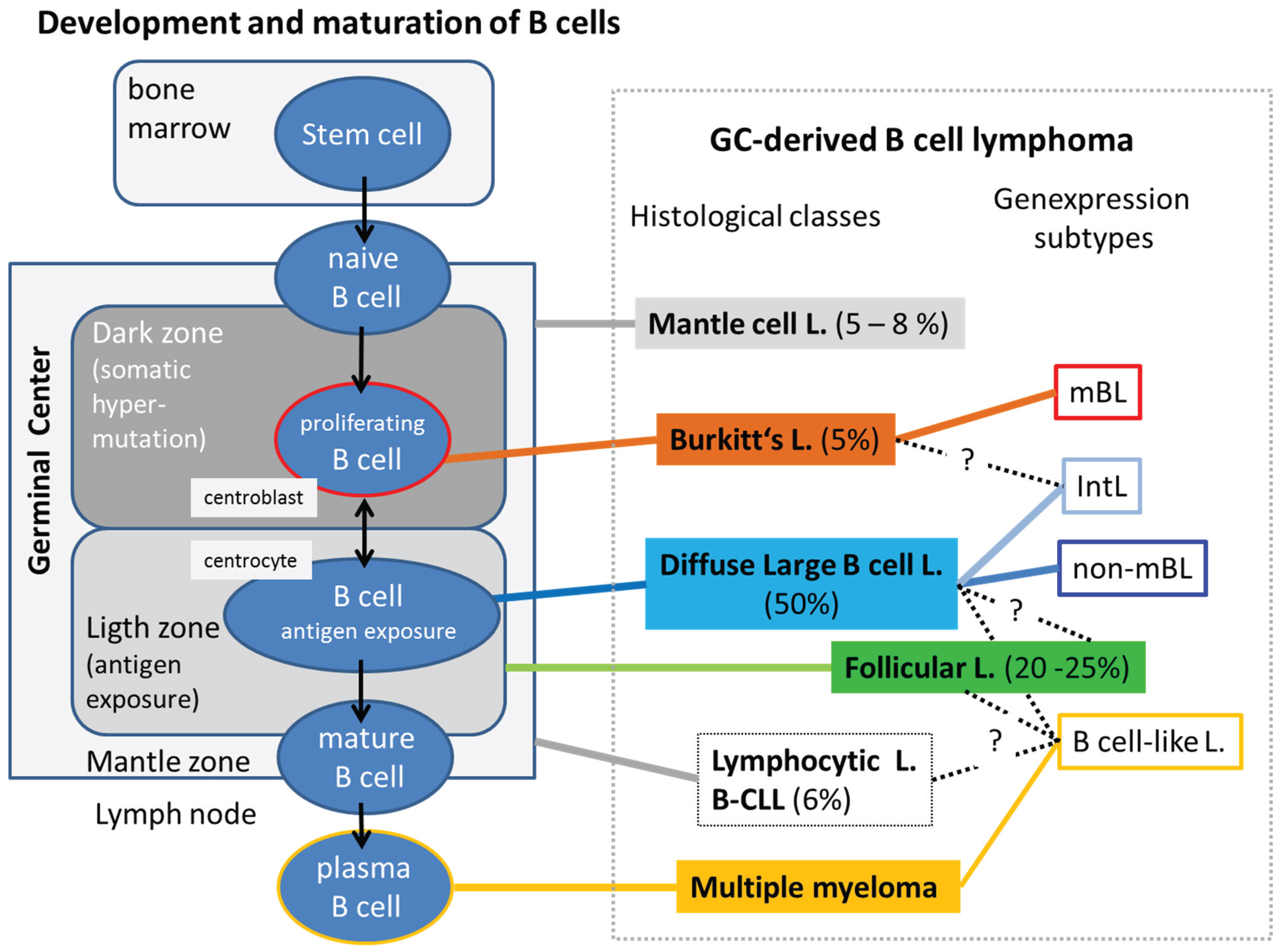
3.1. DNA Methylation of Lymphoma
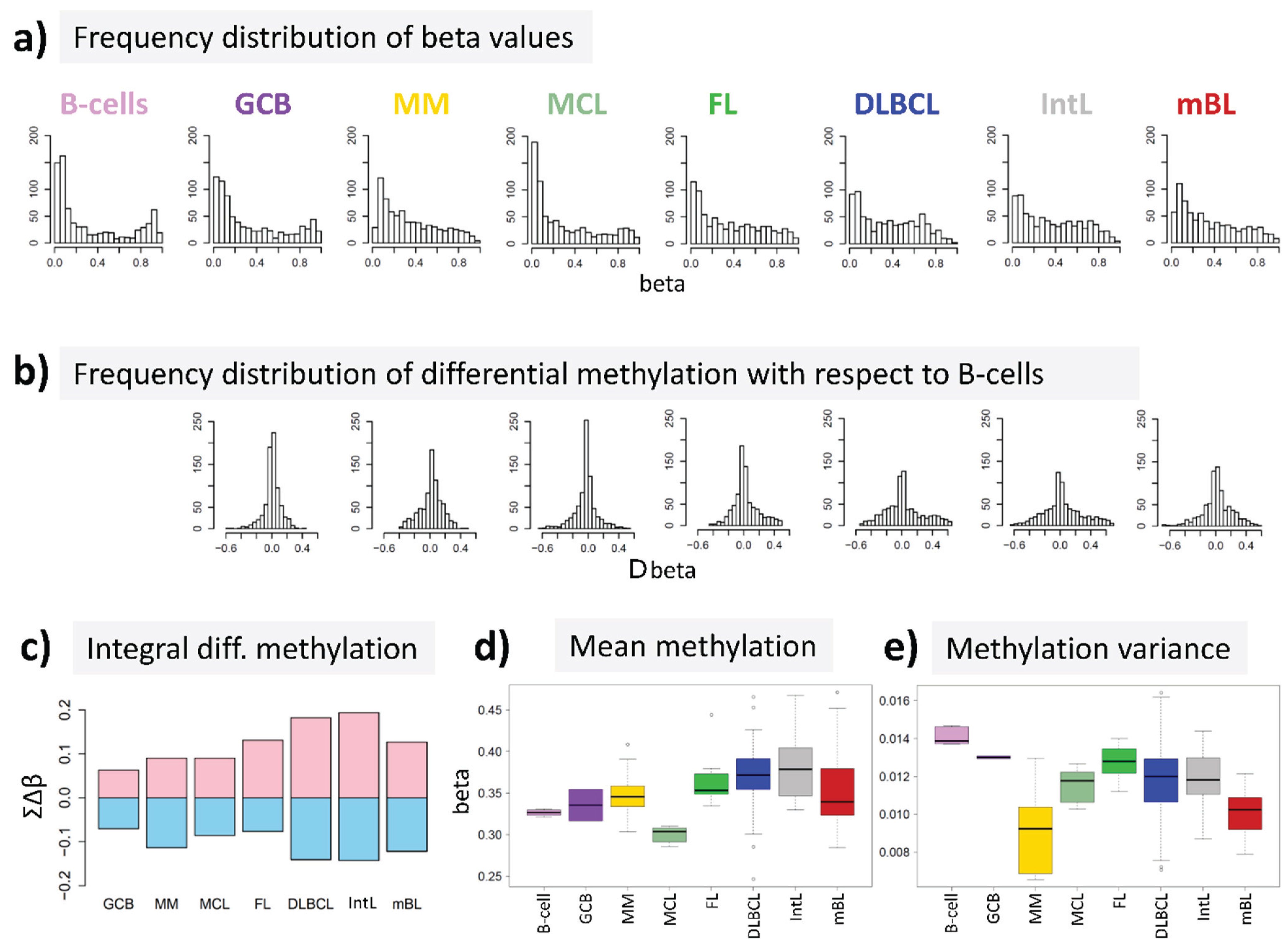
We re-analyzed microarray DNA methylation data published in a previous study on a large number of hematological neoplasms [
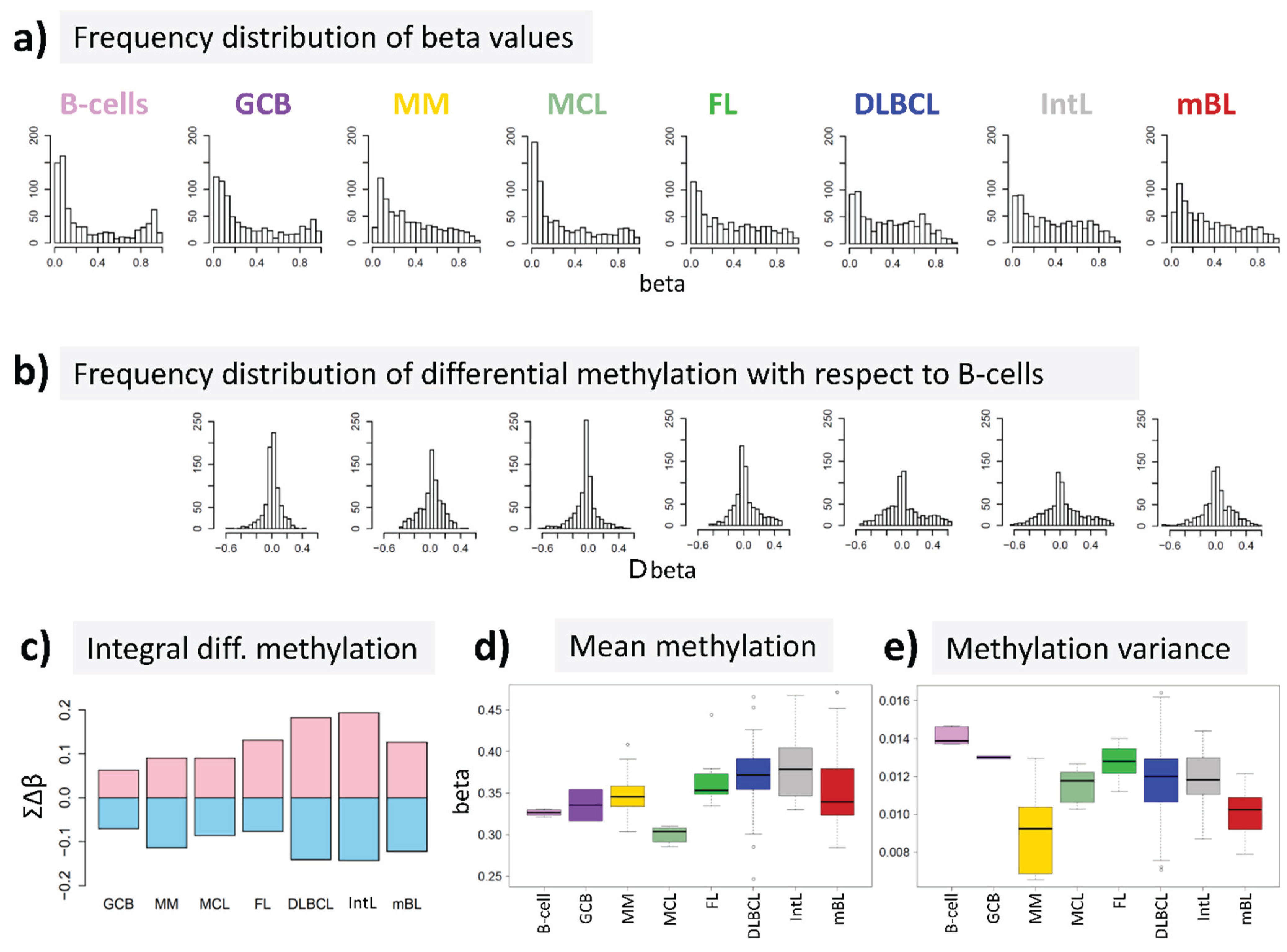
15] to answer the question, whether DNA methylation differs between B-cells and different histological lymphoma classes. For B-cells we found a bimodal shape of the frequency distribution of beta values among the genes studied with maxima near zero (completely de-methylated CpG sites) and unity (completely methylated, see
Figure 2a). The respective distributions of beta values in lymphoma are characterized by a wide loss of this bimodality where especially the fraction of highly methylated genes with beta values near unity markedly decreases. Accordingly, the distributions of beta value alterations of the genes in the different systems compared with their methylation in B-cells were tailed to both, positive and negative values reflecting hypo- and hypermethylation of the respective genes (
Figure 2b). The integral hyper- and hypomethylation of all genes considered reveals the progressively increasing disturbance of DNA methylation in lymphoma being largest in DLBCL and IntL, but being relatively small in MM, FL, MCL and also mBL (
Figure 2c). This trend agrees with the results of previous studies reporting the gain of epigenetic heterogeneity (in terms of differential methylation with respect to the reference state of healthy B-cells) with progressive aggressiveness of lymphoma being largest in DLBCL [
20,
21]. Except for MCL, we find a global hypermethylation of the genes in lymphoma compared with B-cells (
Figure 2d). On the other hand, the variance of beta values in each of the samples strongly decreases in lymphoma mainly due to the decrease or even loss of bimodality reported above (
Figure 2e).
In summary, methylation changes in lymphoma comprise both, hyper- and hypomethylation effects leading to a loss of bimodality of promoter methylation with maxima at low and high beta values and to more balanced methylation landscapes where promoter regions tend to become methylated on intermediate beta-levels.
Figure 2.
DNA methylation summary characteristics of lymphoma and of healthy B and GCB cells. (a) The frequency distribution of the promoter methylation beta values of B-cells shows two maxima referring to almost not- and completely methylated promoters, respectively; The distributions of beta values loose this bimodality to a large degree in lymphoma where weakly and intermediately methylated genes become hyper-methylated and highly methylated genes become hypo-methylated compared with healthy B-cells (b + c); (d) The total methylation level increases and (e) the variability of methylation among the genes in each of the samples decreases.
Figure 2.
DNA methylation summary characteristics of lymphoma and of healthy B and GCB cells. (a) The frequency distribution of the promoter methylation beta values of B-cells shows two maxima referring to almost not- and completely methylated promoters, respectively; The distributions of beta values loose this bimodality to a large degree in lymphoma where weakly and intermediately methylated genes become hyper-methylated and highly methylated genes become hypo-methylated compared with healthy B-cells (b + c); (d) The total methylation level increases and (e) the variability of methylation among the genes in each of the samples decreases.
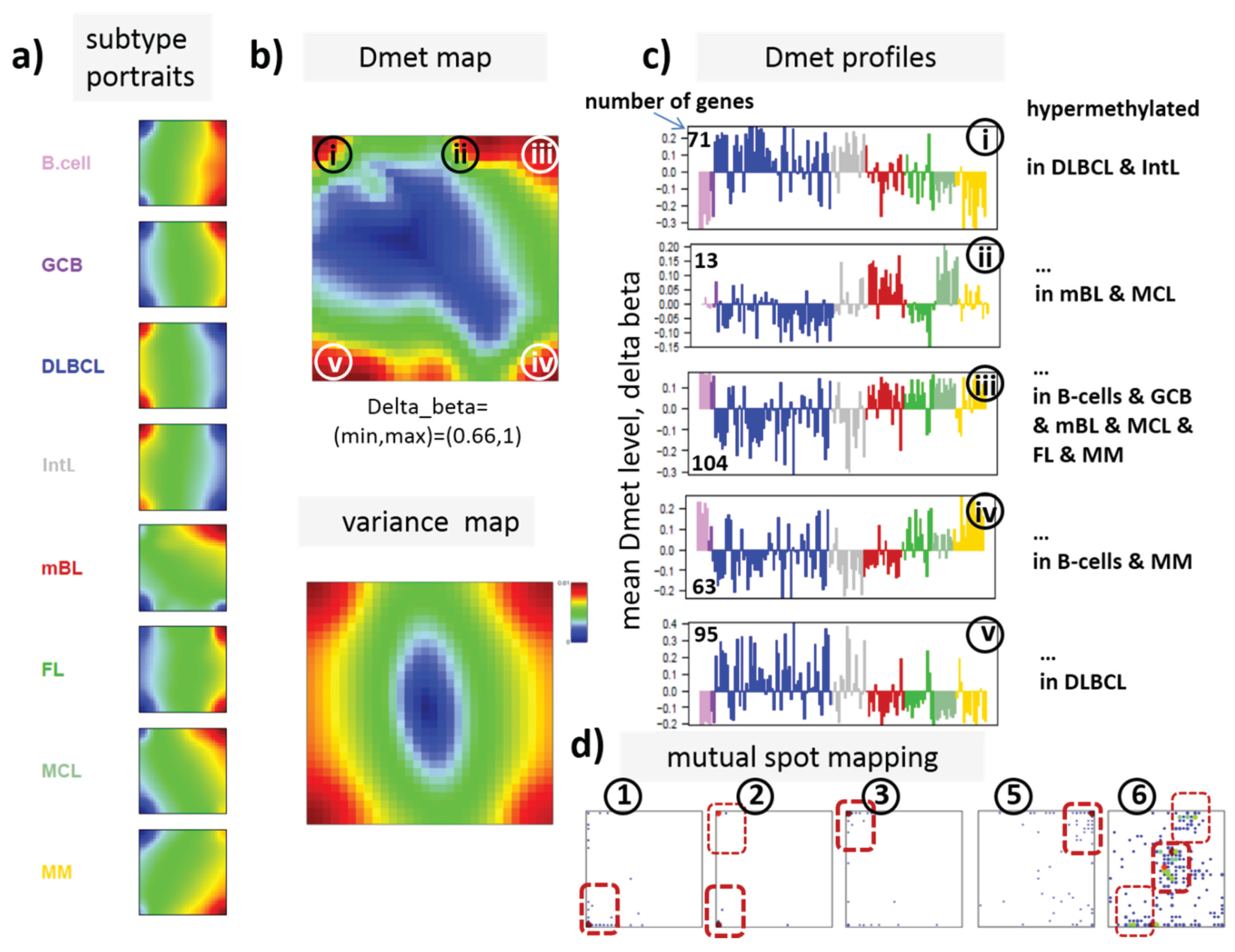
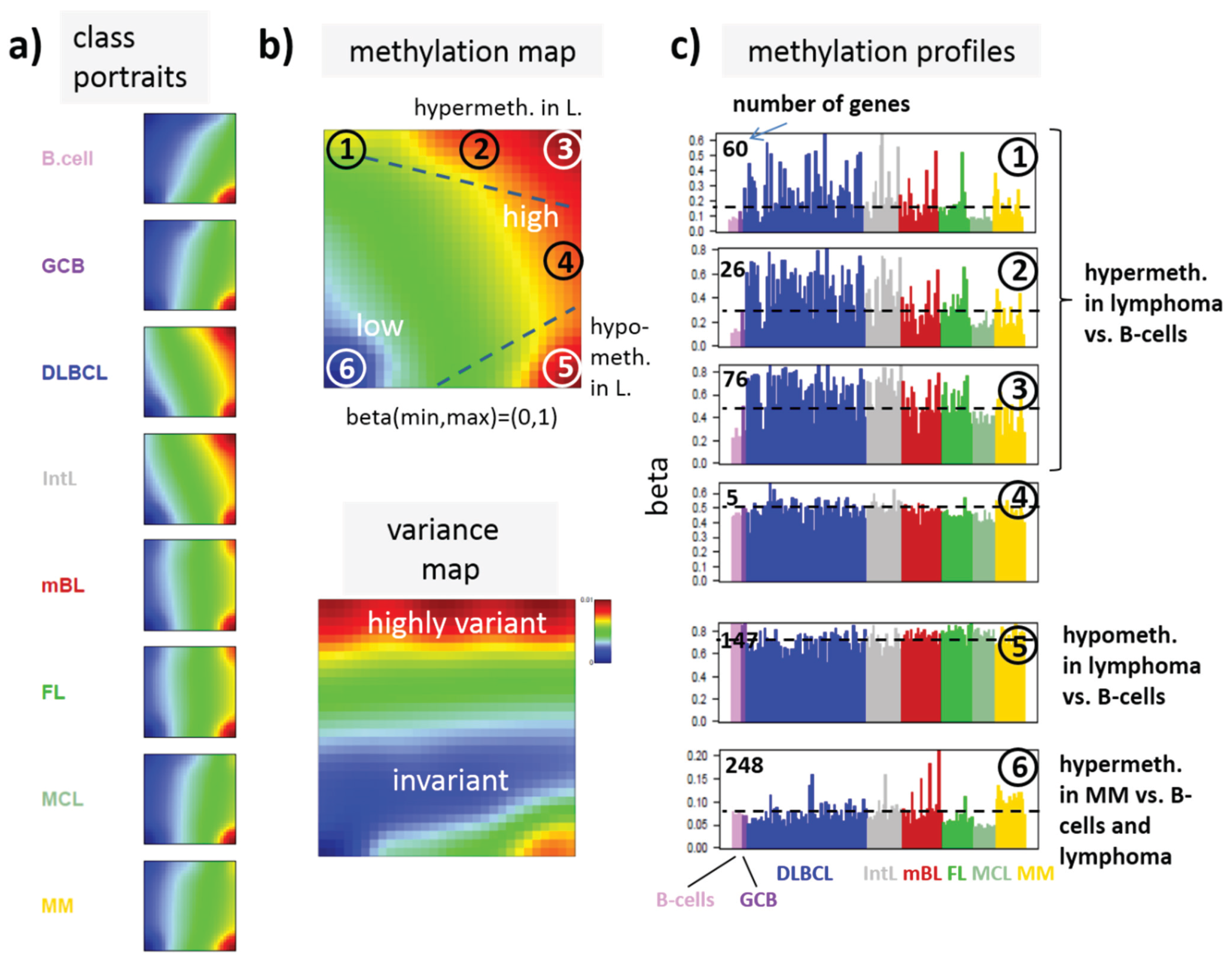
Figure 3.
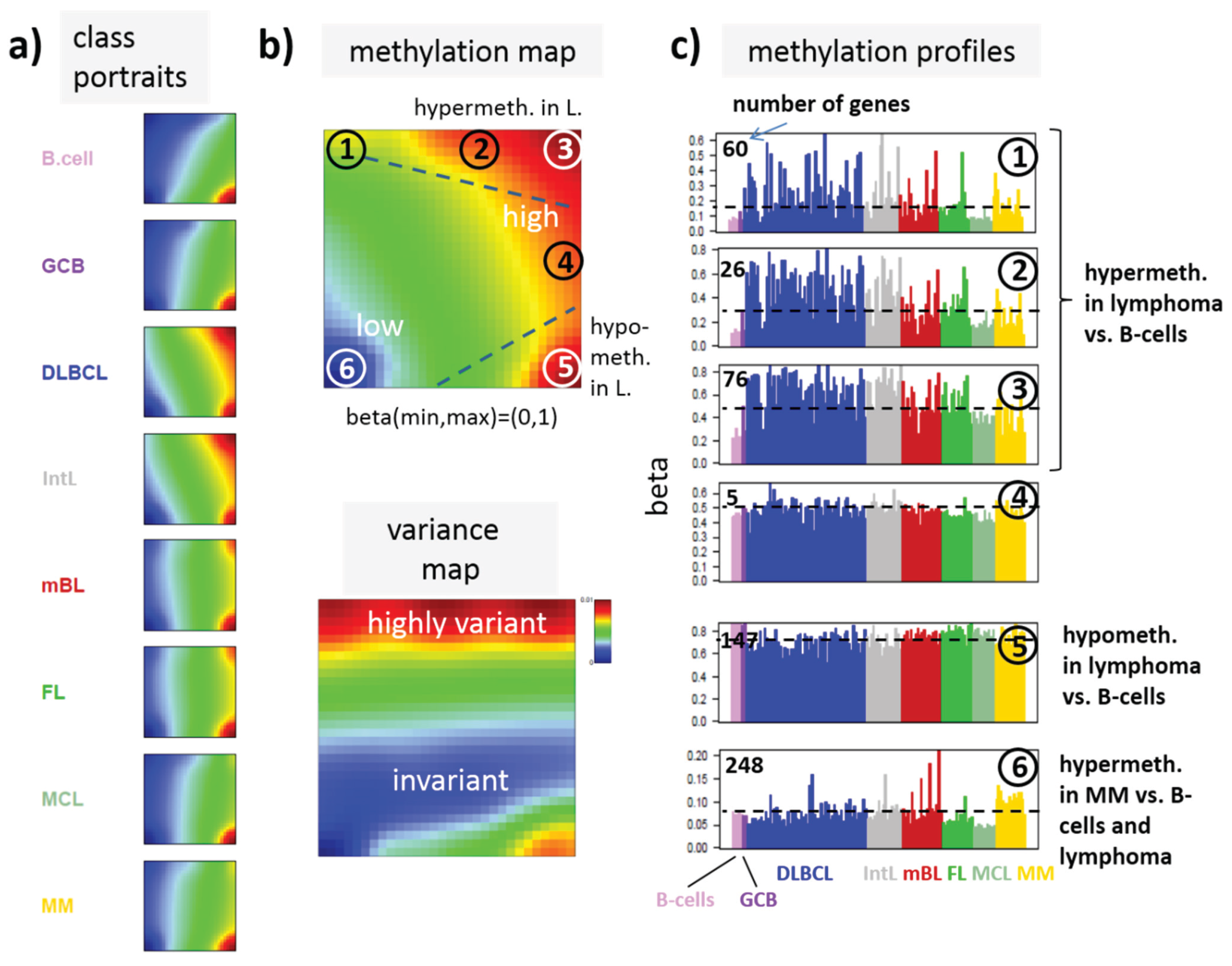
SOM portraying of the DNA methylation landscape of lymphoma (MetSOM). (
a) SOM portraits of histological lymphoma classes and of controls. Red and blue colors assign regions containing genes with high and low methylation levels, respectively; (
b) the methylation overview map summarizes regions hypermethylated in any of the classes compared with any other one in red. The methylation variance map identifies regions of highly variable (red) and almost invariant (blue) beta values; (
c) The methylation profiles show the mean methylation level among the samples of genes taken from the “spot” regions 1–6 assigned in the methylation overview map. Horizontal dashed lines serve as guide for the eye showing the mean methylation level of the respective spot averaged over all samples. Assignments as “hyper-” or “hypomethylated” refer to relative methylations compared with B-cells. Lists of genes in these regions are given in
Table S1.
Figure 3.
SOM portraying of the DNA methylation landscape of lymphoma (MetSOM). (
a) SOM portraits of histological lymphoma classes and of controls. Red and blue colors assign regions containing genes with high and low methylation levels, respectively; (
b) the methylation overview map summarizes regions hypermethylated in any of the classes compared with any other one in red. The methylation variance map identifies regions of highly variable (red) and almost invariant (blue) beta values; (
c) The methylation profiles show the mean methylation level among the samples of genes taken from the “spot” regions 1–6 assigned in the methylation overview map. Horizontal dashed lines serve as guide for the eye showing the mean methylation level of the respective spot averaged over all samples. Assignments as “hyper-” or “hypomethylated” refer to relative methylations compared with B-cells. Lists of genes in these regions are given in
Table S1.
3.2. DNA Methylation Portraying
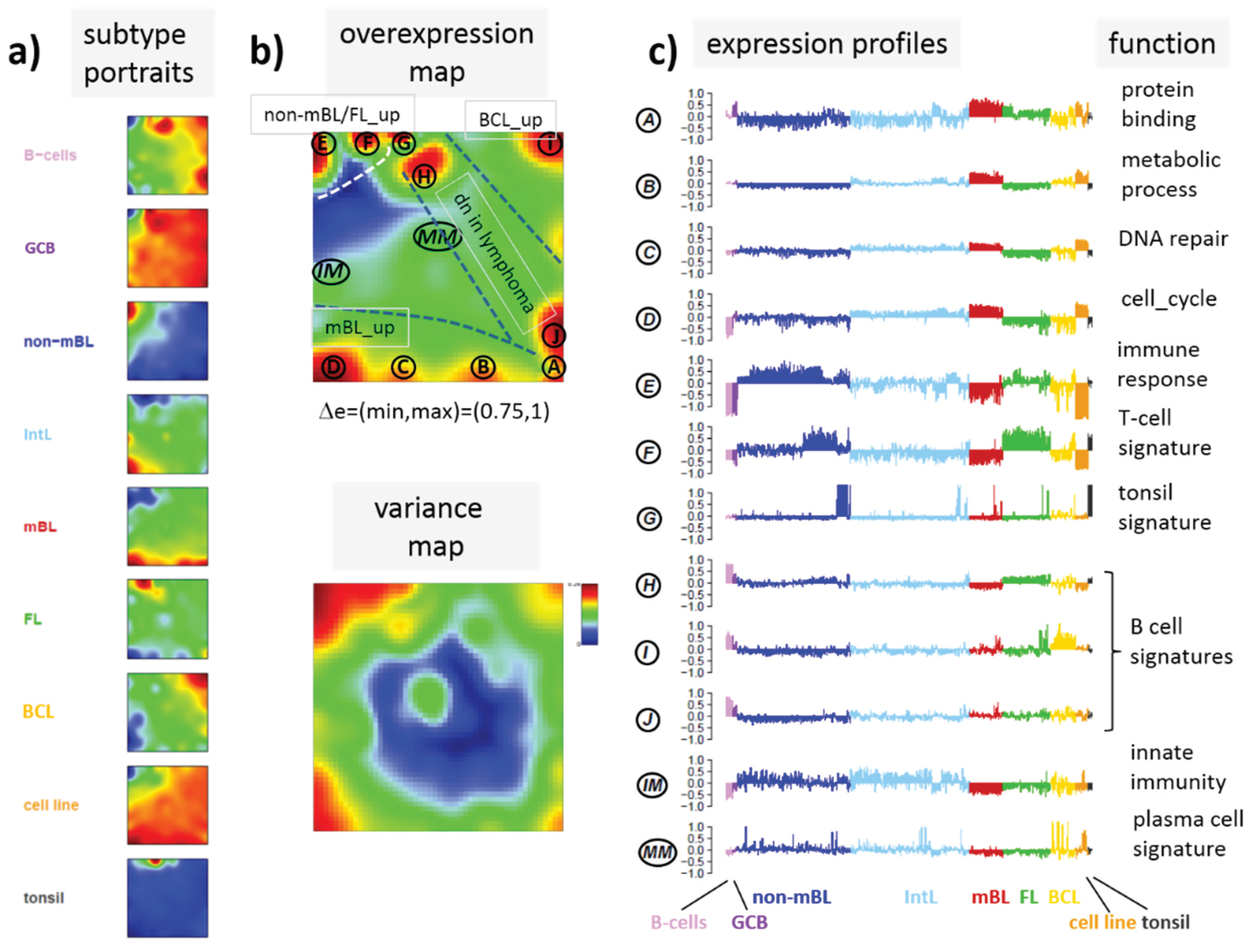
In the next step, SOM data portraying was applied to the methylation data including all lymphoma samples and that of the B and GCB cells serving as reference. The method “projects” the methylation data onto a two-dimensional grid of 50 × 50 pixels. Appropriate color-coding then visualized the methylation landscapes of each sample in terms of its individual methylation portrait shown in
Figure S1. We averaged theses portraits taking into account all samples of each class to identify class-specific methylation signatures.
Figure 3a shows the gallery of these mean portraits for all classes studied. Red and blue regions in the images refer to genes with high and low methylation levels of the probed CpG regions with beta values near one and zero, respectively. The map can be segmented into regions containing genes hyper- and hypomethylated in lymphoma and a region with almost invariantly methylated genes in between (
Figure 3b,c). Note that the SOM algorithm clusters genes with similar methylation profiles among the samples together into the spot-like areas appearing in the methylation maps. Accordingly, groups of signature genes with characteristic methylation profiles can be extracted from these spots assigned using Arabic numbers (
Figure 3c). The methylation maps thus provide genes hyper- and hypomethylated in lymphoma compared with B-cells and also genes with almost invariant methylation levels. For example, genes in region 5 are clearly on high methylation level in B-cells and on lower level and thus hypomethylated in lymphoma.
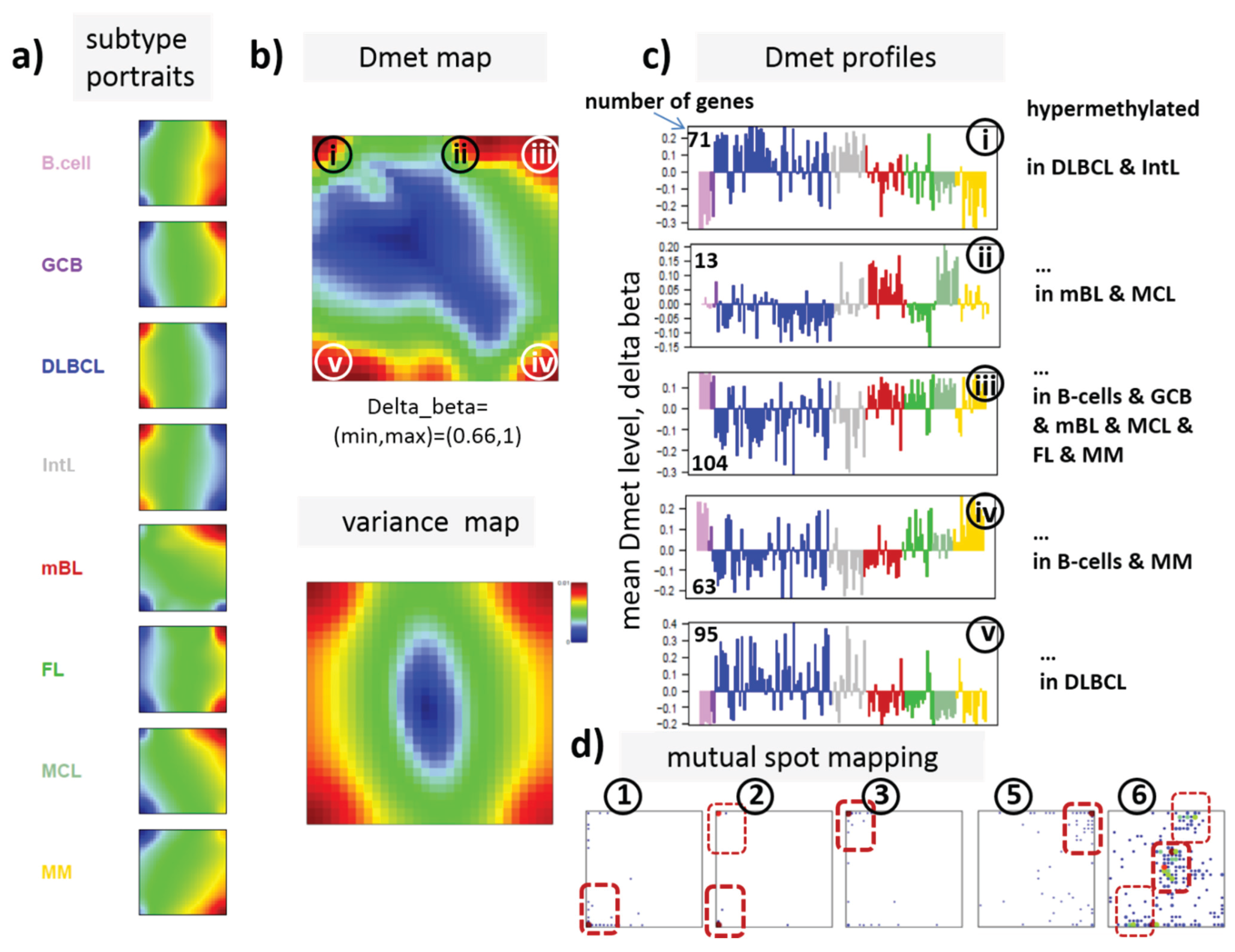
3.3. Portraying of Differential Methylation Better Resolves Differences between the Lymphoma Classes
In our previous work, we showed that the analysis of centralized values better resolves subtle differences between the samples [
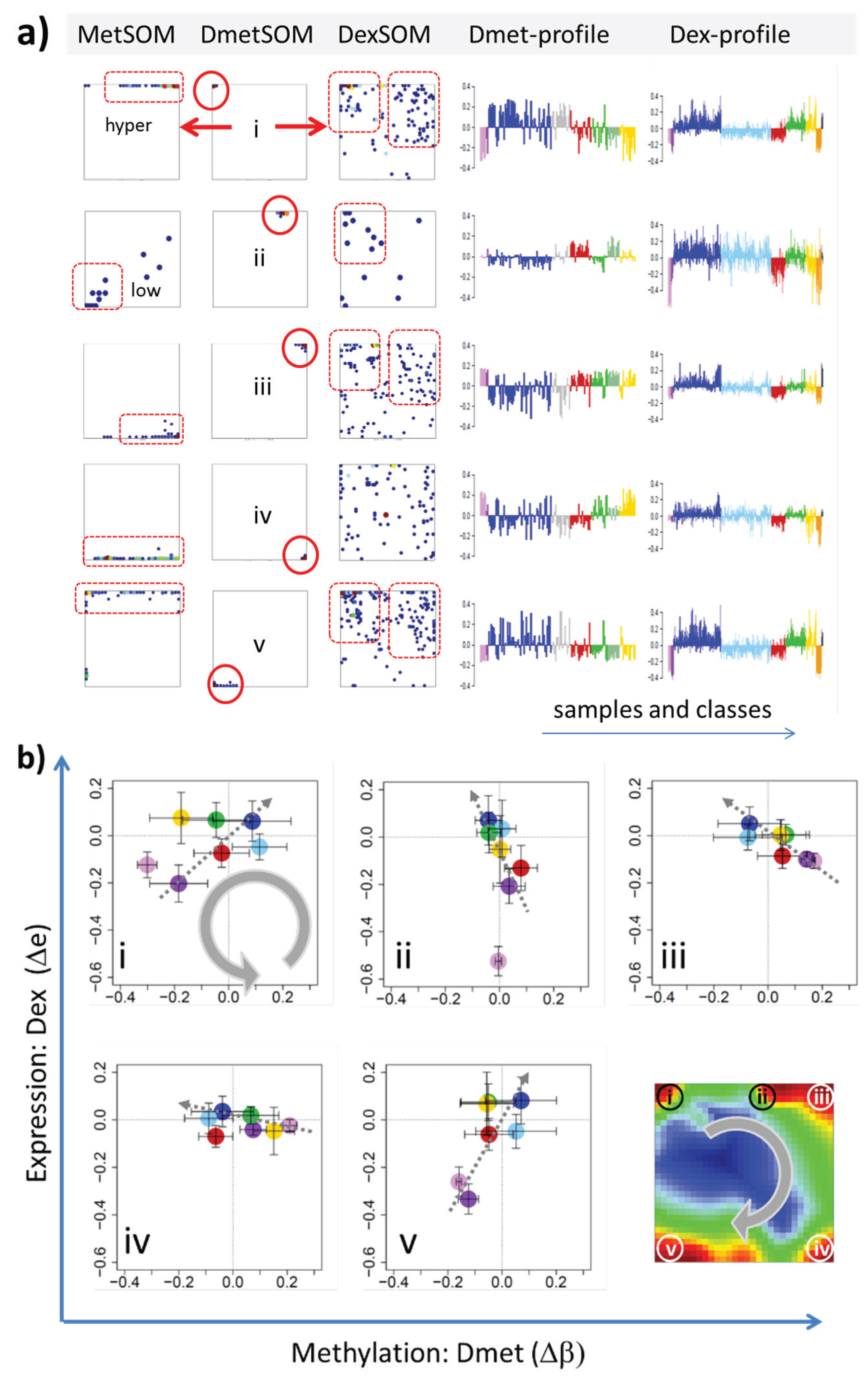
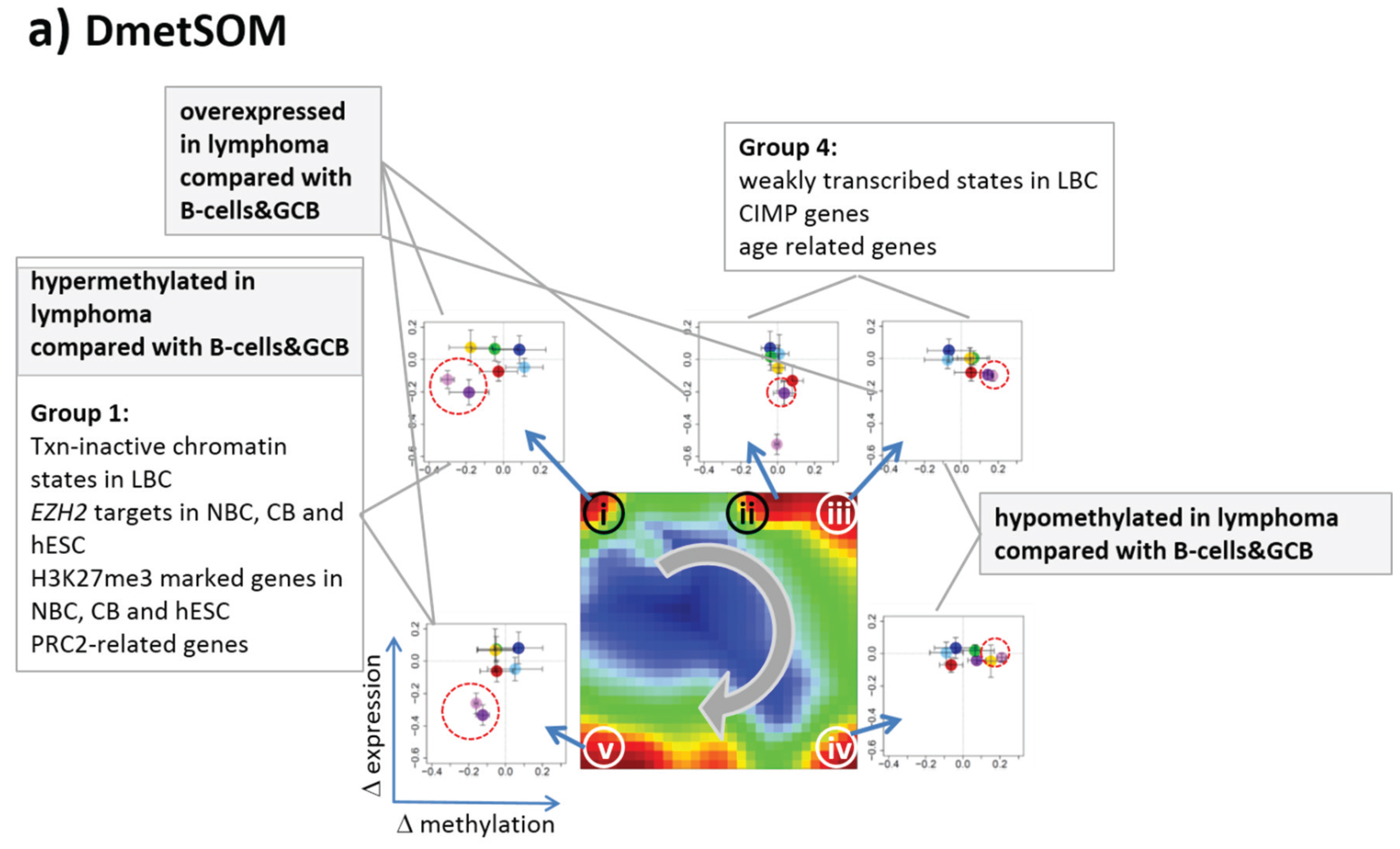
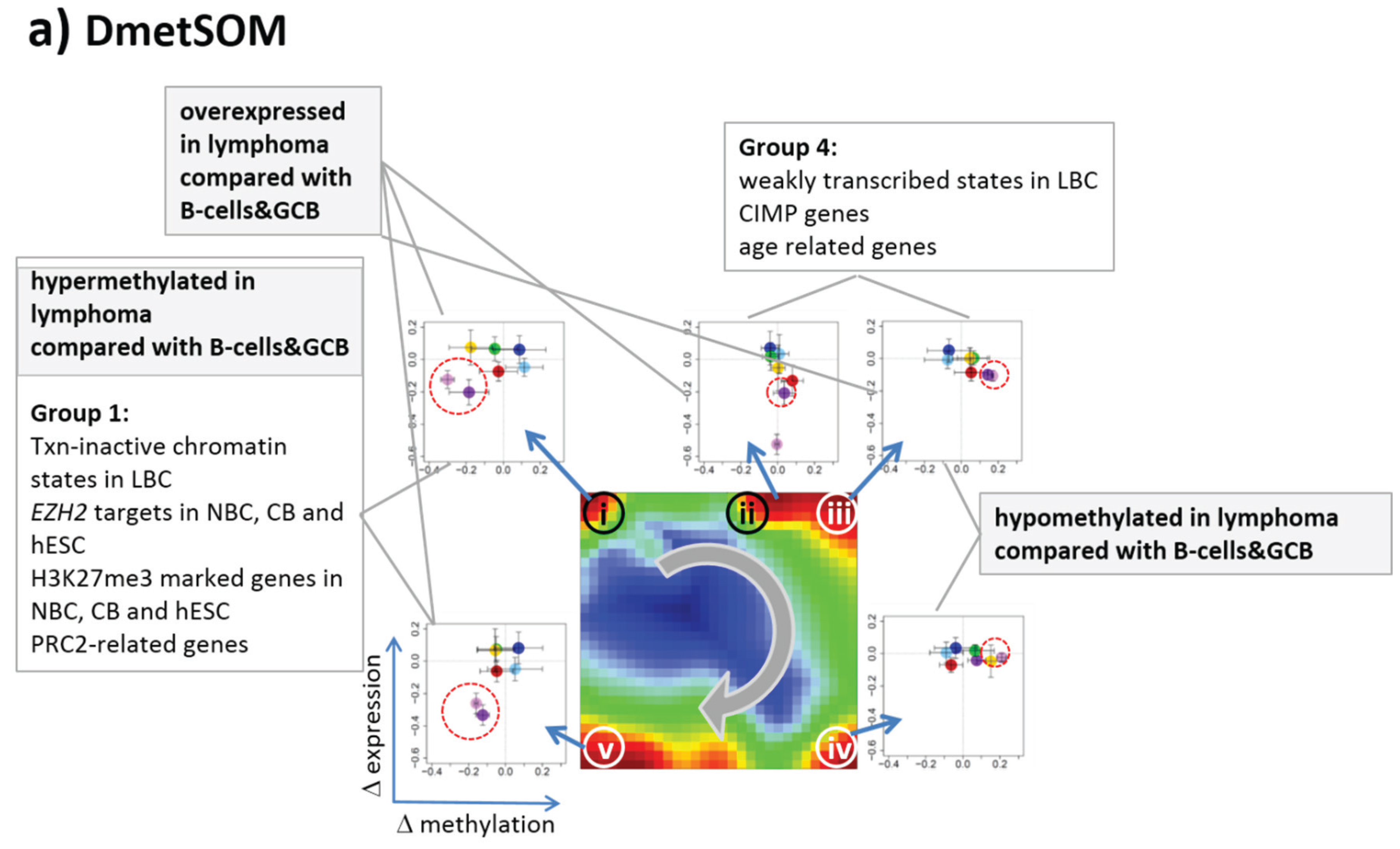
22]. We therefore calculated a second SOM using centralized methylation values (DmetSOM), where the mean beta value of each gene averaged over all samples was subtracted from its actual beta-value. Centralization rather focuses the view on methylation changes between the samples independent of the absolute methylation level of the genes. In the obtained DmetSOM portraits, we identified five spot-clusters numbered i–v, which provide differential methylation profiles reflecting specific hyper- and hypomethylation of selected lymphoma classes compared with B-cells. (
Figure 4a–c). Invariantly methylated genes accumulate in the center of the map, whereas the variable genes occupy different regions near the border in a profile-specific manner. Mapping of the methylation clusters 1–6 obtained from the MetSOM (previous subsection) into the DmetSOM reveals mostly a one-to-one relationship (
Figure 4d). For example, spot v referring to genes specifically hypomethylated in B-cell, mBL and MCL compared to DLBCL distributes over spots 1 and, to a less degree spot 2. This result simply means that most of the genes undergoing hypo- or hypermethylation between the different sample classes show predominantly an initially high or low methylation level, respectively. We therefore restrict our further analysis to the clusters i–v in the MetSOM.
Gene set enrichment analysis provides first ideas about the functional context of the genes in the spot modules (
Table 1). Spots i and v hypermethylated in DLBCL and IntL enrich genes related to the formation of the polycomb repressive complex (PRC2), which controls cellular development and differentiation [
23]. Interestingly, genes from these spots are hypermethylated also in other cancers such as colorectal cancer (CRC) and high and low grade glioma. Vice versa, hypomethylated genes in DLBCL and IntL (spot iii) are also consistently hypomethylated in CRC and glioma suggesting parallels in epigenetic regulation between different cancer types. Genes hypermethylated in B-cells and MM (spot iv) are associated with immune processes, whereas genes hypermethylated in mBL and MCL (spot ii) enrich processes related to cell proliferation and cell cycle activity.
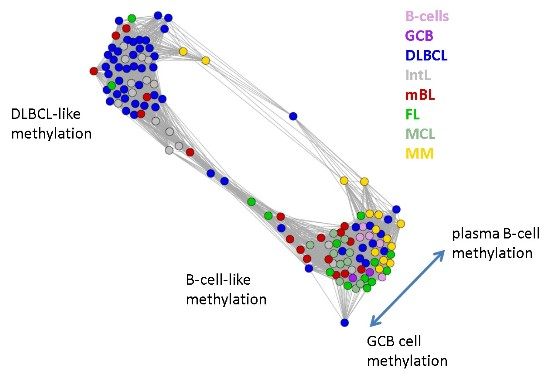
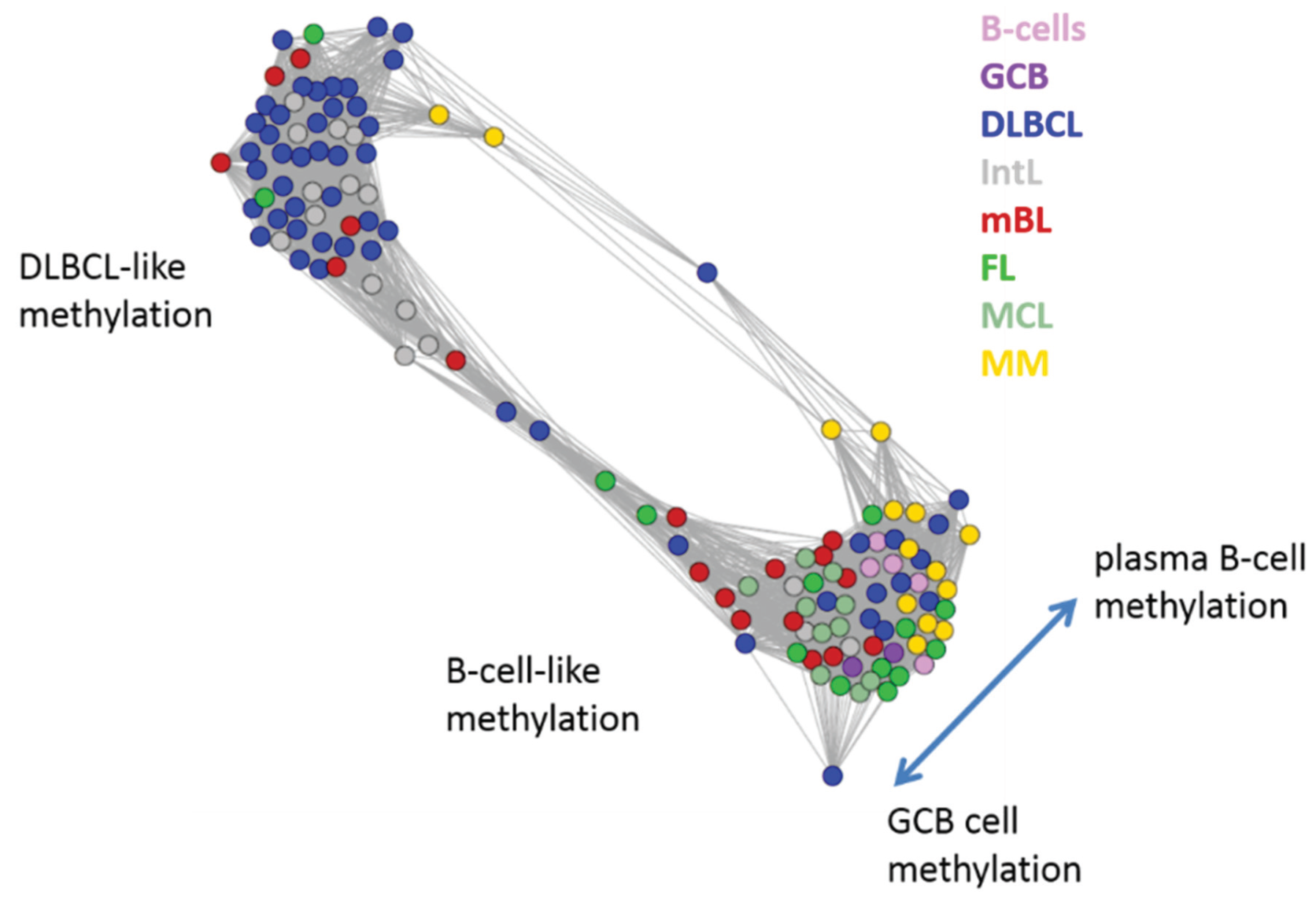
Next, we investigate the diversity landscape of the methylation portraits of the lymphoma and reference samples. The calculated similarity network reveals two main clusters, which can be assigned to samples methylated either similarly to B-cells or to DLBCL (
Figure 5). The essentially two main spot patterns of the mean DmetSOM portraits shown in
Figure 4a directly reflect the separation between two main sample clusters seen in
Figure 5: The samples with DLBCL-like methylation patterns preferentially show red hypermethylation spots in the left part of the portraits (spots i and v, see also
Table 1), whereas the B-cell-like methylation patterns is characterized by red hypermethylation spots in the right part of the map (spots ii to iv). These patterns are strongly anti-correlated (see
Figure S2),
i.e., hypermethylation is opposed by hypomethylation for many genes when compared with mean methylation level averaged over all samples.
Figure 4.
Differential methylation portraying of lymphoma and controls (DmetSOM; see legend of
Figure 3 for a detailed description of the panels (
a–c)). The DmetSOM better resolves differential methylation between the lymphoma classes (compare with the MetSOM in
Figure 3). In part d genes from methylation spots 1–6 of the MetSOM are mapped into the DmetSOM (each dot marks a metagene occupied by at least one gene from each of the spots 1–6, respectively). One finds almost a 1:1 relationship between the spots except for spot 6 which “hides” spot ii. Lists of genes from the regions i–v are given in
Table S2.
Figure 4.
Differential methylation portraying of lymphoma and controls (DmetSOM; see legend of
Figure 3 for a detailed description of the panels (
a–c)). The DmetSOM better resolves differential methylation between the lymphoma classes (compare with the MetSOM in
Figure 3). In part d genes from methylation spots 1–6 of the MetSOM are mapped into the DmetSOM (each dot marks a metagene occupied by at least one gene from each of the spots 1–6, respectively). One finds almost a 1:1 relationship between the spots except for spot 6 which “hides” spot ii. Lists of genes from the regions i–v are given in
Table S2.
Table 1.
Functional context of the differentially methylated gene clusters.
Table 1.
Functional context of the differentially methylated gene clusters.
| Spots | | Regulated Classes | Functional Context |
|---|
| Dmet-Spot | Met-Spot | Mean Met-Level | Dmet up 1 | Dmet down 1 | Enriched Gene Sets 2 |
|---|
| i | 2,3 | intermediate | DLBCL, IntL | B-cell, GCB, FL, MM | CIMP high-vs-low hypermethylated; hypermethylated-in-CRC [24]; Suz12-, Nanog- and Eed-targets [Wang]; Hypermethylated_in-cancer-and-ageing [25]; hypermethylated in primary glioblastoma [26] |
| ii | 6 | low | BL, MCL | DLBCL, FL | Myc-targets [27]; GO_BP: G1/S-transition in mitotic cell cycle; GO_BP: cell cycle |
| iii | 5 | high | B-cell, GCB, mBL, FL, MCL, MM | DLBCL, IntL | Hypomethylated in CRC; CIMP high-vs-low hypomethylated [24]; Hypermethylated in adult brain [28]; Hypomethylated in secondary glioblastoma [26] |
| iv | | | B-cell, MM | DLBCL, IntL, mBL | GO_BP: immune response; hypomethylated in glioma [26]; GO_CC: nuclear chromatin; NKF-beta down in mBL [2]; IL21 targets down [29] |
| v | 1,2 | low, intermediate | DLBCL, IntL | B-cell, GCB, mBL, FL, MCL, MM | Suz12 targets [30]; hypermethylated in grade 3 astrocytoma and grade 2 oligodendroglioma [26]; hypermethylated in low grade glioma [31]; hypermethylated in CRC [24]; low expression TF [32] |
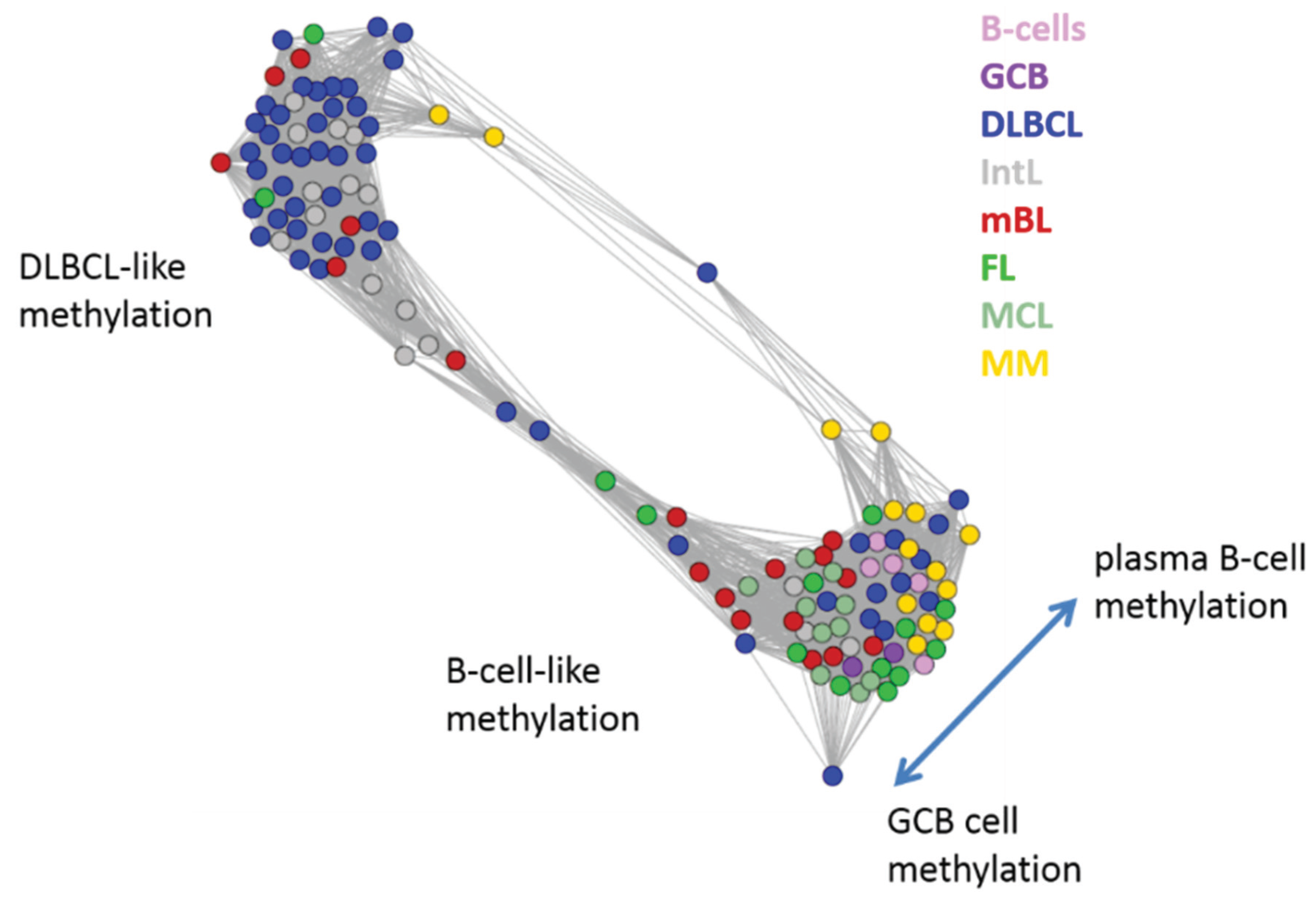
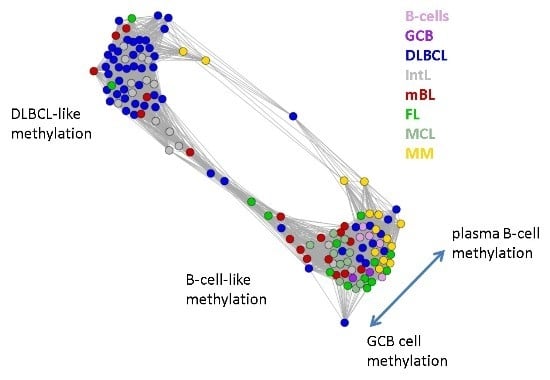
Figure 5.
Similarity network of the methylation landscapes of the lymphoma samples studied. Each circle refers to one sample colored according to its class assignment. Samples with strong correlations between their methylation landscapes were connected by lines (r > 0.5, see [
5] for methodical details). Two main cluster can be distinguished, which include samples of B-cell-like and DLBCL-like methylation.
Figure 5.
Similarity network of the methylation landscapes of the lymphoma samples studied. Each circle refers to one sample colored according to its class assignment. Samples with strong correlations between their methylation landscapes were connected by lines (r > 0.5, see [
5] for methodical details). Two main cluster can be distinguished, which include samples of B-cell-like and DLBCL-like methylation.
The DLBCL-like methylation cluster contains most of the DLBCL (69%) and IntL (81%) samples but also a certain number of FL (14%), mBL (28%) and MM (14%). On the other hand, also the second cluster of B-cell-like methylation contains 25% of the DLBCL and 19% of the IntL samples. Hence, methylation of the lymphoma classes is characterized by a certain degree of fuzziness. The gallery of individual DmetSOM portraits shown in
Figure S1 indicates that, e.g., two of the FL samples show clearly a DLBCL-like methylation characteristics, whereas the majority of the FL are compatible with B cell like methylation patterns. Note also that the B-cell-like methylation cluster reveals a fine structure which separates MM and B-cells on one hand and mBL, GCB cells and MCL on the other hand. This fine structure is related to subtle methylation differences between hypermethylation spots ii–iv (
Figure 4 and
Table 1). Finally note that similarity analysis is based on a relatively small selection of less than 800 genes only, which might distort similarity relationships if relevant groups of genes are under- or over-represented.
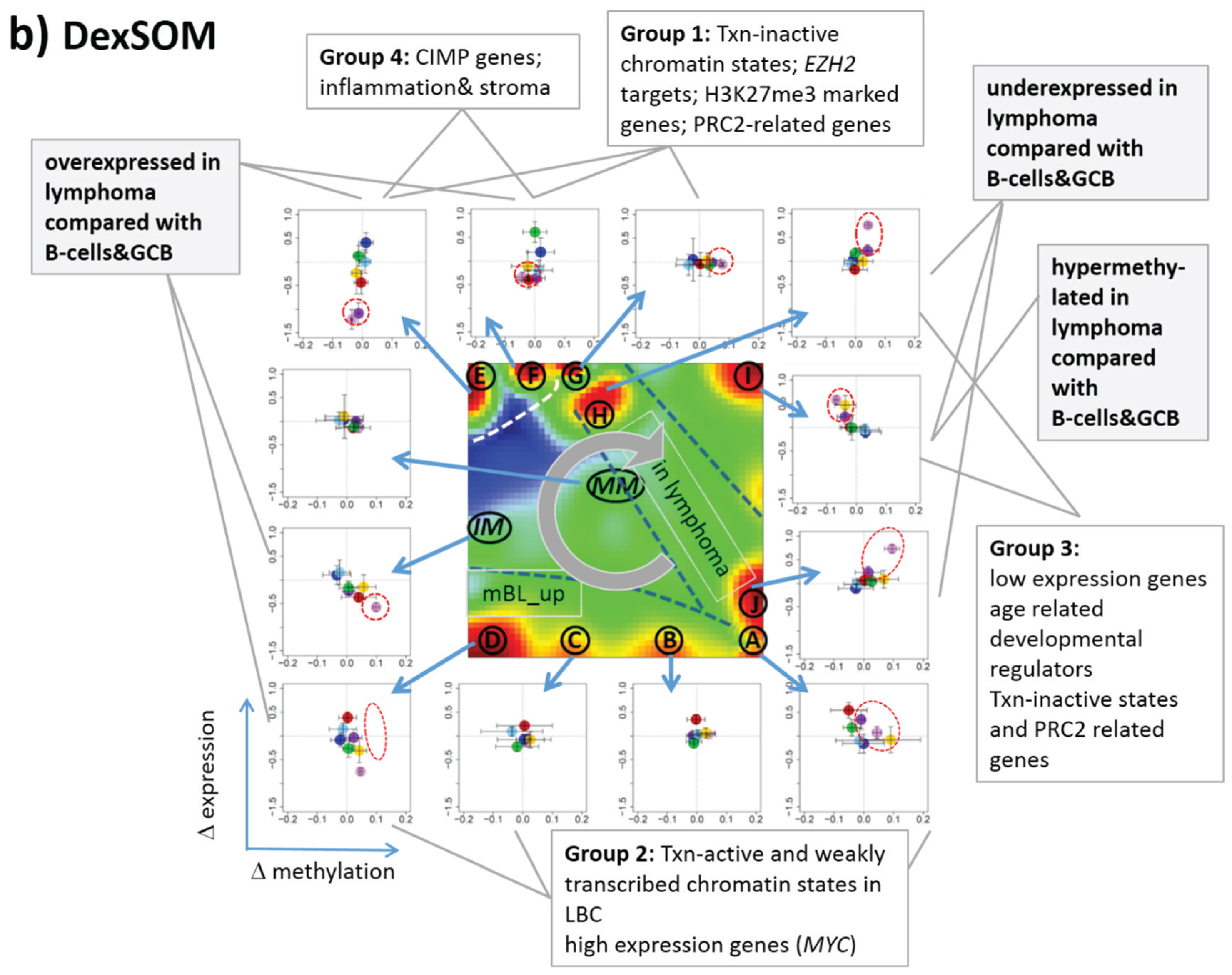
3.5. Mutual Mapping of Expression and Methylation Modules
After separate SOM mapping of DNA methylation and gene expression data we linked both types of analyses in the next step to detect mutual relationships between promoter methylation and gene expression. In a first attempt, we mapped the approximately 800 genes considered on the methylation arrays into the gene expression landscape of lymphoma (DexSOM) and color-coded their methylation level (
Figure S3). No densely populated areas of uniquely methylated genes were found indicating a fuzzy relationship between co-methylated and co-expressed genes. Possibly, this mutual mapping on gene level provides a suitable approach if the methylation assay probes all genes, which are also considered in the expression assay.
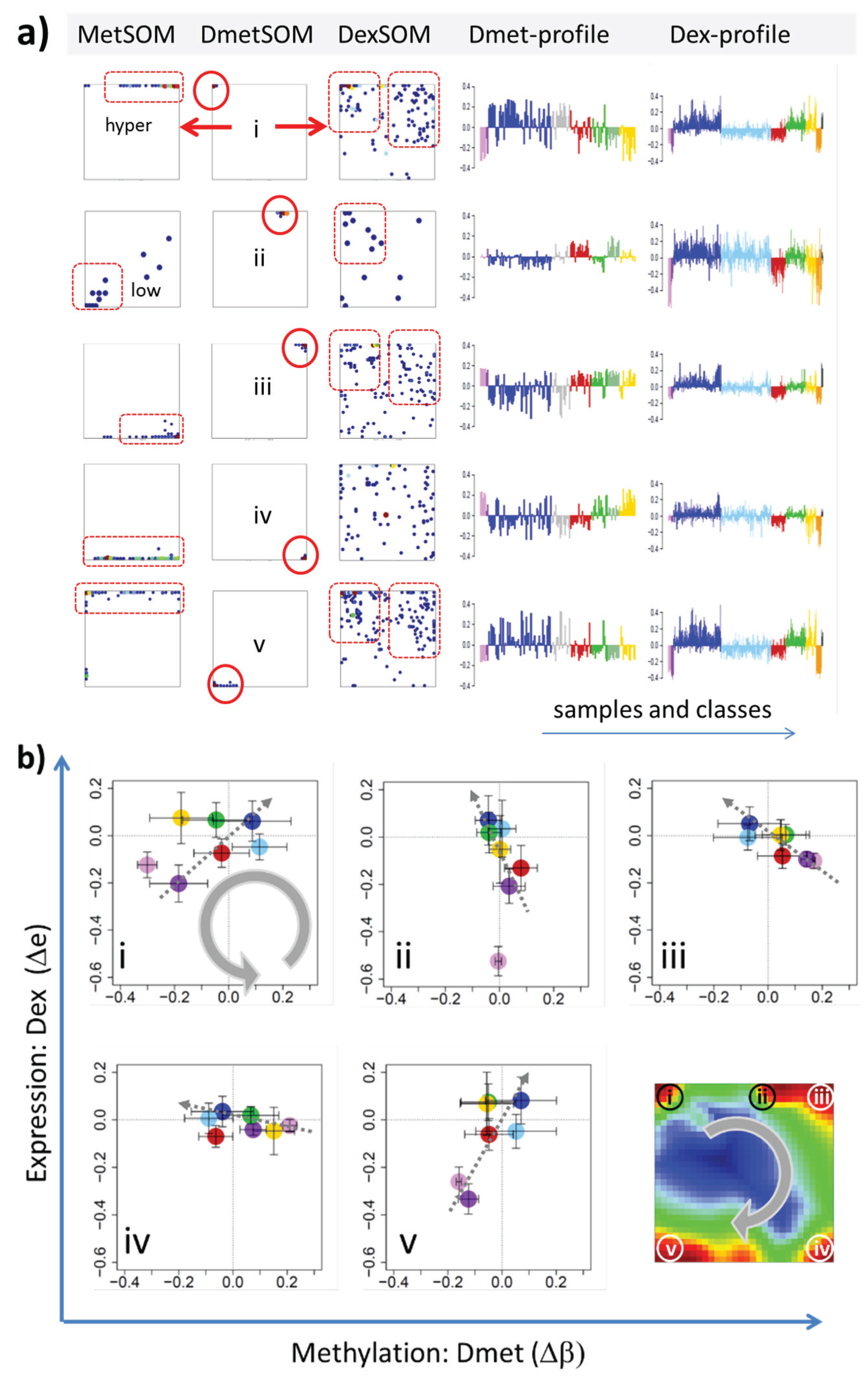
In the next step, we considered groups of co-methylated genes separately: genes of the Dmet-spots i–v (see
Figure 4 and red circles in
Figure 7a) were mapped into the MetSOM and DexSOM where they clearly accumulate in distinct regions as indicated by the dotted red rectangles in
Figure 7a. This result reflects the fact that groups of co-methylated genes are also co-expressed in a class-specific fashion as confirmed also by the respective methylation and expression profiles shown in the right part of
Figure 7a. To better resolve the mutual relationships, we correlate class-averaged mean methylation and expression levels of the gene groups taken from each of the methylation spots i–v in panel b of
Figure 7. Spots i and v are characterized by a positive correlation:
i.e., hypermethylation in DLBCL with respect to B- and GCB cells is accompanied by overexpression in DLBCL with respect to the healthy cell controls. MM and partly FL show concerted coexpression with DLBCL but still similar methylation compared with B and GCB cells. The other lymphoma classes behave similarly however with smaller effects. Genes from spot iii show a negative correlation where differential methylation changes sign compared with spots i and v but differential expression does not. In other words, overexpression in DLBCL is associated with hyper- (spots i and v) and hypo- (spot iii) methylation as well. Recall that all three spots i, iii and v are also functionally related: They enrich genes differentially methylated in other cancer types and related to PRC2 formation. Spot ii (related to proliferation, see
Table 1) collects genes weakly responding to methylation but strongly to differential expression for most of the lymphoma classes. Note that in mBL hypermethylation of spot ii genes associates with underexpression of the respective genes. In contrast, spot iv (related to immune response) weakly responds to expression changes but strongly to differential methylation.
In
Figure S5 we mapped genes differentially methylated in the histological lymphoma classes with respect to B-cells as determined in ref. [
15] into our SOMs. Most of the genes hyper- or hypomethylated in lymphoma accumulate in spot i or iii (and iv), respectively, showing similar correlations with expression data as discussed above. We also studied genes specifically hypermethylated in classical Hodgkin lymphoma compared with B cells determined in ref. [
8] (
Figure S6). Interestingly these genes accumulate in expression spots A, H and MM which mostly associate with healthy GCB-cell functions but lack differential methylation in our data presumably because of the absence of Hodgkin lymphoma cases in our data.
We also mapped the spot-clusters of co-expressed genes extracted from the expression SOM into the methylation SOM to assess mutual correlations (see
supplementary Figure S4). Most of the effects observed are weaker than for the co-methylated gene clusters i–v presumably due to causal relationships between promoter methylation and gene expression leading to the dilution of correlations in the opposite direction. On the other hand, the data clearly reveal expression changes between lymphoma and the reference B and GCB cells, which are accompanied by marked differential methylation effects in both positive and negative directions as well.
Hence, we observe positive and negative correlations between expression and methylation changes by mapping clusters of co-methylated genes into expression space and vice versa. The most pronounced effects are observed between B/GCB cells and DLBCL in correspondence with the sample diversity analysis (
Figure 5), but also the other lymphoma subtypes show gradual and specific effects roughly in the same order as illustrated in
Figure 2c.
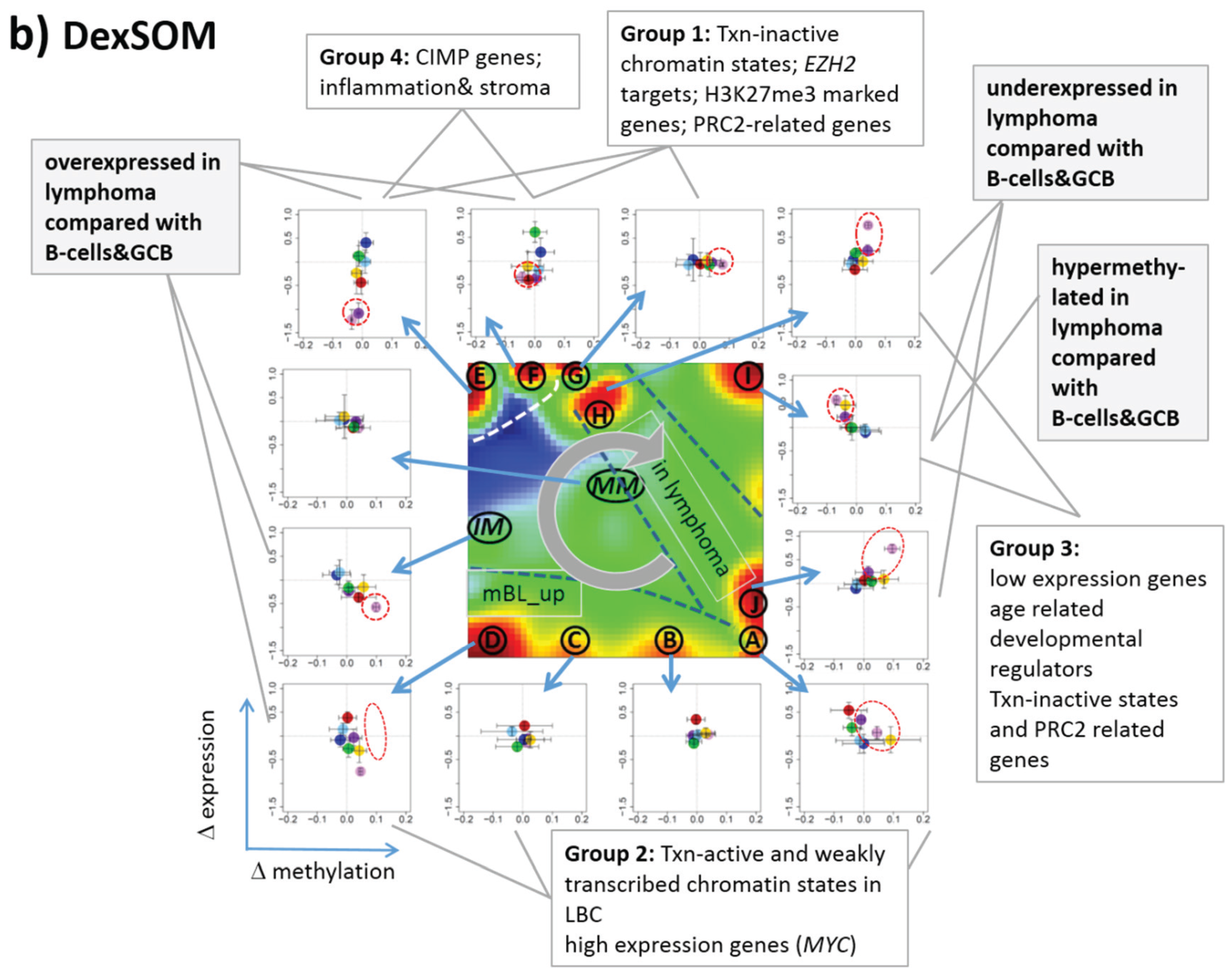
Figure 7.
Mapping of differentially methylated genes into the expression SOM: (a) The Dmet-gene clusters i–v (red circles) were mapped into the Met- and Dex-SOM where they accumulate in specific areas (red rectangles). The red arrows illustrate the mapping direction; The Dmet- and Dex-profiles reveal class-specific correlations between methylation and expression data which are plotted in panel (b) for genes taken from each of the Dmet-spots i–v. The class-averaged mean methylation and expression levels of the gene groups were plotted in x and y directions, respectively, each class represented by a colored dot. The error bars indicate the standard deviation of the sample data of each class. The dotted arrows point from the GCB cell to the DLBCL dots thus serving as indicator for the slope of the mutual association between the methylation and expression data. Note that the clockwise arrangement of the spots i–v in the DmetSOM transforms into counter-clockwise arrangement of the data cloud in the correlation plots (see Discussion section).
Figure 7.
Mapping of differentially methylated genes into the expression SOM: (a) The Dmet-gene clusters i–v (red circles) were mapped into the Met- and Dex-SOM where they accumulate in specific areas (red rectangles). The red arrows illustrate the mapping direction; The Dmet- and Dex-profiles reveal class-specific correlations between methylation and expression data which are plotted in panel (b) for genes taken from each of the Dmet-spots i–v. The class-averaged mean methylation and expression levels of the gene groups were plotted in x and y directions, respectively, each class represented by a colored dot. The error bars indicate the standard deviation of the sample data of each class. The dotted arrows point from the GCB cell to the DLBCL dots thus serving as indicator for the slope of the mutual association between the methylation and expression data. Note that the clockwise arrangement of the spots i–v in the DmetSOM transforms into counter-clockwise arrangement of the data cloud in the correlation plots (see Discussion section).
![]()
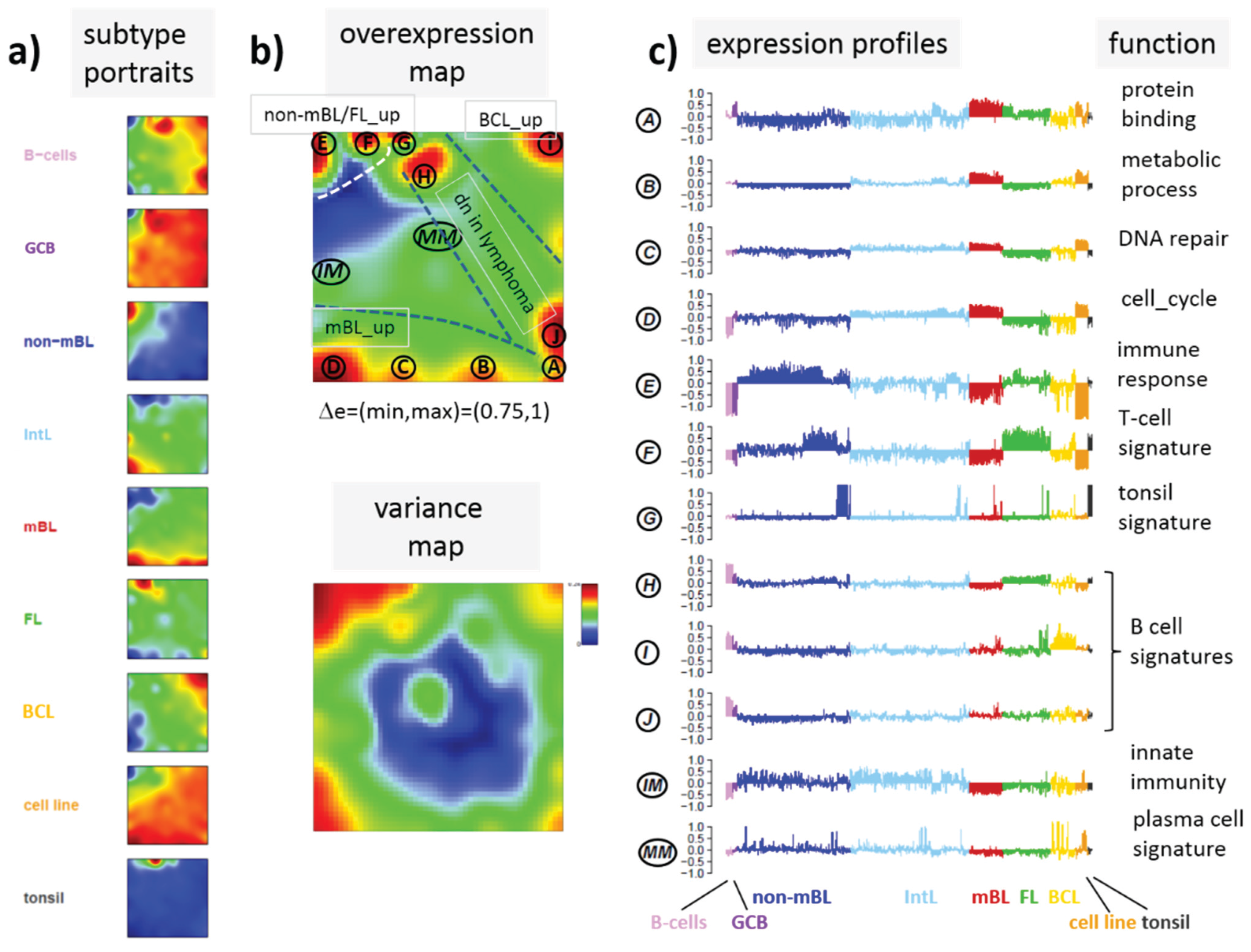
3.6. Mapping of Functional Gene Sets Indicates a Distinguished Role of PRC2 Genes
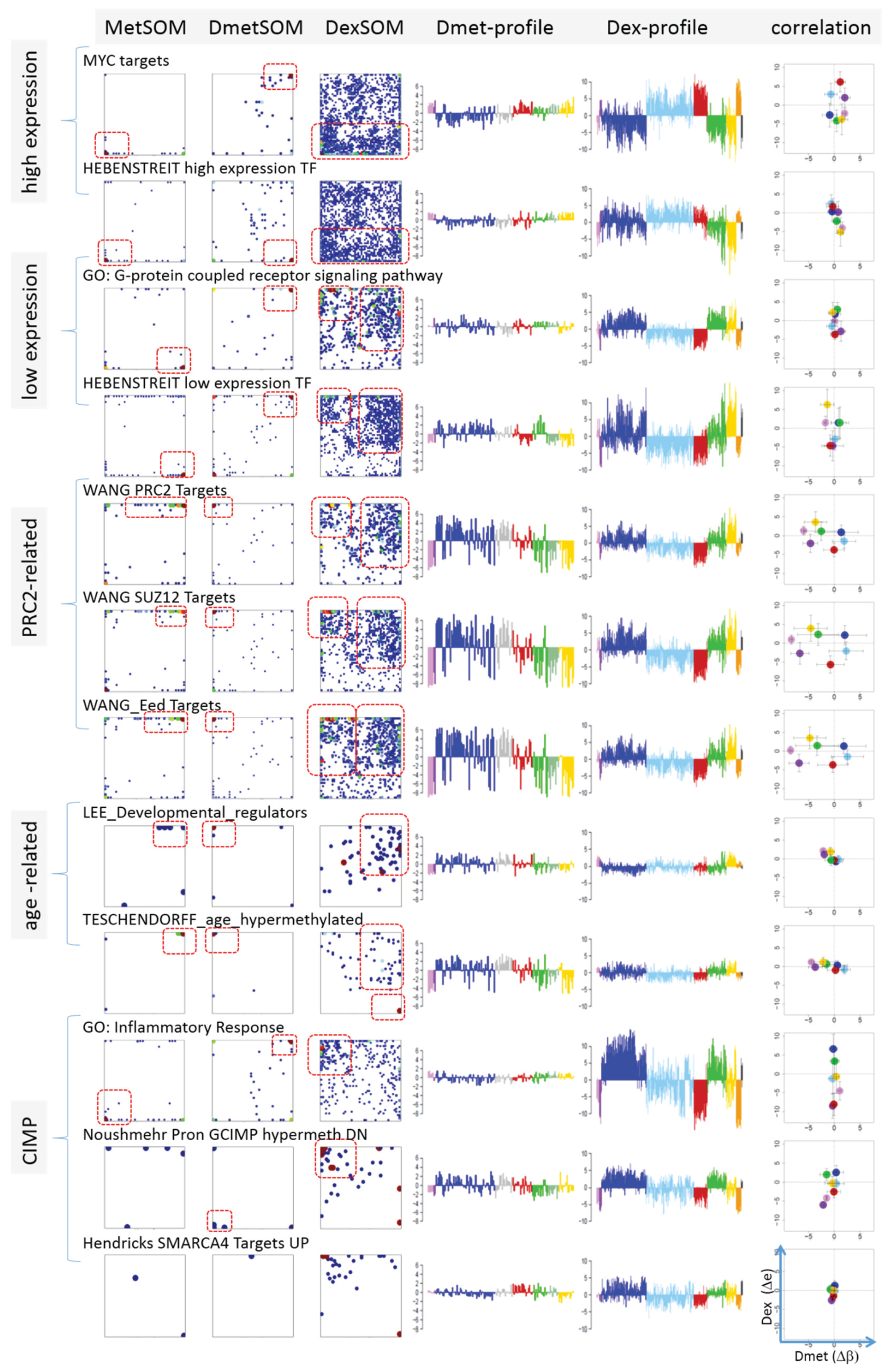
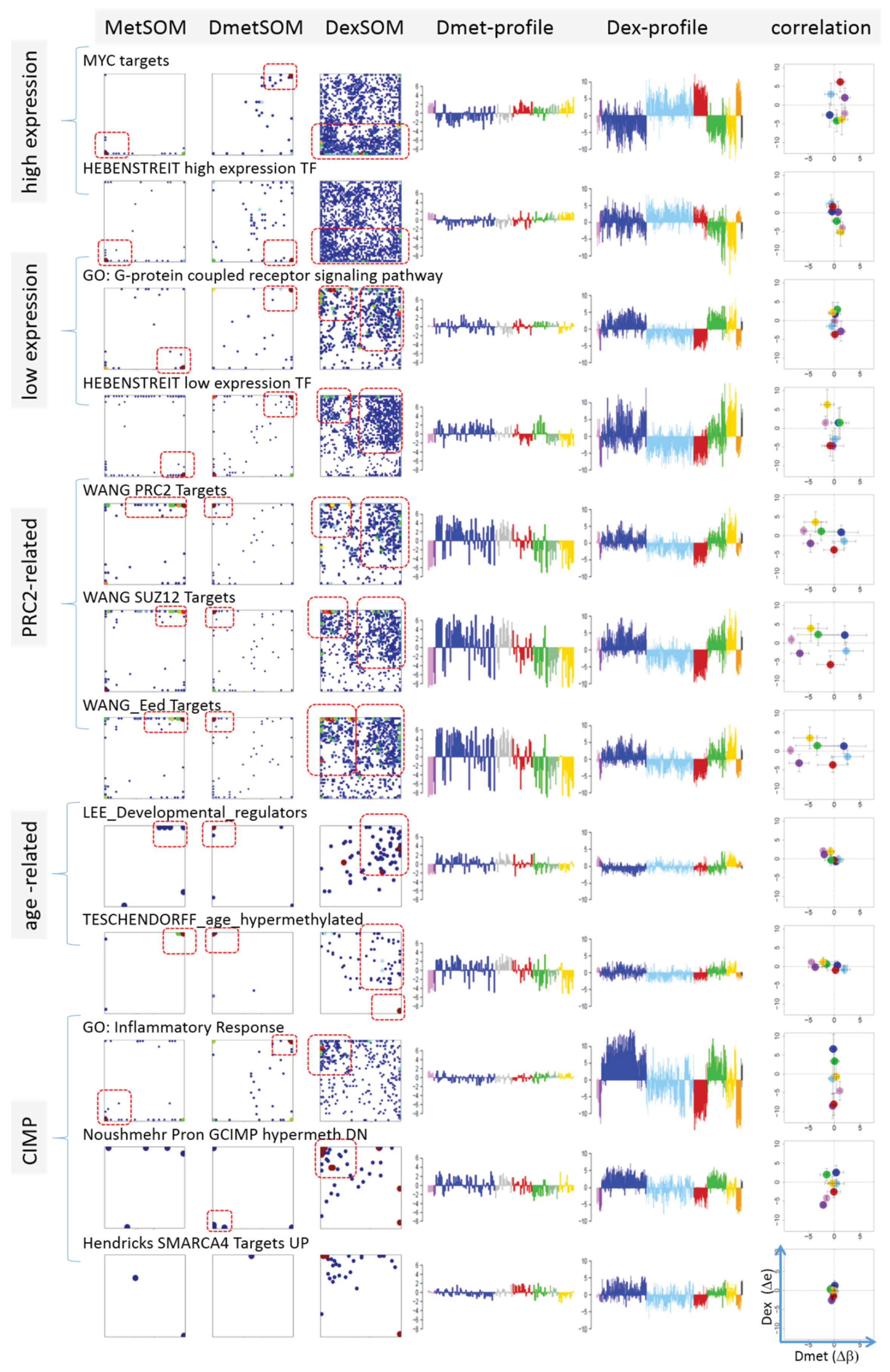
Next, we analyzed a series of functional gene sets in an analogous fashion as the spot modules in the previous subsection (
Figure 8). The obtained characteristics can be grouped into different patterns.
MYC targets [
33] and transcription factors (TF) associated with high gene expression levels [
32] give rise to large expression differences between the lymphoma classes but almost negligible methylation effects. TFs associated with low expression levels and G-protein receptors show a similar relationship between expression and methylation changes where the expression levels of the lymphoma classes however swap their order in the correlation plot. The latter effect can be directly extracted from the areas of highest population densities of the genes in the DexSOM: The “high expression” genes enrich the lower part of the DexSOM whereas the “low expression genes” preferentially occupy areas near the left and right upper corners of the map (see the dotted red rectangles in
Figure 8). The third group of PRC2-related genes gives rise to marked class-specific expression and methylation changes. Interestingly, the expression characteristics of the PRC2-group and of the “low expression” group are almost identical whereas their methylation characteristics differ largely in amplitude. It seems that the PRC2-related gene sets specifically select genes which change expression and methylation in a lymphoma-specific fashion whereas the low expression gene sets contain genes which show main effects in the expression domain only. This difference can be rationalized by the fact that a large fraction of these genes is affected “indirectly” by downstream co-regulation of gene expression without alterations of promoter methylation.
The next group of “age related genes” can be interpreted as a subgroup of the PRC2-related and low expression genes which occupies essentially only the right upper region of the DexSOM. In the correlation plot, one sees that this restriction strongly reduces the variance of the expression values between the lymphoma classes whereas the alterations of methylation are similar to the PRC2-related gene sets. This result implies that PRC2-related genes are governed by more diverse regulation mechanisms of gene expression than the age-related genes. Note however that the gene set “developmental regulators” being part of the “age-related” group also collects genes referring to the formation of the polycomb complex [
34]. These genes were obtained from gene expression measurements whereas the “ageing-associated hypermethylated genes” [
35] are extracted from DNA methylation studies which explains the larger response of the latter ones in the methylation dimension.
The last “CIMP”-group genes accumulate in the top left region of the DexSOM. They consequently share similarities with the groups of “PCR2-related” and “low expression genes” whose genes also accumulate in this region of the map. The methylation effect of the gene sets’ “inflammatory response” is small but more pronounced for the GCIMP-gene set extracted from glioma data [
36]. Other CIMP- and GCIMP-related gene sets obtained in colorectal and in brain cancer studies, respectively, also respond in the methylation dimension (see
Table 1).
In summary, we found two main combined methylation/expression patterns exemplified by the “high-expression” and “PCR2-related” groups where only the latter is characterized by both expression and methylation changes. The latter group can be further split into CIMP-like and “age” related genes.
Figure 8.
Mapping of selected functional gene sets into the methylation and expression SOM (see legend of
Figure 7 for assignments). Gene sets were taken from [
30,
32,
33,
34,
35,
36,
37,
38]. The combined methylation-expression data group into five different patterns as indicated by the brackets and the designations given at the left part of the figure. An enlarged version of the correlation plots for selected gene sets is given in
Figure S7.
Figure 8.
Mapping of selected functional gene sets into the methylation and expression SOM (see legend of
Figure 7 for assignments). Gene sets were taken from [
30,
32,
33,
34,
35,
36,
37,
38]. The combined methylation-expression data group into five different patterns as indicated by the brackets and the designations given at the left part of the figure. An enlarged version of the correlation plots for selected gene sets is given in
Figure S7.
3.7. EZH2 Targets Strongly Deregulate in Lymphoma
EZH2 is the catalytic subunit of the PRC2 and mediates transcriptional repression through its histone methyltransferase activity that trimethylates H3K27 [
39,
40].
EZH2 is upregulated in normal GCB cells and it is implicated in lymphomagenesis. Its targets overlapped extensively between GCB cells and embryonic stem cells [
39] and are preferentially trimethylated lysine 27 on histone H3- (H3K27me3, about 70%) and thus transcriptional inactive.
EZH2 in normal GCB cells favors cellular proliferation by directly repressing several tumor suppressor genes, creates a repression state similar to that found in stem cells that might foster self-renewal and prevent premature differentiation, and maintains and stabilizes a transcriptional repression program already in place in naïve B-cells [
37].
We analyzed
EZH2 targets and H3K27me3-marked genes determined in hESC, (naïve) B and GCB (centroblasts) cells obtained by means of ChIP-chip experiments [
39]. The respective sets of genes were mapped into our different SOMs (
Figure S8). The methylation and expression characteristics of both
EZH2 target and H3K27me3-marked genes in hESC, B and GCB cells among the systems studied here are very similar and closely resemble those of the PRC2-related genes (compare
Figure S8 with
Figure 8). These genes show both low expression and methylation in mBL, B and GCB cells and increased expression in DLBCL, IntL, FL and MM. This result suggests that transcription is repressed in H3K27me3-marked
EZH2 targets in healthy B and GCB cells, and that repression of these targets is mostly maintained in mBL but at least partly turned into activation in DLBCL, IntL, FL and MM. These expression changes are paralleled by hypomethylation in B and GCB cells and in MM on one hand and hypermethylation in DLBCL and IntL on the other hand. Stratification of genes with respect to anti-correlation between expression of
EZH2 and that of its targets [
39] specifically selects genes from DexSOM spots E and F supporting this view because
EZH2-mediated trimethylation of H3K27 is expected to inactivate the expression of the
EZH2 target genes. Interestingly,
de-novo EZH2 targets in centroblasts compared with naïve B-cells (NB) reverses expression levels and to a less degree also methylation levels in MM, B and GCB cells. It was suggested that
EZH2 upregulation during the transition from NB cells to centroblasts reactivates a stem cell-like repression program, which is not present in NB cells and possibly featuring increased self-renewal and proliferative potential [
39].
Velichutina
et al. [
39] also compared absolute expression levels between
EZH2 targets and non-targets serving as reference. We found similar results, particularly a reduced mean gene expression of
EZH2 targets in all sample classes studied if one considers all
EZH2 targets identified in B-cells (
Figure S9). The difference of expression levels between targets and non-targets however largely reduces if one considers only
EZH2 targets showing negative correlation of their expression levels with expression of the
EZH2 gene. This difference even changes sign for targets showing positive correlations with the
EZH2 gene expression. This result shows that total gene expression markedly varies between different groups of genes where the origin of this effect is not clear. The total gene expression level of
EZH2 targets changing in concert with
EZH2 is the largest, for anticorrelated targets it is intermediate, and for targets with low correlation it is at the lowest level. Transcriptome-wide MYC hyperactivation of the first group, and chromatin reorganization and promoter methylation of the second and third groups, are potential and mutually overlapping factors that modulate total expression (see below).
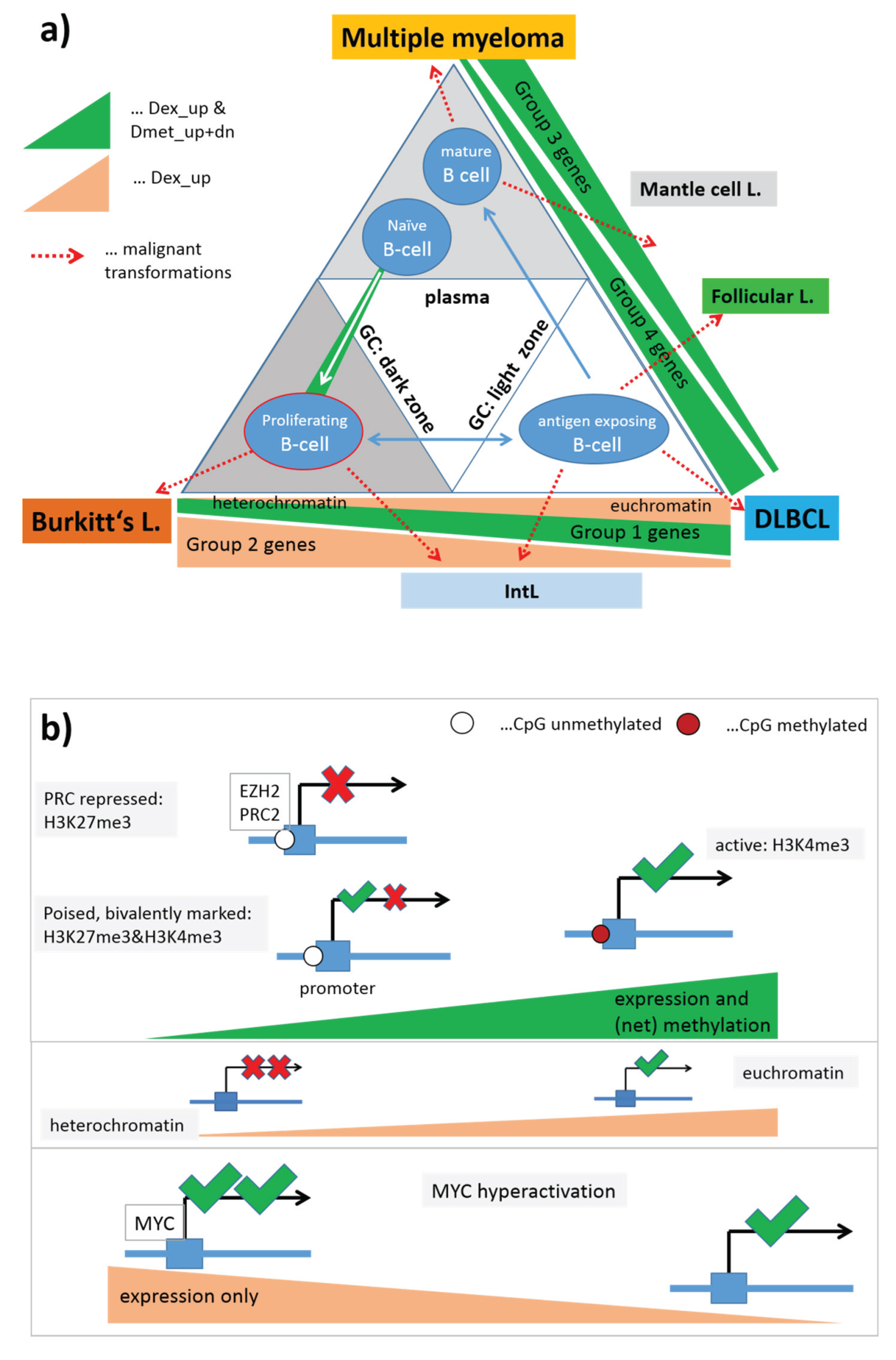
3.8. Chromatin States and Their Remodeling
Higher-order chromatin structure is emerging as an important regulator of gene expression. Alterations of gene expression programs can be induced by the remodeling of chromatin states which, for example, facilitate transcription in open regions of euchromatin, but prevent gene expression in dense packed regions of heterochromatin. These different states of chromatin conformation are governed by the arrangement of nucleosomes being the central structural elements of DNA packing in the nucleus. In turn, the arrangement of nucleosomes is modulated by chemical modifications of distinct amino acids in the side chains of the histone units forming the nucleosomes. A whole battery of such modifications and their combinatorial patterns are able to tune the transcriptional activity of the affected genes by influencing the functional state of gene’s structural elements such as enhancers and promoters, and also stages of the transcriptional process such as transcriptional elongation, transition, activation and repression [
41].
To get insight into the possible mechanism of chromatin remodeling in lymphoma, we make use of the chromatin states identified in GM12878 lymphoblastoid cells (LBC) which imitate immature lymphocytes [
38]. The chromatin states were calculated from ChIP-Seq data of a series of histone modifications using a hidden Markov model [
42]. We mapped the respective chromatin regions of each state on the human genome and collected the genes included in each of the 11 chromatin states into one gene set, and then mapped them into the lymphoma methylation and expression SOMs to assess their methylation and expression characteristics as described above (
Figure S10).
We found close correspondence between the methylation/expression properties of groups of chromatin states and the groups of functional gene sets identified above: Genes with chromatin states strongly promoting transcription (active Txn-states), namely the states “active promoters”, “transcriptional elongation” and “transcriptional transition”, “weak transcription” and also the state “weak promoter” closely resemble the characteristics of the “high expression” gene sets shown in
Figure 8. Contrarily, transcriptionally inactive states (“poised promoters” and “repressed promoters”) share close similarity with the PCR2-related gene sets. The state “heterochromatin” resembles the “low expression” gene sets. Note that the Txn-inactive states and “heterochromatin” show a nearly mirror symmetrical profile of gene expression compared with the Txn-active states with low expression levels in mBL and IntL and high levels in BCL (MM), non-mBL and FL. The state “strong enhancers” forms a separate group which differs from the functional gene sets considered. Its methylation and expression profiles virtually agree with that of the active Txn-states except for the expression level in mBL which turns from high activity in the latter states into low activity in the former one and vice versa for FL.
Interestingly, the transcriptional inactive chromatin states (and also PRC2-related genes) show the largest variability of DNA methylation between the classes with lowest levels in healthy GCB and B-cells, intermediate levels in MM, FL and mBL, and high levels in DLBCL and IntL. Thus, they resemble the order of overall methylation variability shown in
Figure 2. These methylation changes were paralleled by positively correlated alterations of gene expression. Genes located in heterochromatin show virtually the same class-dependence of gene expression but almost no variation in methylation. Hence, genes becoming activated in heterochromatin are obviously affected by other mechanisms not associated with methylation changes of their promoters.
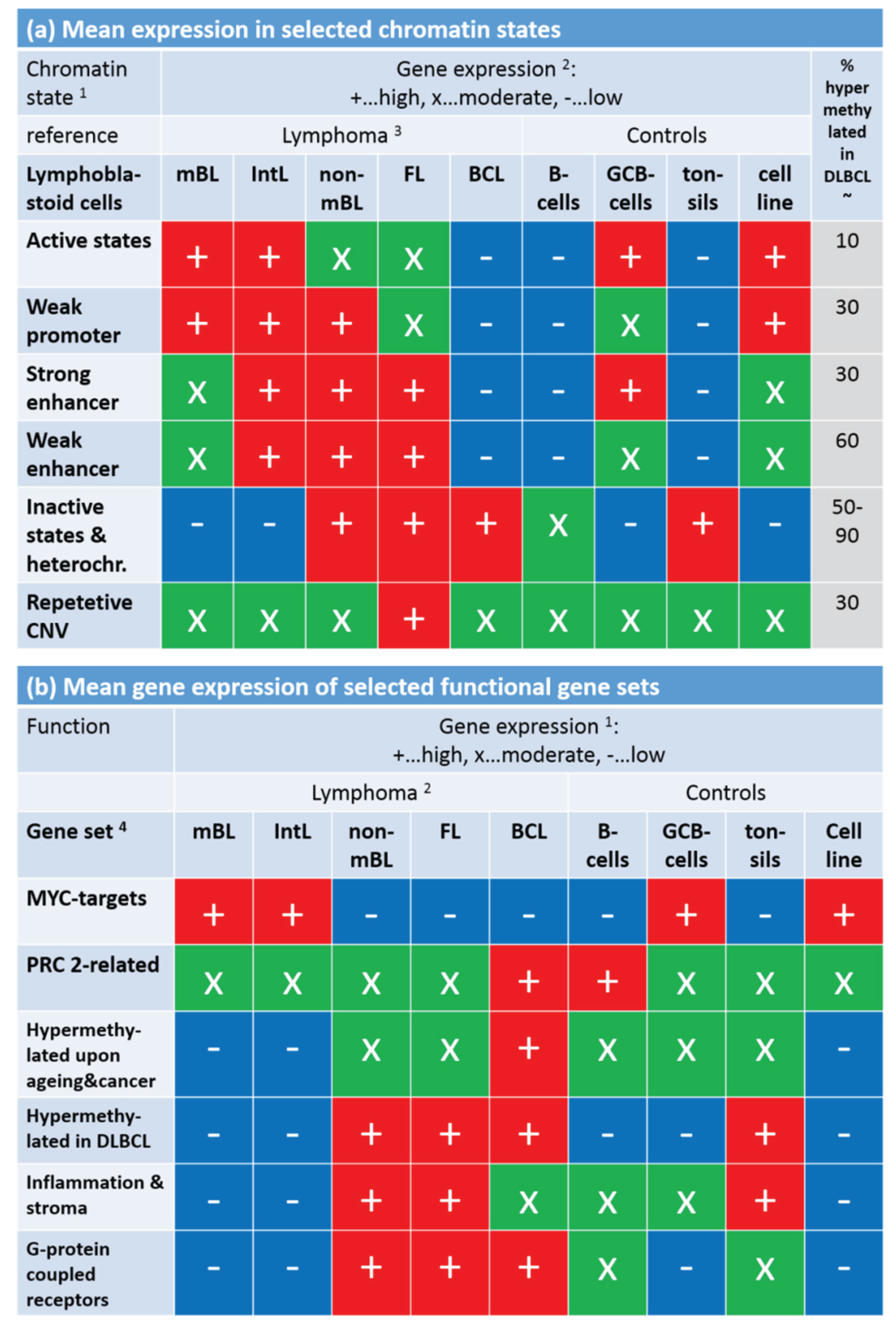
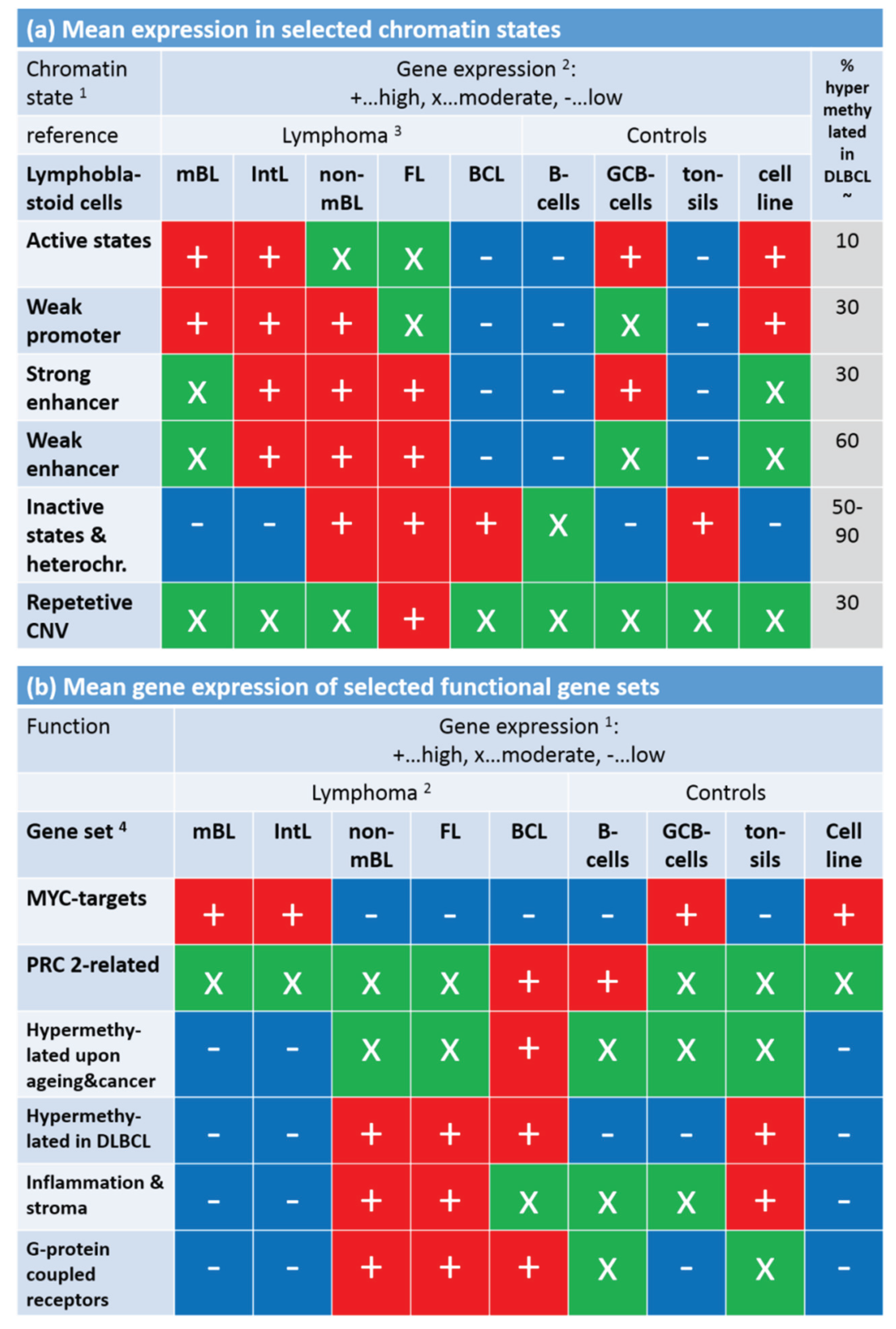
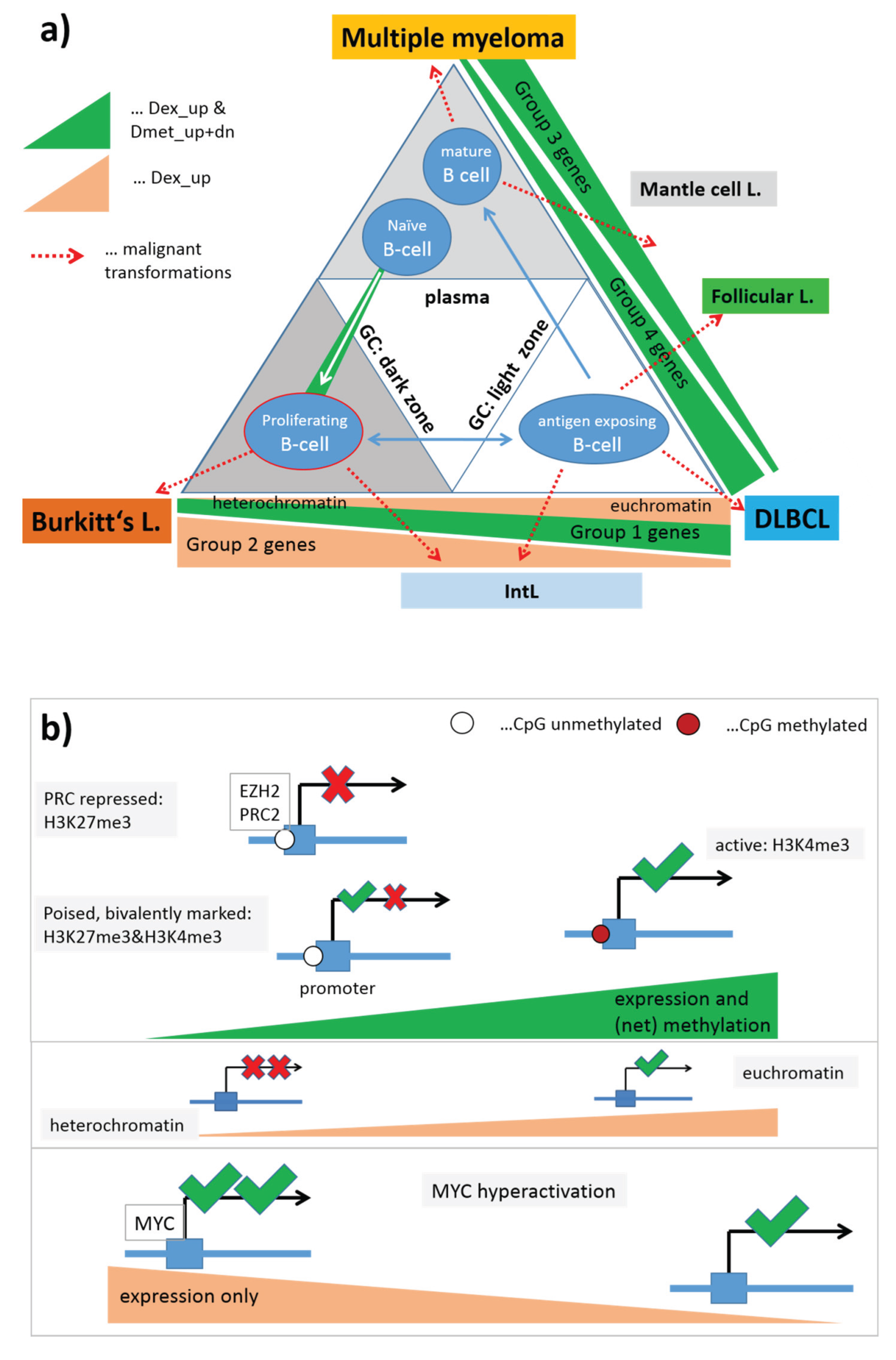
Note that the assignment of chromatin states refers to the lymphoblastoid cell line but not to the lymphoma classes studied here. Generally, one expects that Txn-active states show higher gene expression levels than Txn-inactive states and heterochromatin. This trivial relationship implies using the mean transcriptional activity of the chromatin states in lymphoma as a measure to estimate the correspondence between the nominal chromatin state referring to lymphoblastoid cells and the real one in lymphoma. For an overview, we stratified the expression levels of the chromatin states in the different lymphoma classes into high, moderate and low levels based on the GSZ-profiles shown in
Figure S10 and visualized them in
Figure 9a: The expression level observed in GCB cells, mBL and IntL is indeed high in Txn-active chromatin states and low in Txn-inactive chromatin states. Thus, the real expression levels agree with the nominal ones suggesting global correspondence between the chromatin states in the reference cells and that in mBL and IntL. Contrarily, the expression levels in BCL, FL and non-mBL disagree with the expression levels expected for the nominal chromatin states: High expression levels in the lymphoma subtypes were observed for genes assigned to inactive states in the reference cells and reduced expression levels were observed for genes in states of high transcriptional activity of the lymphoblastoid cells. Hence, the expression levels observed in mBL and IntL, in GCB cells and the cancer cell line are in correspondence with the chromatin states in lymphoblastoid cells, whereas the expression levels observed in B-cells, BCL, FL, non-mBL and also tonsils contradict them. This switching of gene activity between these two groups of samples suggests remodeling of chromatin in BCL, FL and also non-mBL compared with lymphoblastoid cells and thus also with mBL, IntL and GCB cells. Note also that the activity patterns of B-cells, tonsils and also of BCL differs from that of GCB cells suggesting remodeling of chromatin between healthy (pre- and post-GC) B-cells and GCB cells. Moreover, the similar expression patterns of B-cells and of BCL supports the plasma cell characteristics of BCL differing from the characteristics of the GC-derived lymphoma subtypes.
To assess the relationship between DNA hypermethylation in lymphoma and the chromatin states, we calculated the percentage of overlap-genes from the different chromatin states also found in the set “hypermethylated in DLBCL” taken from [
15]. The overlap of hypermethylated genes is only about 10% for transcriptional active states but much higher (50%–90%) for transcriptional inactive states. Hence, activation of the latter states in DLBCL/non-mBL and FL seems to be accompanied by hyper-methylation of a large fraction of genes being inactive in lymphoblastoid cells, mBL and IntL.
Finally, we transferred the expression levels of selected gene sets discussed above into the tabular form for direct comparison with that of the chromatin states (
Figure 9b).
MYC target genes are expressed in parallel with Txn-active states among the systems studied. This agreement suggests that the
MYC targets are found predominantly in chromatin regions active in mBL, IntL, GCB cells, the cancer cell line and lymphoblastoid cells (95% overlap between
MYC targets and active promoters). In contrast, gene sets related to inflammation and G-protein receptor activity both hypermethylated in DLBCL accumulate in chromatin states inactive in the reference system but activated in DLBCL, IntL, FL and also BCL.
In summary, gene sets referring to distinct chromatin states in the reference cells show well distinguished expression and DNA methylation characteristics either agreeing or disagreeing with the expression level expected in the nominal chromatin states. Disagreement indicates chromatin remodeling in IntL and non-mBL and especially in B-cells and BCL compared with mBL and GCB cells. Hence, one can distinguish three groups of samples showing characteristic expression patterns of genes assigned to different chromatin states. They comprise (i) mBL, GCB cells and cancer cell lines; (ii) BCL, MM and (pre- and post-GC) B cells and (iii) IntL, DLBCL and FL.
Figure 9.
Mean gene expression level of selected chromatin states in the lymphoma and reference systems (part
a) and of selected gene sets (part
b). Gene expression was stratified into high (red), moderate (green) and low (blue) levels using the respective GSZ-profiles.
1 Chromatin states were defined in [
42] with respect to the associated histone marks (see, e.g.,
Figure 1b in [
42]). The most characteristic marks are in active states (e.g., active promoters): H3K4me3/me2, H3K27ac, H3K9ac; weak promoter: H3K4me3/me2; strong enhancer: H3K4me1/me2, H3K27ac, H3K9ac; weak enhancer: H3K4me1/me2; inactive states (e.g., inactive and poised promoters): H3K27me3, H3K4me2; heterochromatin: no mark; repetitive CNV: all marks;
2 gene expression levels of the chromatin states (
Figure S10) and functional gene sets (
Figure 8);
3 assignment of lymphoma classes refers to the expression classes introduced in
Figure 6;
4 Gene sets were taken from [
33] (MYC), [
34] (PRC developmental regulators), [
35] (Hypermethylated upon ageing and cancer), [
15] (hypermethylated in DLBCL, [
3,
43] (inflammation and stroma) and GO (G-protein coupled receptor activity and signaling pathway); see also
Figure 8.
Figure 9.
Mean gene expression level of selected chromatin states in the lymphoma and reference systems (part
a) and of selected gene sets (part
b). Gene expression was stratified into high (red), moderate (green) and low (blue) levels using the respective GSZ-profiles.
1 Chromatin states were defined in [
42] with respect to the associated histone marks (see, e.g.,
Figure 1b in [
42]). The most characteristic marks are in active states (e.g., active promoters): H3K4me3/me2, H3K27ac, H3K9ac; weak promoter: H3K4me3/me2; strong enhancer: H3K4me1/me2, H3K27ac, H3K9ac; weak enhancer: H3K4me1/me2; inactive states (e.g., inactive and poised promoters): H3K27me3, H3K4me2; heterochromatin: no mark; repetitive CNV: all marks;
2 gene expression levels of the chromatin states (
Figure S10) and functional gene sets (
Figure 8);
3 assignment of lymphoma classes refers to the expression classes introduced in
Figure 6;
4 Gene sets were taken from [
33] (MYC), [
34] (PRC developmental regulators), [
35] (Hypermethylated upon ageing and cancer), [
15] (hypermethylated in DLBCL, [
3,
43] (inflammation and stroma) and GO (G-protein coupled receptor activity and signaling pathway); see also
Figure 8.
![]()





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}