vries, B.B.A.; Wiegers, A.M.; Smits, A.P.T.; Mohkamsing, S.; Duivenvoorden, H.J.; Fryns, J.-P.; Curfs, L.M.G.; Halley, D.J.J.; Oostra, B.A.; van den Ouweland, A.M.W.; et al. Mental status of females with an FMR1 gene full mutation. Am. J. Hum. Genet. 1996, 58, 1025–1032. [Google Scholar] [PubMed]
- Rajan-Babu, I.S.; Teo, C.R.; Lian, M.; Lee, C.G.; Law, H.Y.; Chong, S.S. Single-tube methylation-specific duplex-PCR assay for rapid and accurate diagnosis of Fragile X Mental Retardation 1-related disorders. Expert Rev. Mol. Diagn. 2015, 15, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Loomis, E.W.; Eid, J.S.; Peluso, P.; Yin, J.; Hickey, L.; Rank, D.; McCalmon, S.; Hagerman, R.J.; Tassone, F.; Hagerman, P.J. Sequencing the unsequenceable: Expanded CGG-repeat alleles of the fragile X gene. Genome Res. 2013, 23, 121–128. [Google Scholar] [CrossRef] [PubMed]
- United Kingdom Genetic Testing Network (UKGTN) testing criteria for Fragile X Mental Retardation Syndrome. Available online: www.ukgtn.nhs.uk (accessed on 5 September 2016).
- Allingham-Hawkins, D.J.; Babul-Hirji, R.B.; Chitayat, D.; Holden, J.J.A.; Yang, K.T.; Lee, C.; Hudson, R.; Gorwill, H.; Nolin, S.L.; Glicksman, A.; et al. Fragile X premutation is a significant risk factor for premature ovarian failure: The International Collaborative POF in Fragile X Study—preliminary data. Am. J. Med. Genet. 1999, 83, 322–325. [Google Scholar] [CrossRef]
- Ennis, S.; Ward, D.; Murray, A. Nonlinear association between CGG repeat number and age of menopause in FMR1 premutation carriers. Eur. J. Hum. Genet. 2006, 14, 253–255. [Google Scholar] [CrossRef] [PubMed]
- Ruth, K.S.; Bennett, C.E.; Schoemaker, M.J.; Weedon, M.N.; Swerdlow, A.J.; Murray, A. Length of FMR1 repeat alleles within the normal range does not substantially affect the risk of early menopause. Hum. Reprod. 2016, 31, 2396–2403. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.E.; Conway, G.S.; Macpherson, J.N.; Jacobs, P.A.; Murray, A. Intermediate sized CGG repeats are not a common cause of idiopathic premature ovarian failure. Hum. Reprod. 2010, 25, 1335–1338. [Google Scholar] [CrossRef] [PubMed]
- Murray, A.; Schoemaker, M.J.; Bennett, C.E.; Ennis, S.; Macpherson, J.N.; Jones, M.; Morris, D.H.; Orr, N.; Ashworth, A.; Jacobs, P.A. Population-based estimates of the prevalence of FMR1 expansion mutations in women with early menopause and primary ovarian insufficiency. Genet. Med. 2014, 16, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Murray, A.; Ennis, S.; MacSwiney, F.; Webb, J.; Morton, N.E. Reproductive and menstrual history of females with fragile X expansions. Eur. J. Hum. Genet. 2000, 8, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Jacquemont, S.; Hagerman, R.J.; Leehey, M.A.; Hall, D.A.; Levine, R.A.; Brunberg, J.A.; Zhang, L.; Jardini, T.; Gane, L.W.; Harris, S.W.; et al. Penetrance of the Fragile X-associated Tremor/Ataxia Syndrome in a premutation carrier population. JAMA 2004, 291, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Inaba, Y.; Schwartz, C.E.; Bui, Q.M.; Li, X.; Skinner, C.; Field, M.; Wotton, T.; Hagerman, R.J.; Francis, D.; Amor, D.J.; et al. Early detection of Fragile X Syndrome: Applications of a novel approach for improved quantitative methylation analysis in venous blood and newborn blood spots. Clin. Chem. 2014, 60, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Aliaga, S.M.; Slater, H.R.; Francis, D.; Du Sart, D.; Li, X.; Amor, D.J.; Alliende, A.M.; Santa Maria, L.; Faundes, V.; Morales, P.; et al. Identification of males with cryptic Fragile X alleles by methylation-specific quantitative melt analysis. Clin. Chem. 2016, 62, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Rifé, M.; Badenas, C.; Mallolas, J.; Jiménez, L.; Cervera, R.; Maya, A.; Glover, G.; Rivera, F.; Milà, M. Incidence of Fragile X in 5,000 consecutive newborn males. Genetic Testing 2003, 7, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Tassone, F.; Iong, K.P.; Tong, T.-H.; Lo, J.; Gane, L.W.; Berry-Kravis, E.; Nguyen, D.; Mu, L.Y.; Laffin, J.; Bailey, D.B.; et al. FMR1 CGG allele size and prevalence ascertained through newborn screening in the United States. Genome Med. 2012. [Google Scholar] [CrossRef] [PubMed]
- Toledano-Alhadef, H.; Basel-Vanagaite, L.; Magal, N.; Davidov, B.; Ehrlich, S.; Drasinover, V.; Taub, E.; Halpern, G.J.; Ginott, N.; Shohat, M. Fragile-X carrier screening and the prevalence of premutation and full-mutation carriers in Israel. Am. J. Hum. Genet. 2001, 69, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Berkenstadt, M.; Ries-Levavi, L.; Cuckle, H.; Peleg, L.; Barkai, G. Preconceptual and prenatal screening for fragile X syndrome: Experience with 40 000 tests. Prenat. Diagn. 2007, 27, 991–994. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, R.; Mohkamsing, S.; De Vries, B.; Devys, D.; van den Ouweland, A.; Mandel, J.-L.; Galjaard, H.; Oostra, B. Rapid antibody test for fragile X syndrome. Lancet 1995, 345, 1147–1148. [Google Scholar] [CrossRef]
- Lozano, R.; Martinez-Cerdeno, V.; Hagerman, R.J. Advances in the understanding of the gabaergic neurobiology of FMR1 expanded alleles leading to targeted treatments for Fragile X Spectrum disorder. Curr. Pharm. Des. 2015, 21, 4972–4979. [Google Scholar] [CrossRef] [PubMed]
- Kumari, D.; Usdin, K. Sustained expression of FMR1 mRNA from reactivated fragile X syndrome alleles after treatment with small molecules that prevent trimethylation of H3K27. Hum. Mol. Genet. 2016. [Google Scholar] [CrossRef] [PubMed]

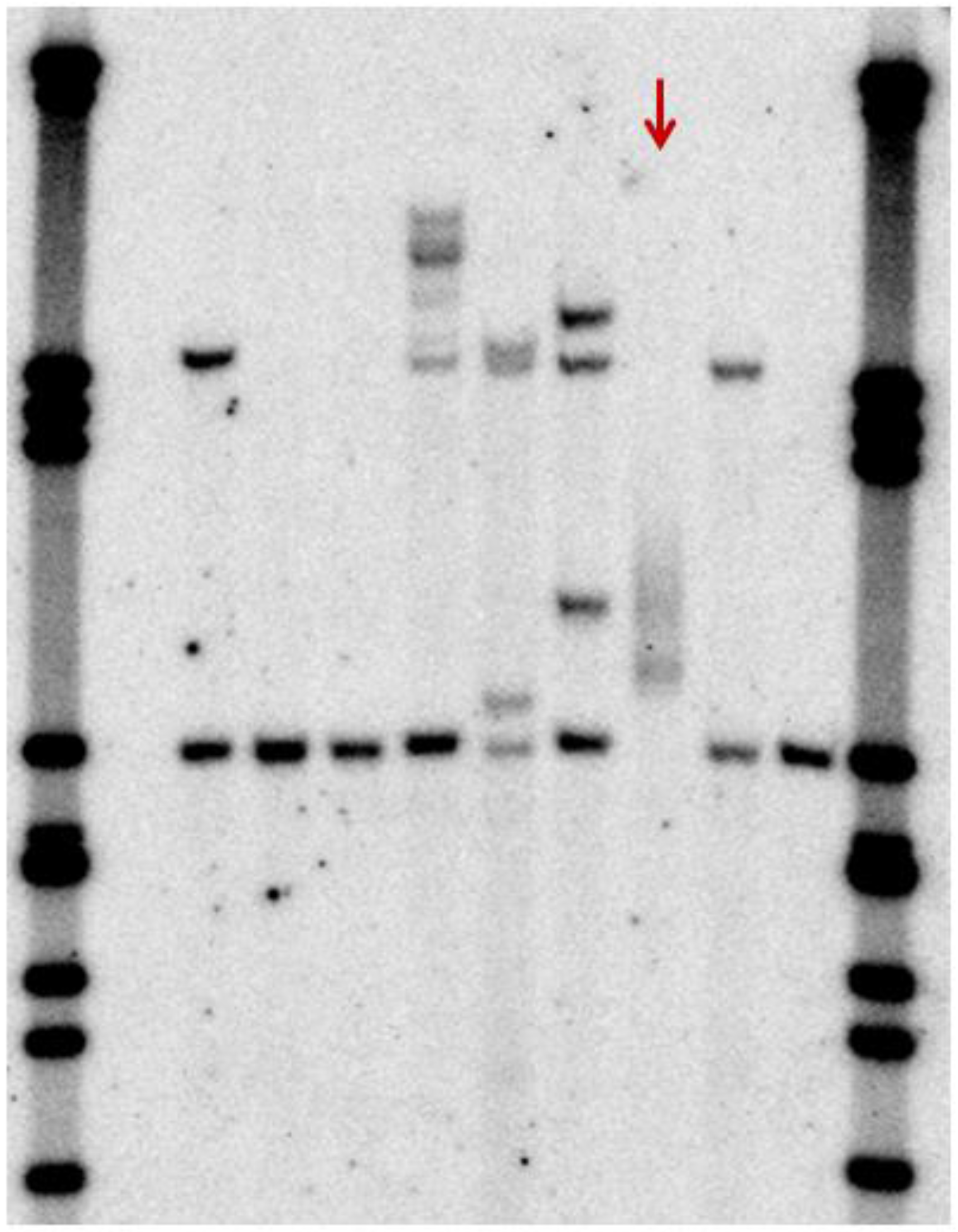

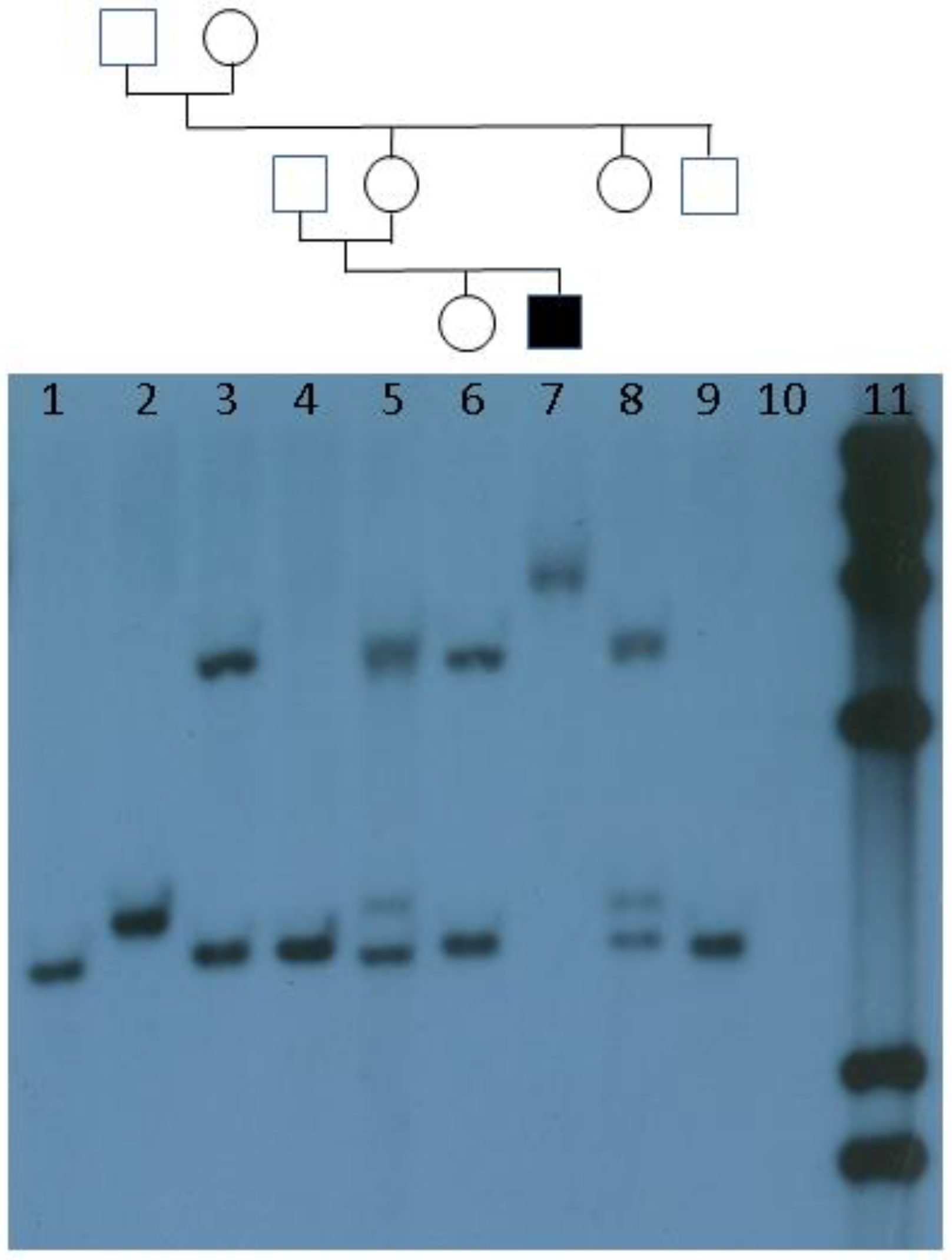{kind=link}
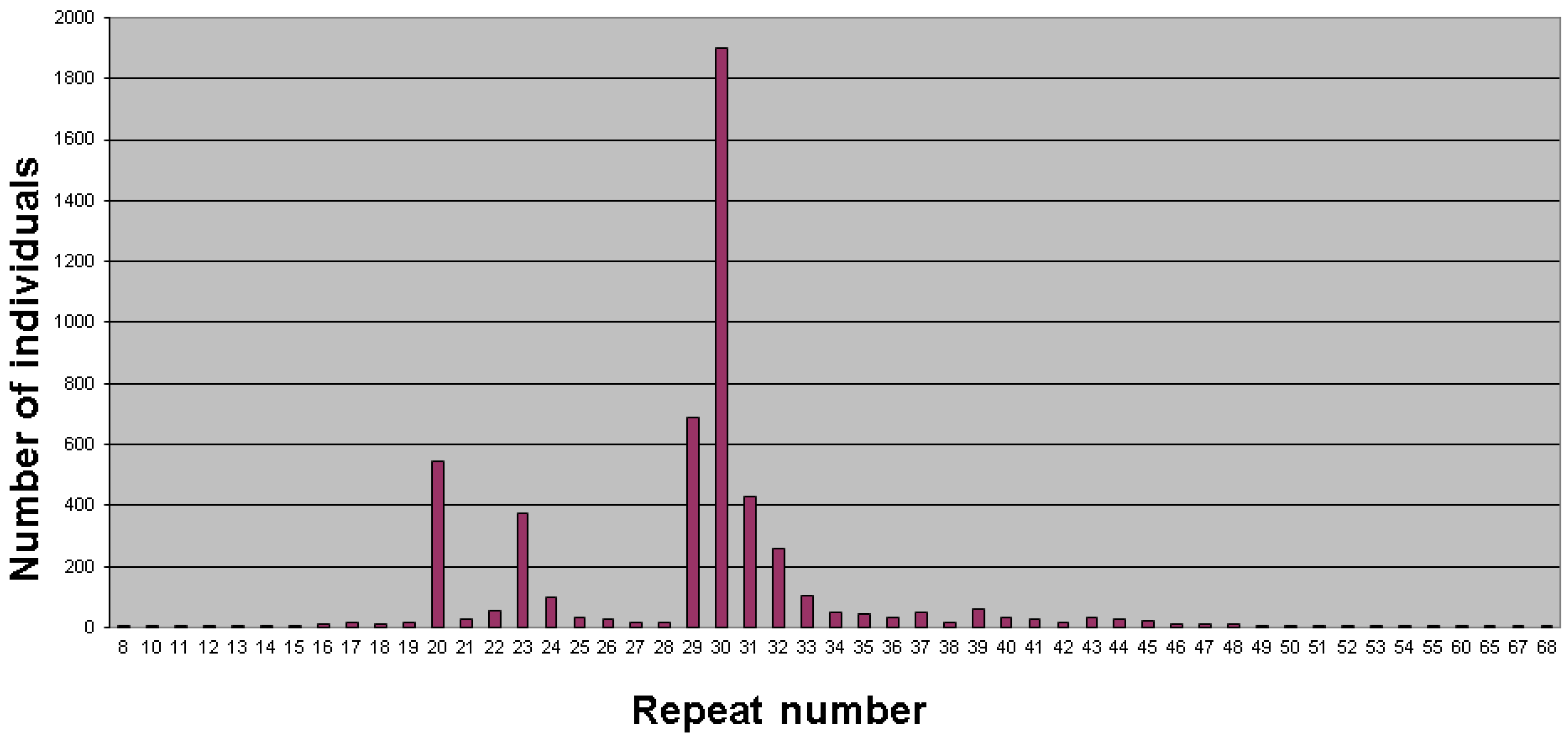{kind=link}
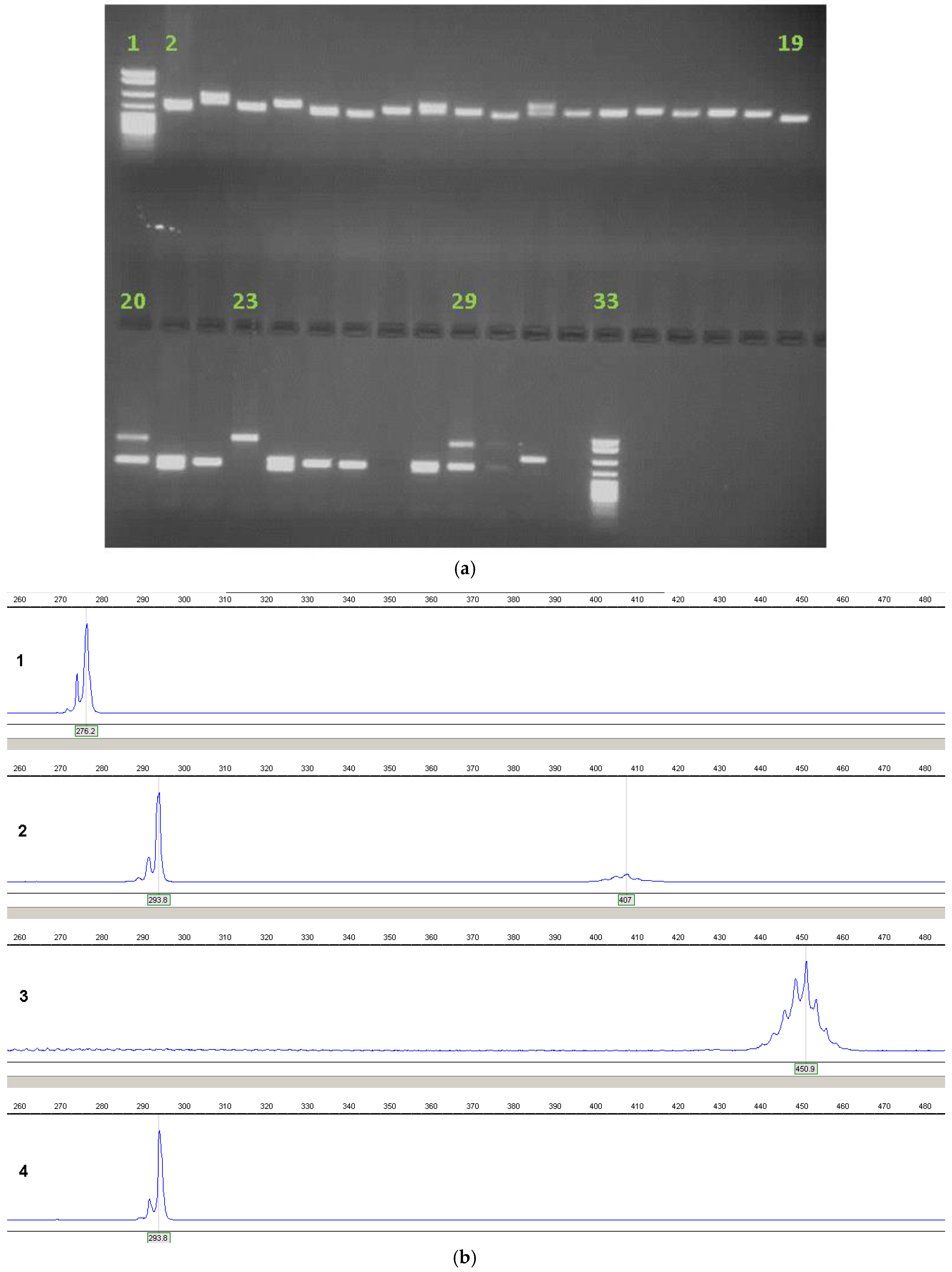{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Allele | UK | USA | ||
|---|---|---|---|---|
| Repeat size | Methylation status | Repeat size | Methylation status | |
| Normal | 0–45 | Regular | 0–44 | Regular |
| Intermediate | 46–58 | Regular | 45–54 | Regular |
| Premutation | 59–200 * | Regular | 55–200 * | Regular |
| Full mutation | >200 * | Hypermethylated * | >200 * | Hypermethylated * |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Macpherson, J.N.; Murray, A. Development of Genetic Testing for Fragile X Syndrome and Associated Disorders, and Estimates of the Prevalence of FMR1 Expansion Mutations. Genes 2016, 7, 110. https://doi.org/10.3390/genes7120110
Macpherson JN, Murray A. Development of Genetic Testing for Fragile X Syndrome and Associated Disorders, and Estimates of the Prevalence of FMR1 Expansion Mutations. Genes. 2016; 7(12):110. https://doi.org/10.3390/genes7120110
Chicago/Turabian StyleMacpherson, James N., and Anna Murray. 2016. "Development of Genetic Testing for Fragile X Syndrome and Associated Disorders, and Estimates of the Prevalence of FMR1 Expansion Mutations" Genes 7, no. 12: 110. https://doi.org/10.3390/genes7120110
APA StyleMacpherson, J. N., & Murray, A. (2016). Development of Genetic Testing for Fragile X Syndrome and Associated Disorders, and Estimates of the Prevalence of FMR1 Expansion Mutations. Genes, 7(12), 110. https://doi.org/10.3390/genes7120110