
Transition and Transversion Mutations Are Biased towards GC in Transposons of Chilo suppressalis (Lepidoptera: Pyralidae)

Abstract
:1. Introduction
2. Material and Methods
2.1. Sample Collection and DNA Isolation
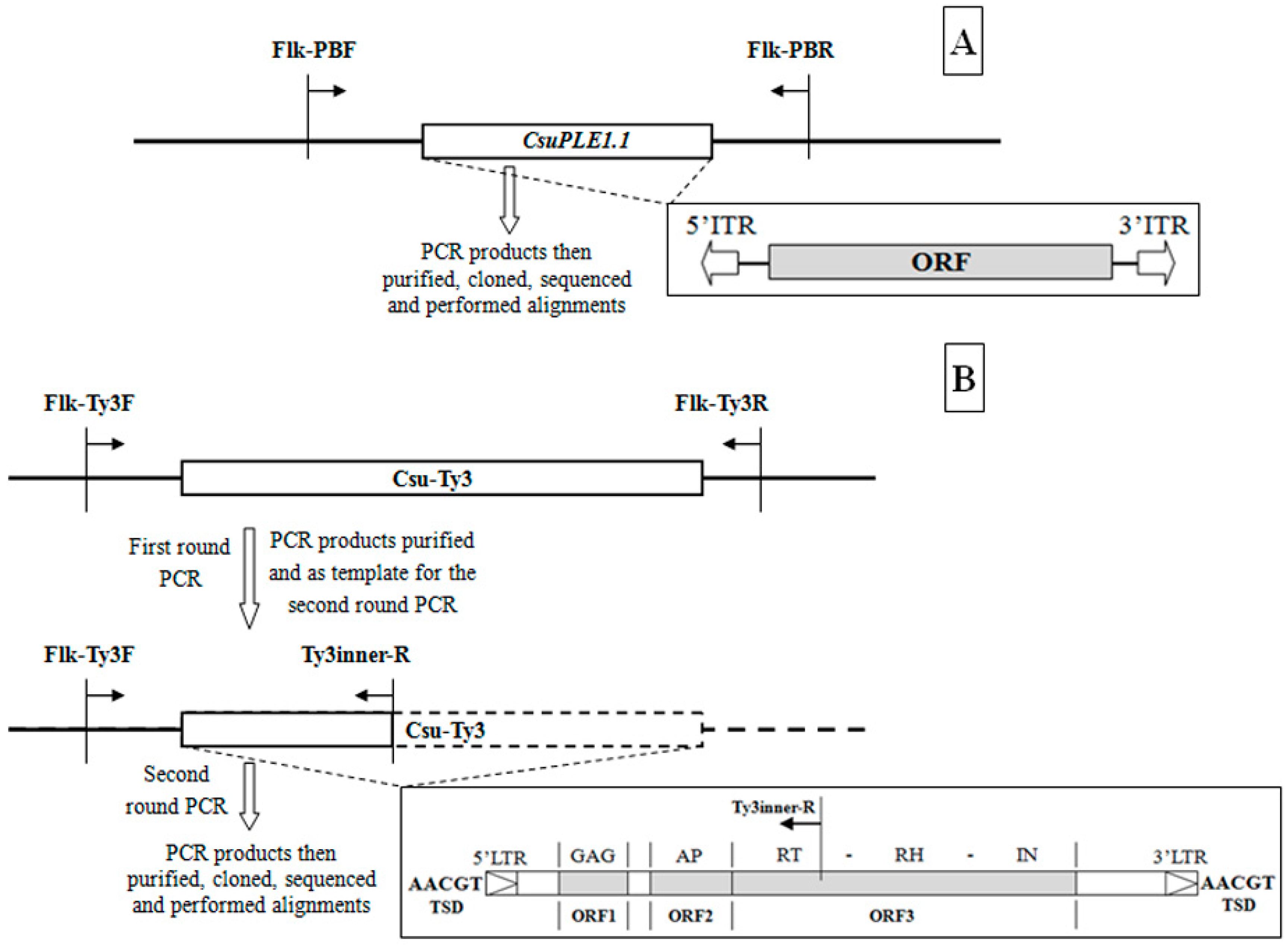
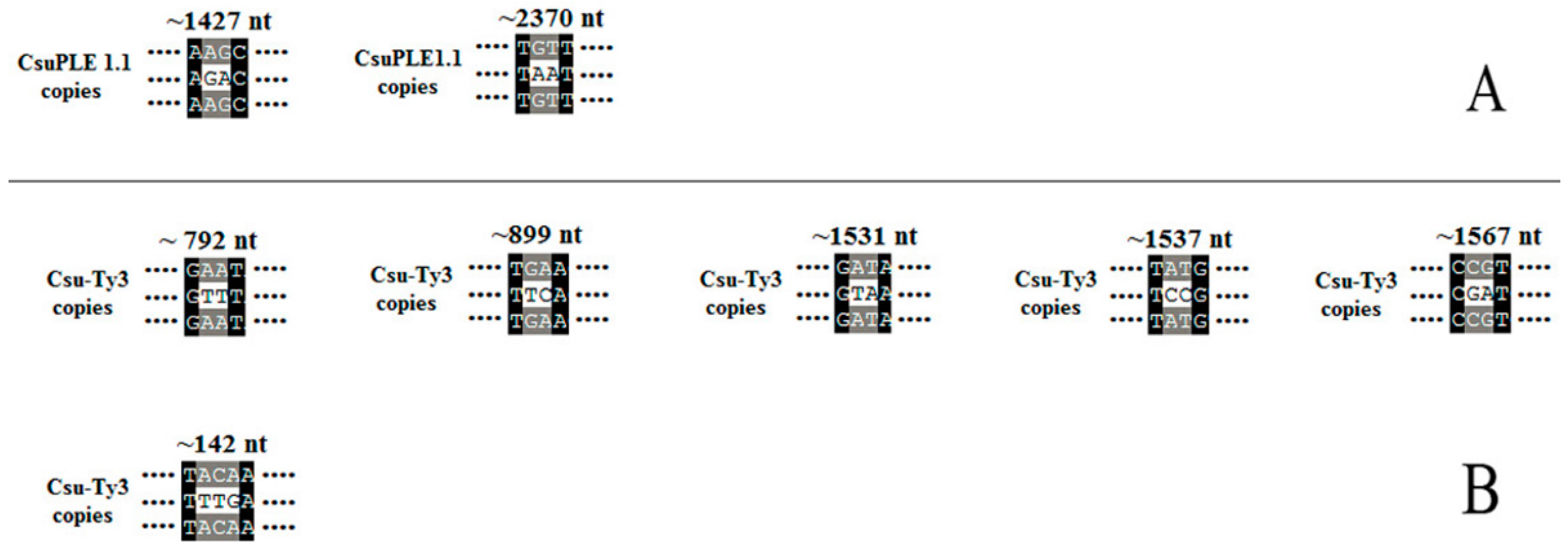
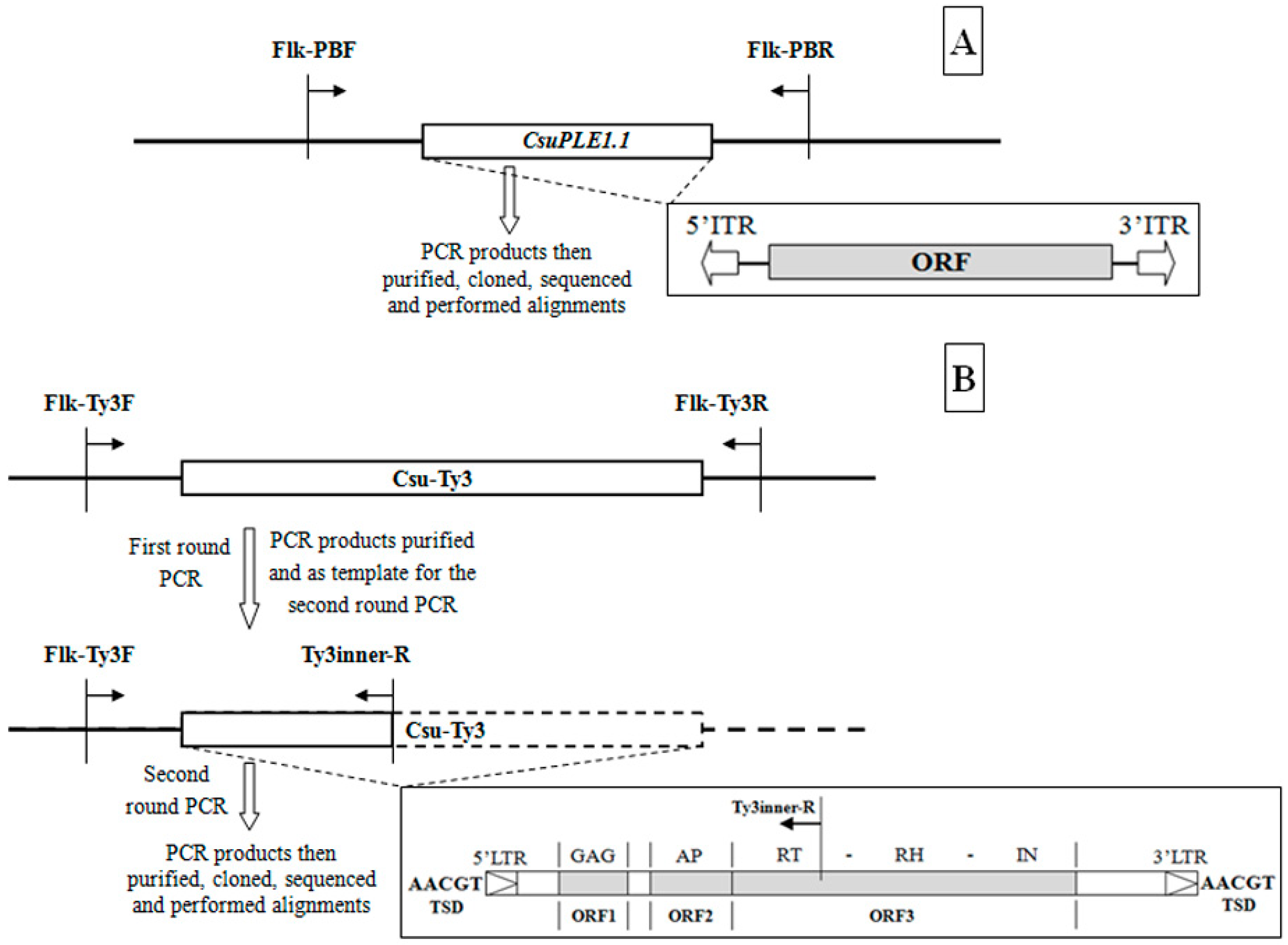
2.2. Determination of the CsuPLE1.1 Copy at the Same Locus in Different Individuals
2.3. Determination of the Csu-Ty3 Copy at the Same Locus in Different Individuals
2.4. Substitution, Deletion and Insertion Mutations Analyses
2.5. Statistical Analysis
3. Results
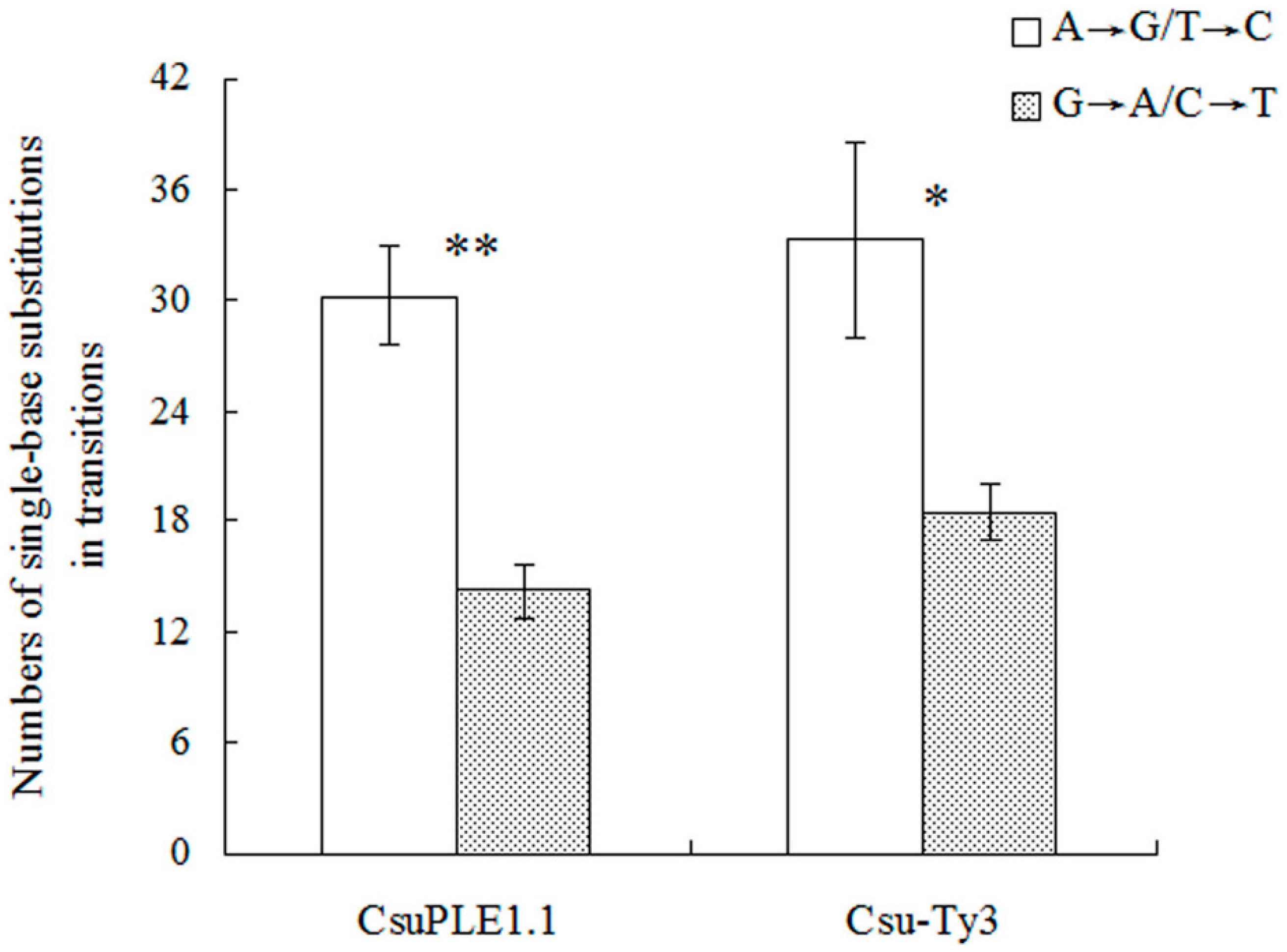
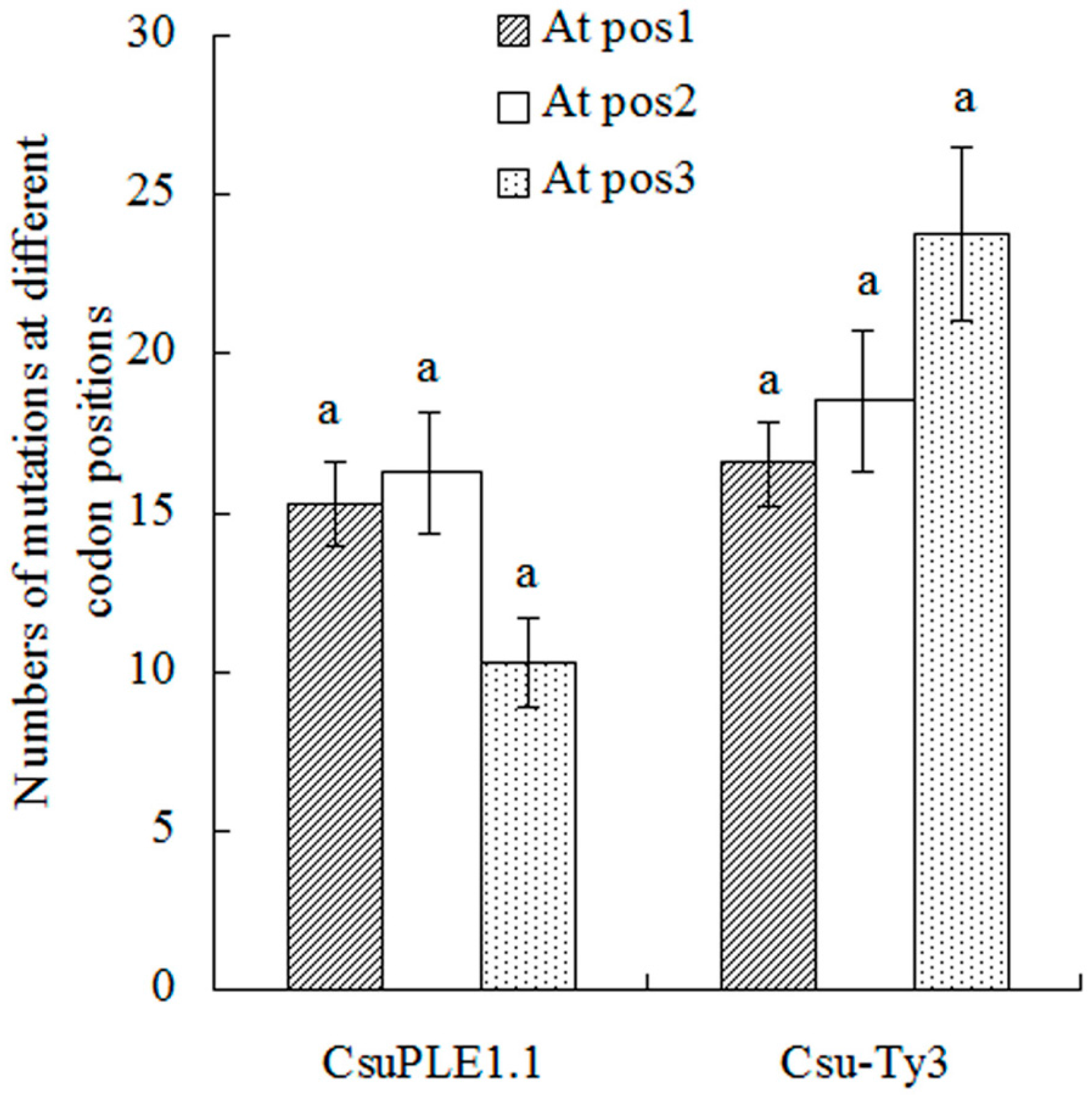
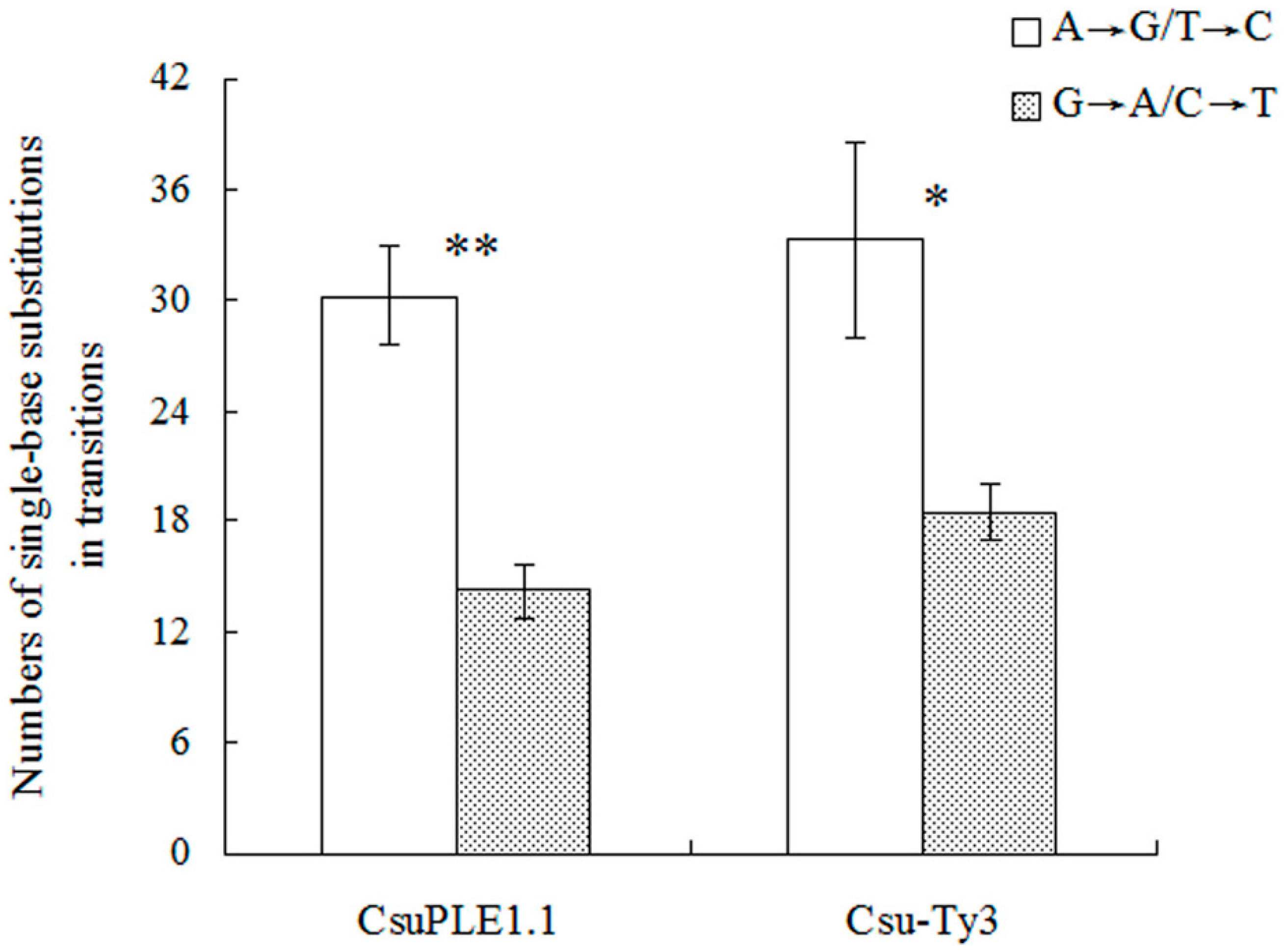
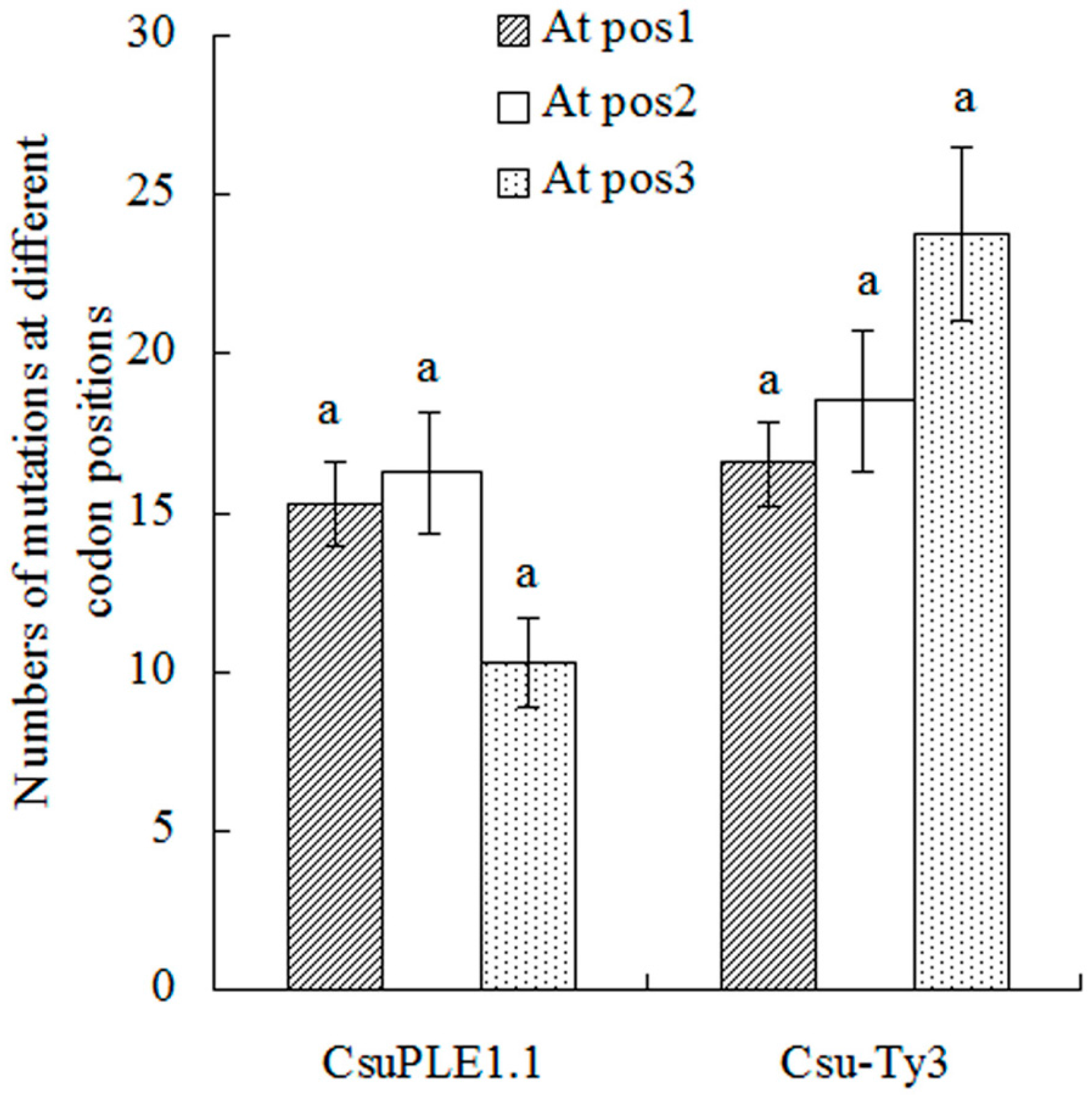
3.1. Single-Base Substitution Mutations in CsuPLE1.1 Copies and Csu-Ty3 Elements
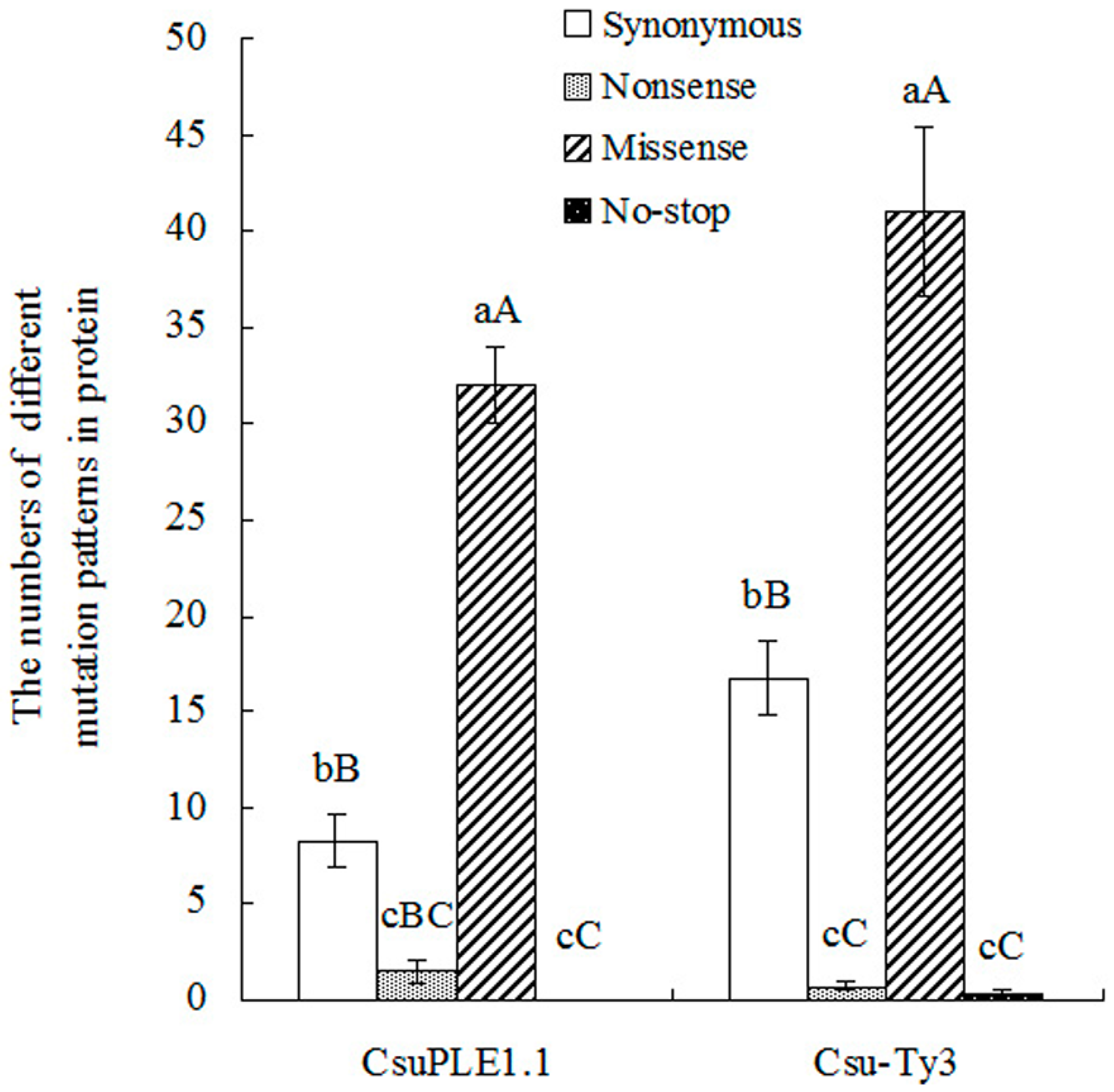
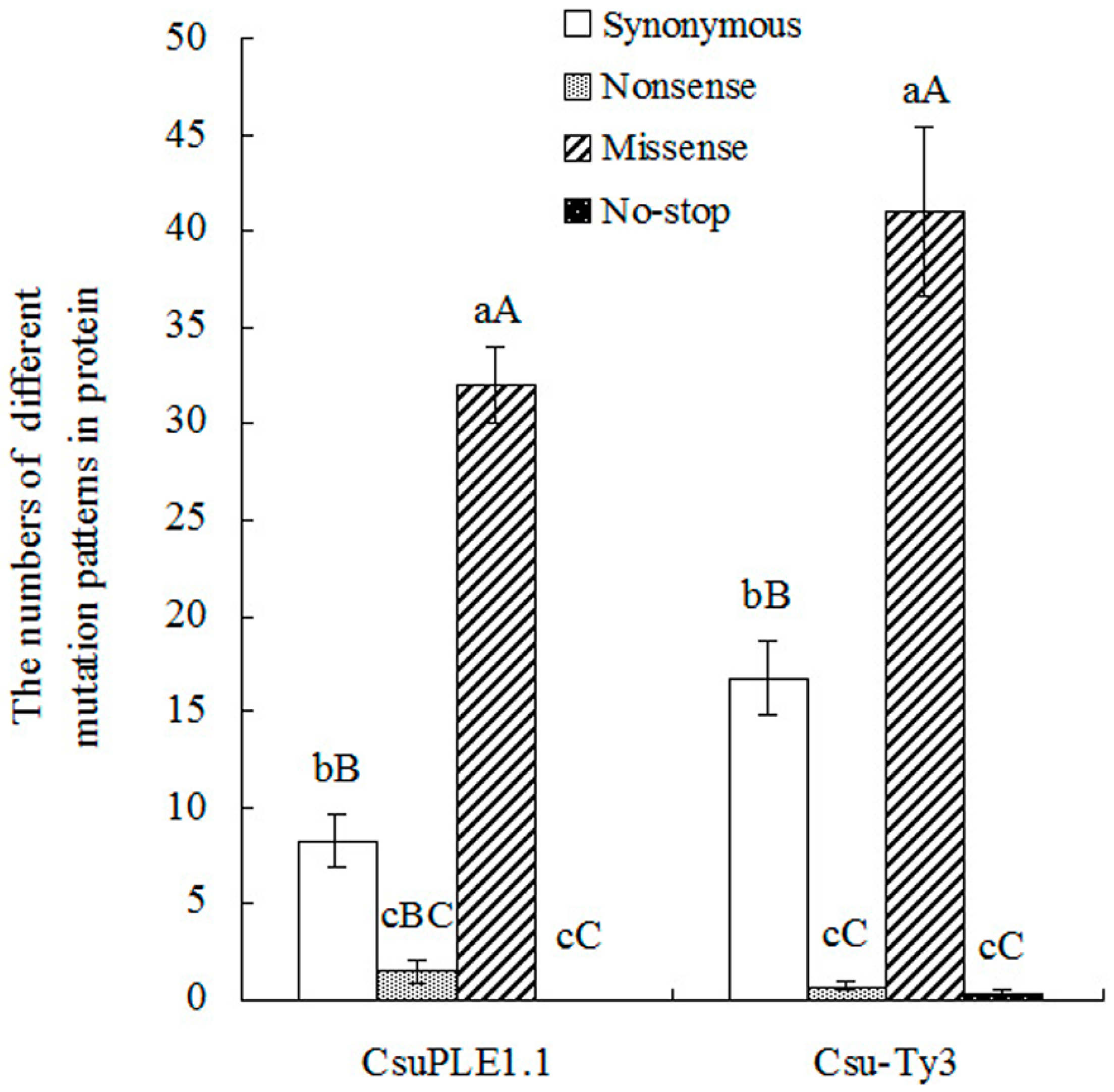
3.2. Two-Base or More Substitution Mutations in CsuPLE1.1 Copies and Csu-Ty3 Elements
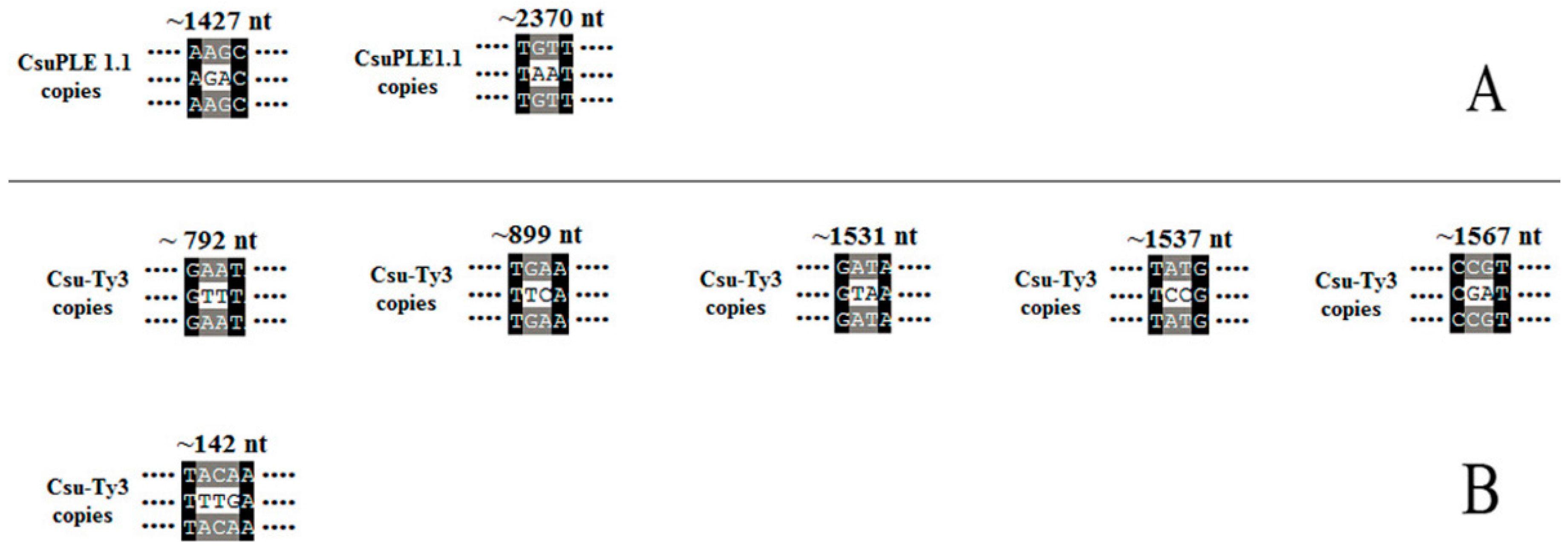
3.3. Deletion and Insertion Mutations in CsuPLE1.1 Copies and Csu-Ty3 Elements
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kimura, M. Estimation of evolutionary distances between homologous nucleotide sequences. Proc. Natl. Acad. Sci. USA 1981, 78, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Topal, M.D.; Fresco, J.R. Complementary base pairing and the origin of substitution mutations. Nature 1976, 263, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Gojobori, T.; Li, W.H.; Graur, D. Patterns of nucleotide substitution in pseudogenes and functional genes. J. Mol. Evol. 1982, 18, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Boerwinkle, E. Neighboring-nucleotide effects on single nucleotide polymorphisms: A study of 2.6 million polymorphisms across the human genome. Genome Res. 2002, 12, 1679–1686. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Gerstein, M. Patterns of nucleotide substitution, insertion and deletion in the human genome inferred from pseudogenes. Nucleic Acids Res. 2003, 31, 5338–5348. [Google Scholar] [CrossRef] [PubMed]
- Petrov, D.A.; Hartl, D.L. Patterns of nucleotide substitution in Drosophila and mammalian genomes. Proc. Natl. Acad. Sci. USA 1999, 96, 1475–1479. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Li, W.H. The size distribution of insertions and deletions in human and rodent pseudogenes suggests the logarithmic gap penalty for sequence alignment. J. Mol. Evol. 1995, 40, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Ophir, R.; Graur, D. Patterns and rates of indel evolution in processed pseudogenes from humans and murids. Gene 1997, 205, 191–202. [Google Scholar] [CrossRef]
- Saitou, N.; Ueda, S. Evolutionary rates of insertion and deletion in noncoding nucleotide sequences of primates. Mol. Biol. Evol. 1994, 11, 504–512. [Google Scholar] [PubMed]
- Feschotte, C.; Pritham, E.J. DNA transposons and the evolution of eukaryotic genomes. Annu. Rev. Genet. 2007, 41, 331–368. [Google Scholar] [CrossRef] [PubMed]
- Feschotte, C. Transposable elements and the evolution of regulatory networks. Nat. Rev. Genet. 2008, 9, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.H.; Li, X.H.; Han, Z.J.; Guo, H.F.; Yang, Q.; Wu, M.; Zhang, Z.C.; Liu, B.S.; Qian, L.; Fang, J.C. Molecular characterization of the piggyBac-like element, a candidate marker for phylogenetic research of Chilo suppressalis (Walker) in China. BMC Mol. Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Li, X.H.; Luo, G.H.; Zhang, Z.C.; Liu, B.S.; Fang, J.C. Coloning and characterization of Ty3/gypsy retrotransposon in Chilo suppressalis (Lepidoptera: Pyralidae). Chin. J. Rice. Sci. 2014, 28, 314–321. (In Chinese) [Google Scholar]
- Hershberg, R.; Petrov, D.A. Evidence that mutation is universally biased towards AT in bacteria. PLoS Genet. 2010, 6, e1001115. [Google Scholar] [CrossRef] [PubMed]
- Saze, H.; Kakutani, T. Differentiation of epigenetic modifications between transposons and genes. Curr. Opin. Plant. Biol. 2011, 14, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Lippman, Z.; Gendrel, A.V.; Black, M.; Vaughn, M.W.; Dedhia, N.; McCombie, W.R.; Lavine, K.; Mittal, V.; May, B.; Kasschau, K.D.; et al. Role of transposable elements in heterochromatin and epigenetic control. Nature 2004, 430, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yazaki, J.; Sundaresan, A.; Cokus, S.; Chan, S.W.; Chen, H.; Henderson, I.R.; Shinn, P.; Pellegrini, M.; Jacobsen, S.E.; et al. Genome-wide high-resolution mapping and functional analysis of DNA methylation in arabidopsis. Cell 2006, 126, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Singer, T.; Yordan, C.; Martienssen, R.A. Robertson’s Mutator transposons in A. thaliana are regulated by the chromatin-remodeling gene decrease in DNA Methylation (DDM1). Genes Dev. 2001, 15, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Mirouze, M.; Reinders, J.; Bucher, E.; Nishimura, T.; Schneeberger, K.; Ossowski, S.; Cao, J.; Weigel, D.; Paszkowski, J.; Mathieu, O. Selective epigenetic control of retrotransposition in Arabidopsis. Nature 2009, 461, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Martienssen, R.; Baron, A. Coordinate suppression of mutations caused by Robertson’s mutator transposons in maize. Genetics 1994, 136, 1157–1170. [Google Scholar] [PubMed]
- Martienssen, R. Transposons, DNA methylation and gene control. Trends. Genet. 1998, 14, 263–264. [Google Scholar] [CrossRef]
- Iengar, P. An analysis of substitution, deletion and insertion mutations in cancer genes. Nucleic Acids Res. 2012, 40, 6401–6413. [Google Scholar] [CrossRef] [PubMed]
- Wakeley, J. The excess of transitions among nucleotide substitutions: New methods of estimating transition bias underscore its significance. Trends Ecol. Evol. 1996, 11, 158–162. [Google Scholar] [CrossRef]
- Smith, D.B.; Simmonds, P. Characteristics of nucleotide substitution in the hepatitis C virus genome: Constraints on sequence change in coding regions at both ends of the genome. J. Mol. Evol. 1997, 45, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Keller, I.; Bensasson, D.; Nichols, R.A. Transition-transversion bias is not universal: A counter example from grasshopper pseudogenes. PLoS Genet. 2007, 3, e22. [Google Scholar] [CrossRef] [PubMed]
- Takano-Shimizu, T. Local changes in GC/AT substitution biases and in crossover frequencies on Drosophila chromosomes. Mol. Biol. Evol. 2001, 18, 606–619. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patterns | Numbers of Substitutions * (Means ± SE) | |
|---|---|---|
| CsuPLE1.1 | Transition | 44.5 ± 3.52 A |
| Transversion | 18.25 ± 1.44 B | |
| Csu-Ty3 | Transition | 51.75 ± 6.63 A |
| Transversion | 24.5 ± 1.66 B | |
| Transition Patterns | Numbers of Substitutions * (Means ± SE) | |
|---|---|---|
| CsuPLE1.1 | A→G | 15.75 ± 1.7 aA |
| G→A | 7.25 ± 0.95 bB | |
| T→C | 14.5 ± 1.32 aAB | |
| C→T | 7 ± 1.58 bB | |
| Csu-Ty3 | A→G | 17.75 ± 2.78 aA |
| G→A | 10.25 ± 1.25 abA | |
| T→C | 15.5 ± 2.53 abA | |
| C→T | 8.25 ± 0.48 bA | |
| Transposon | Region | 1 Base Deletion | 2 or more Base Deletion | 1 Base Insertion | 2 or More Base Insertion |
|---|---|---|---|---|---|
| CsuPLE1.1 | Inside of the ORF region | 1415_1417: one T deletion 1531_1533: one A deletion 1735_1737: one G deletion | 750_761: 10 nt deletion 1530_1533: AA deletion 1606_1732: 125 nt deletion 2118_2128: 9 nt deletion | 1532_1533: one A insertion | 1798_1799: AGGTATA insertion |
| Outside of the ORF region | 49_51: one A deletion 50_52: one T deletion 77_79: one T deletion 556_558: one A deletion 2312_2314: one T deletion | 79_91: 11 nt deletion | 50_51: one A insertion 129_130: one A insertion 548_549: one G insertion 2313_2314: one T insertion | 207_208: ACG insertion 539_540: CCTGCCT insertion 2313_2314: TT insertion 2313_2314: TTT insertion 2313_2314: TC insertion | |
| Csu-Ty3 | Inside of the ORF region | 1133_1135: one G deletion 1389_1391: one G deletion 1397_1399: one T deletion 1646_1648: one A deletion 1810_1812: one T deletion | 65_1029: 963 nt deletion 1539_1567: 27 nt deletion | 272_273: one C insertion 555_556: one A insertion 576_577: one G insertion 1852_1853: one A insertion | 896_897: TTCA insertion 1163_1164: TTAT insertion 1529_1530: 30 nt insertion |
| Outside of the ORF region | 47_49: one A deletion 128_130: one A deletion 729_731: one A deletion | 696_701: CTTT deletion 695_701: TCTTT deletion | 19_20: one A insertion 200_201: one T insertion | 139_140: TGTGA insertion |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, G.-H.; Li, X.-H.; Han, Z.-J.; Zhang, Z.-C.; Yang, Q.; Guo, H.-F.; Fang, J.-C. Transition and Transversion Mutations Are Biased towards GC in Transposons of Chilo suppressalis (Lepidoptera: Pyralidae). Genes 2016, 7, 72. https://doi.org/10.3390/genes7100072
Luo G-H, Li X-H, Han Z-J, Zhang Z-C, Yang Q, Guo H-F, Fang J-C. Transition and Transversion Mutations Are Biased towards GC in Transposons of Chilo suppressalis (Lepidoptera: Pyralidae). Genes. 2016; 7(10):72. https://doi.org/10.3390/genes7100072
Chicago/Turabian StyleLuo, Guang-Hua, Xiao-Huan Li, Zhao-Jun Han, Zhi-Chun Zhang, Qiong Yang, Hui-Fang Guo, and Ji-Chao Fang. 2016. "Transition and Transversion Mutations Are Biased towards GC in Transposons of Chilo suppressalis (Lepidoptera: Pyralidae)" Genes 7, no. 10: 72. https://doi.org/10.3390/genes7100072
APA StyleLuo, G.-H., Li, X.-H., Han, Z.-J., Zhang, Z.-C., Yang, Q., Guo, H.-F., & Fang, J.-C. (2016). Transition and Transversion Mutations Are Biased towards GC in Transposons of Chilo suppressalis (Lepidoptera: Pyralidae). Genes, 7(10), 72. https://doi.org/10.3390/genes7100072