
Genotype-Epigenotype Interaction at the IGF2 DMR

Abstract
:1. Introduction
2. Materials and Methods
Human Subjects
3. Results
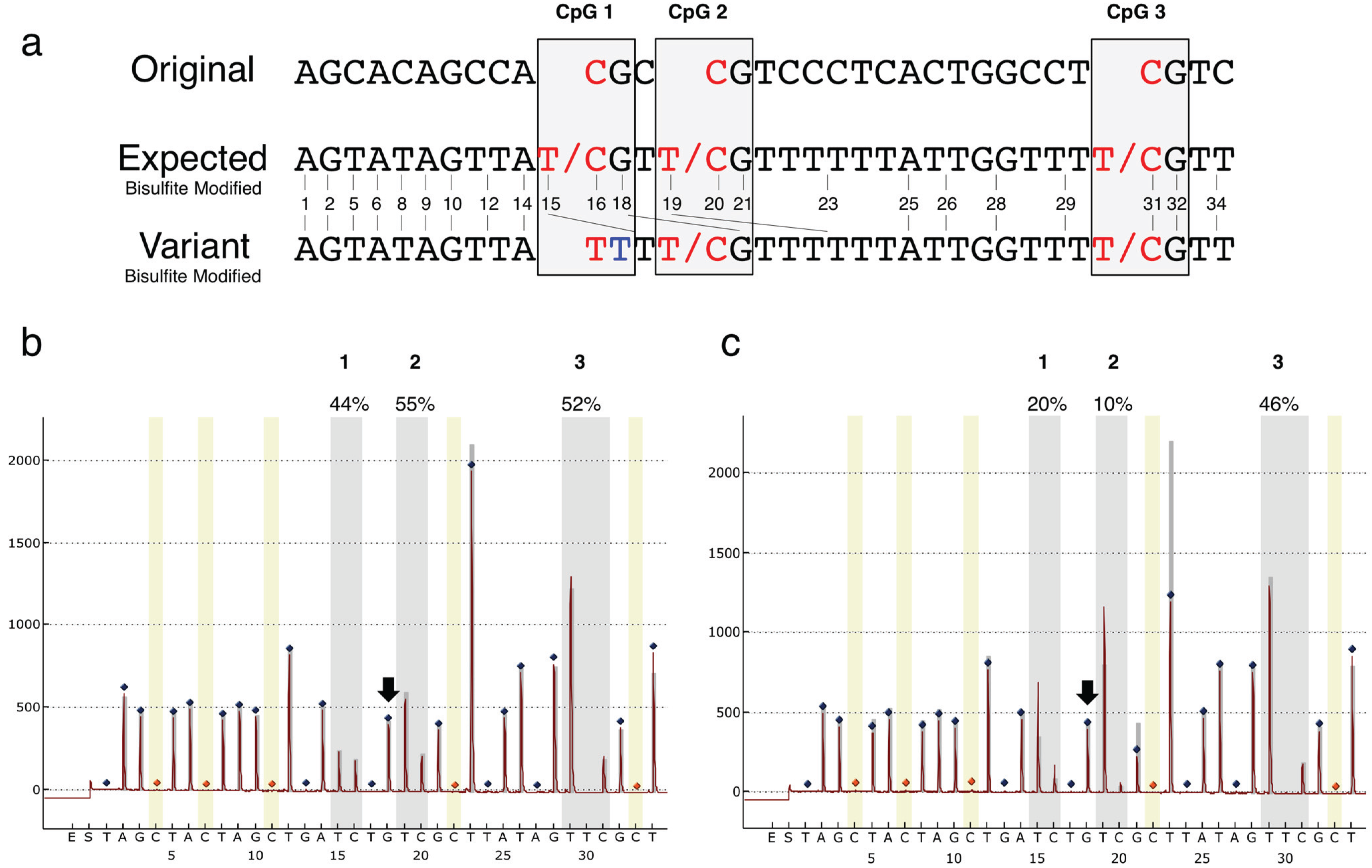

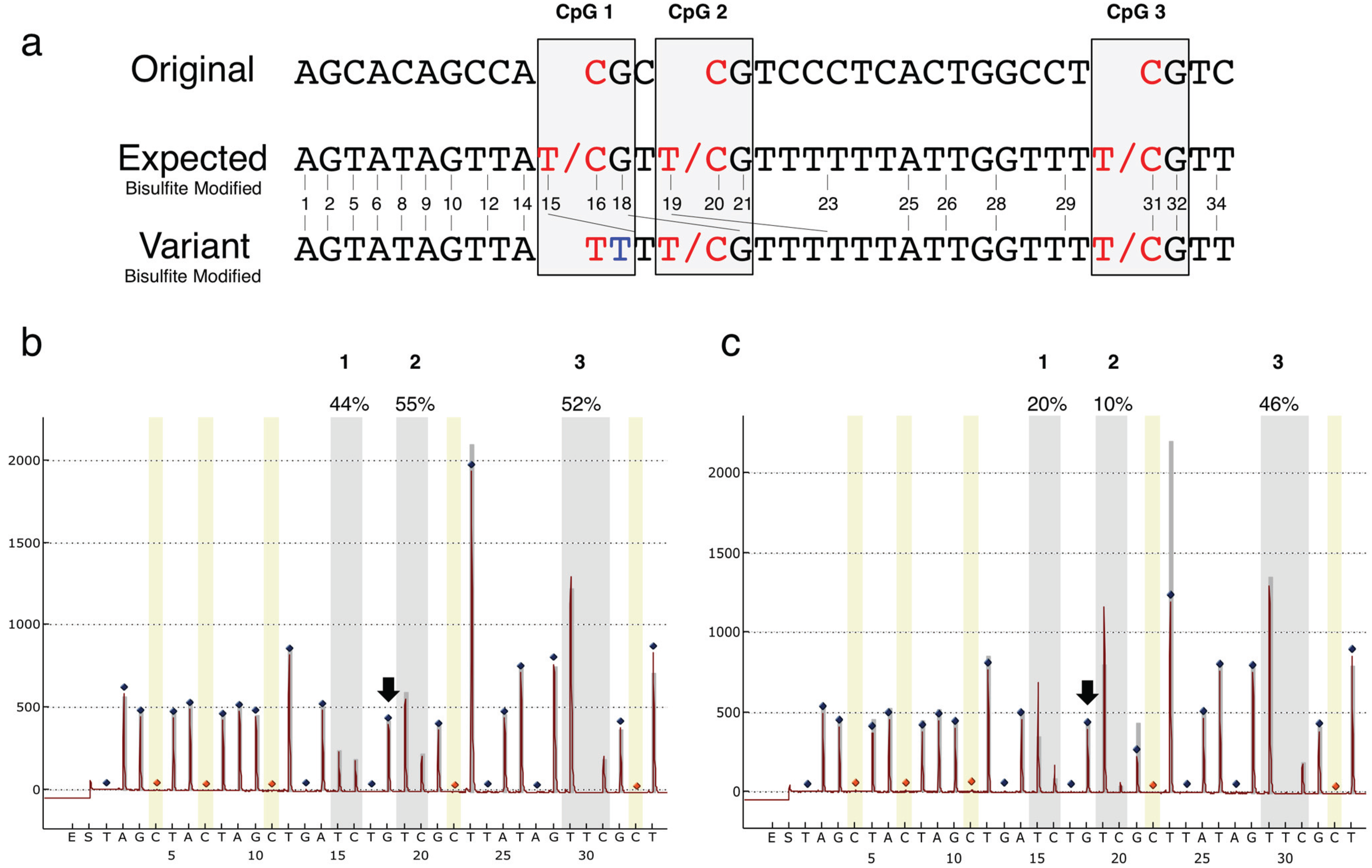{kind=link}
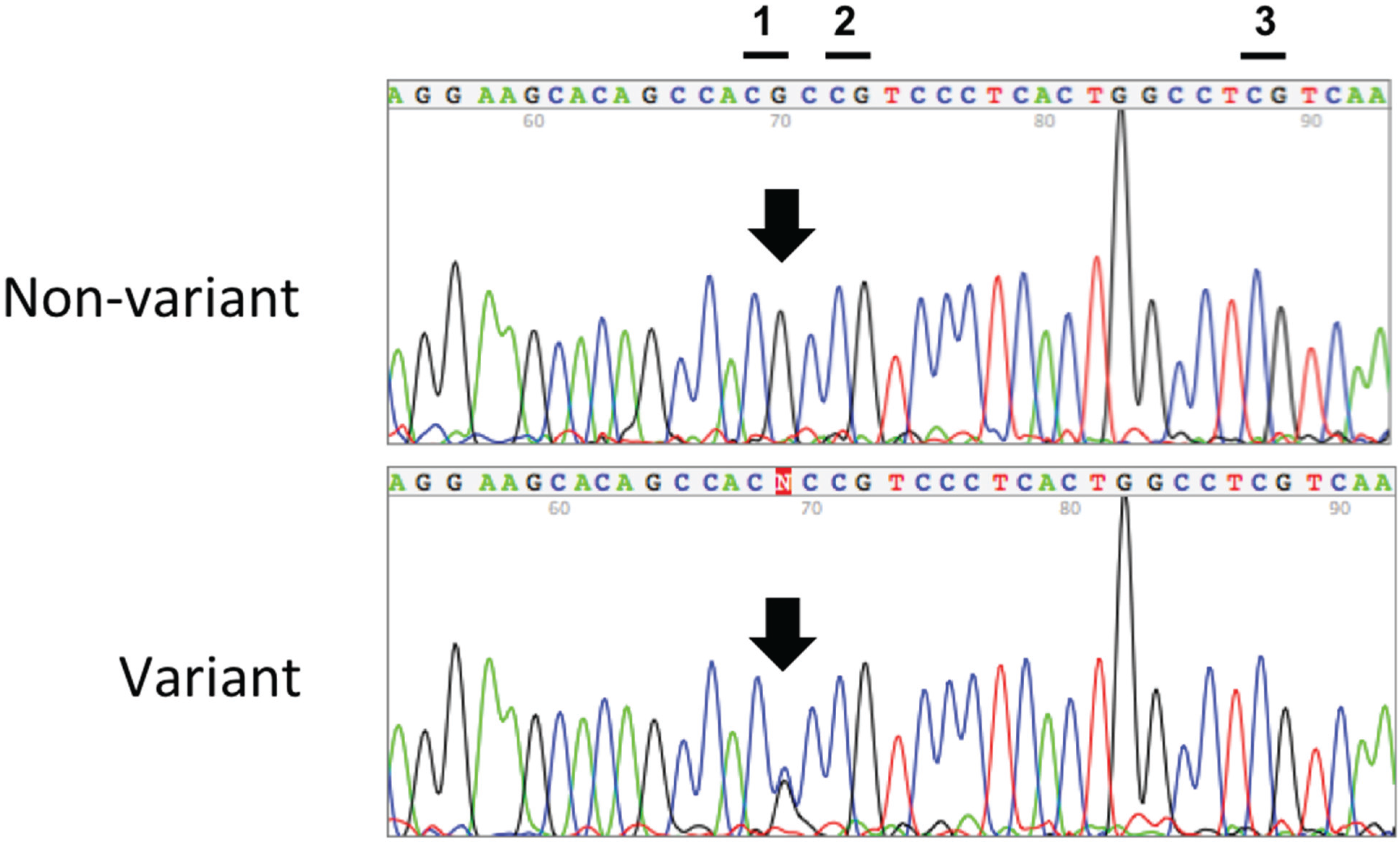
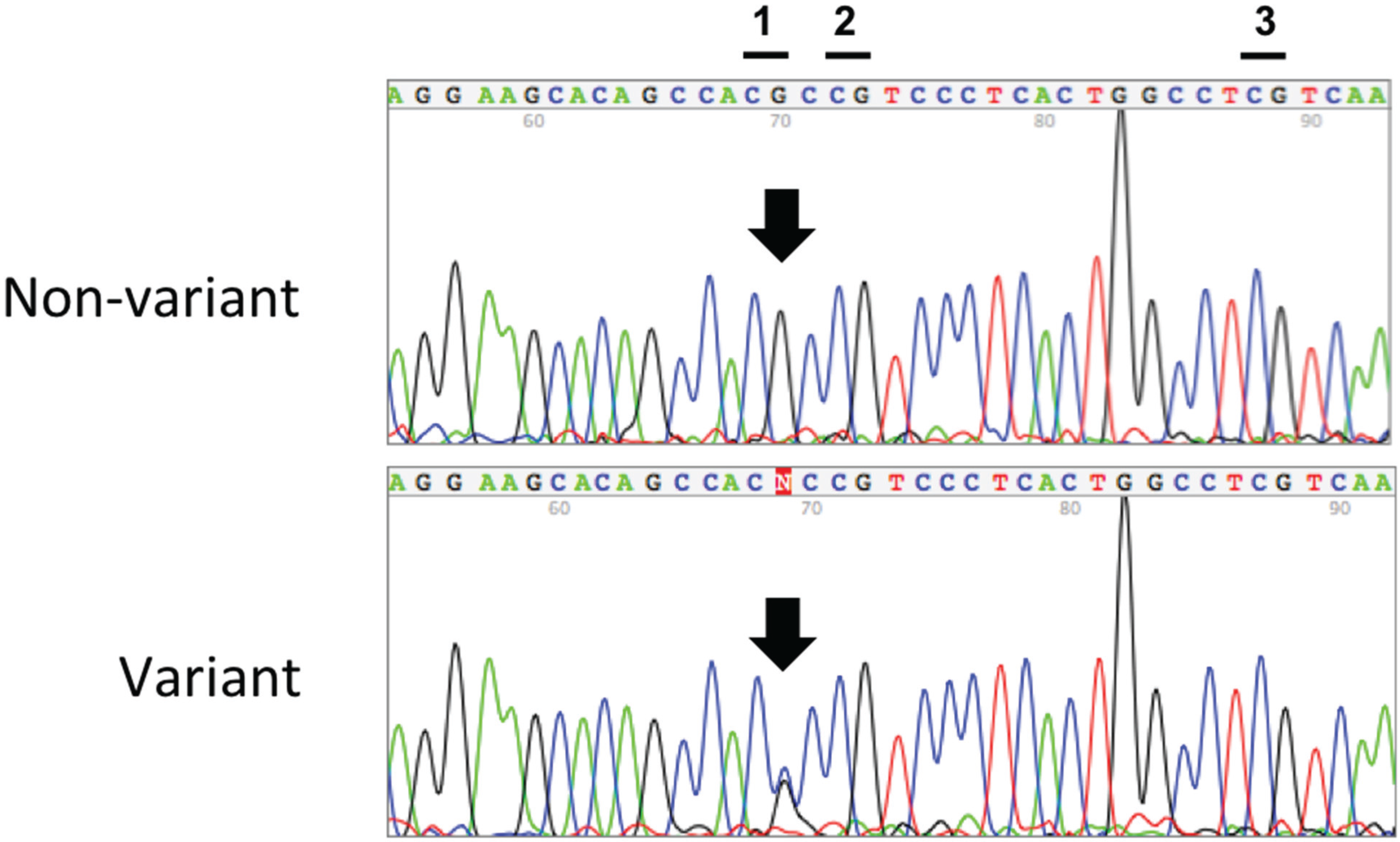{kind=link}
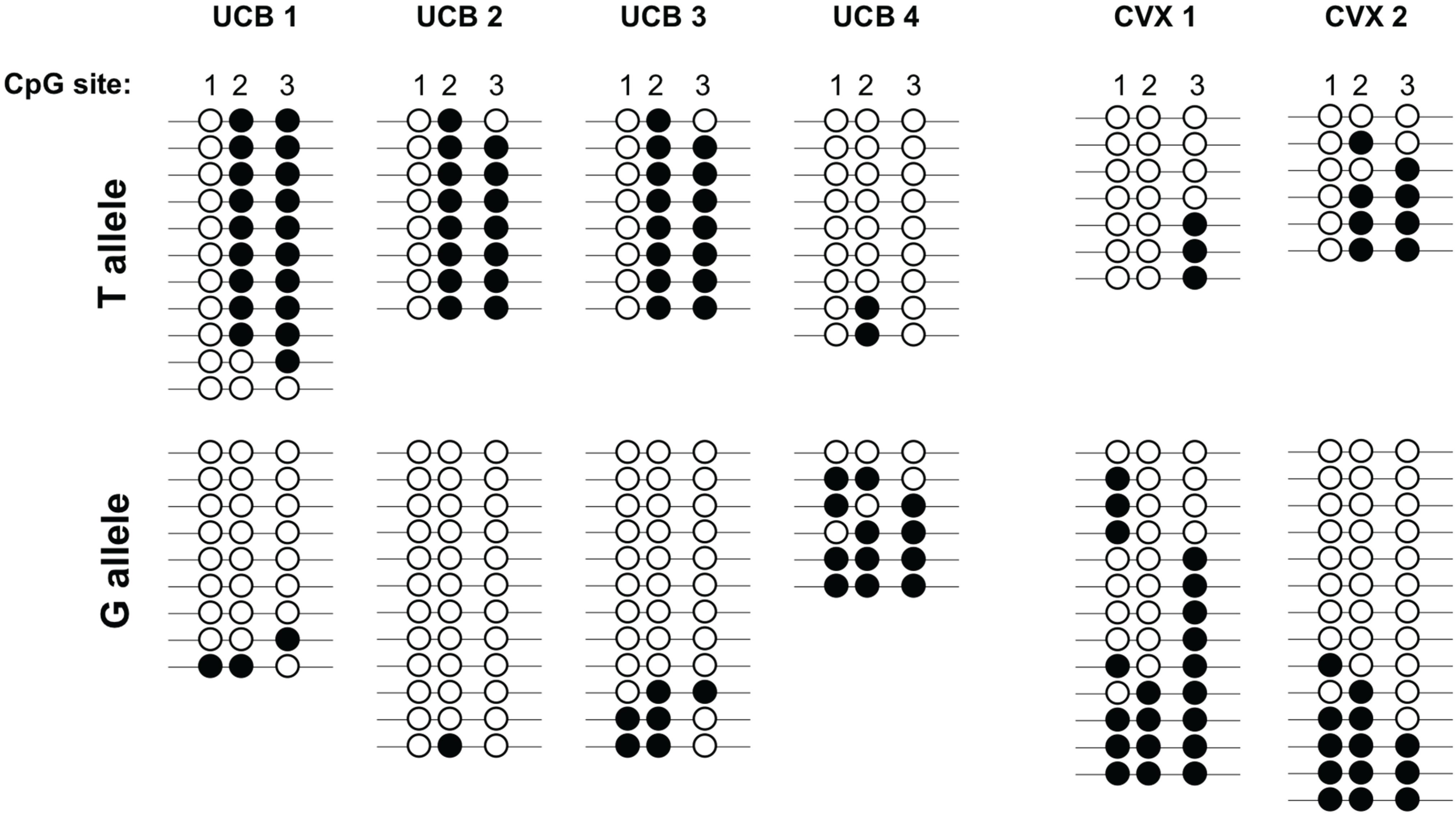{kind=link}
| rs116779517 | WT (%) | Heterozygote (%) |
|---|---|---|
| Caucasian | 510 (99) | 7 (1) |
| African American | 445 (92) | 39 (8) |
| Asian/Pacific Islander | 18 (100) | 0 (0) |
| Native American | 4 (100) | 0 (0) |
| Multiracial | 10 (100) | 0 (0) |
| Other | 53 (96) | 2 (4) |
| Don't Know | 5 (100) | 0 (0) |
| Missing | 9 (82) | 2 (18) |
| Total | 1054 (95) | 50 (5) |
| rs116779517 | WT (%) | Heterozygote (%) |
|---|---|---|
| Non-neoplastic | 88 (90) | 10 (10) |
| Neoplastic | 59 (87) | 9 (13) |
| Total | 147 (89) | 19 (11) |
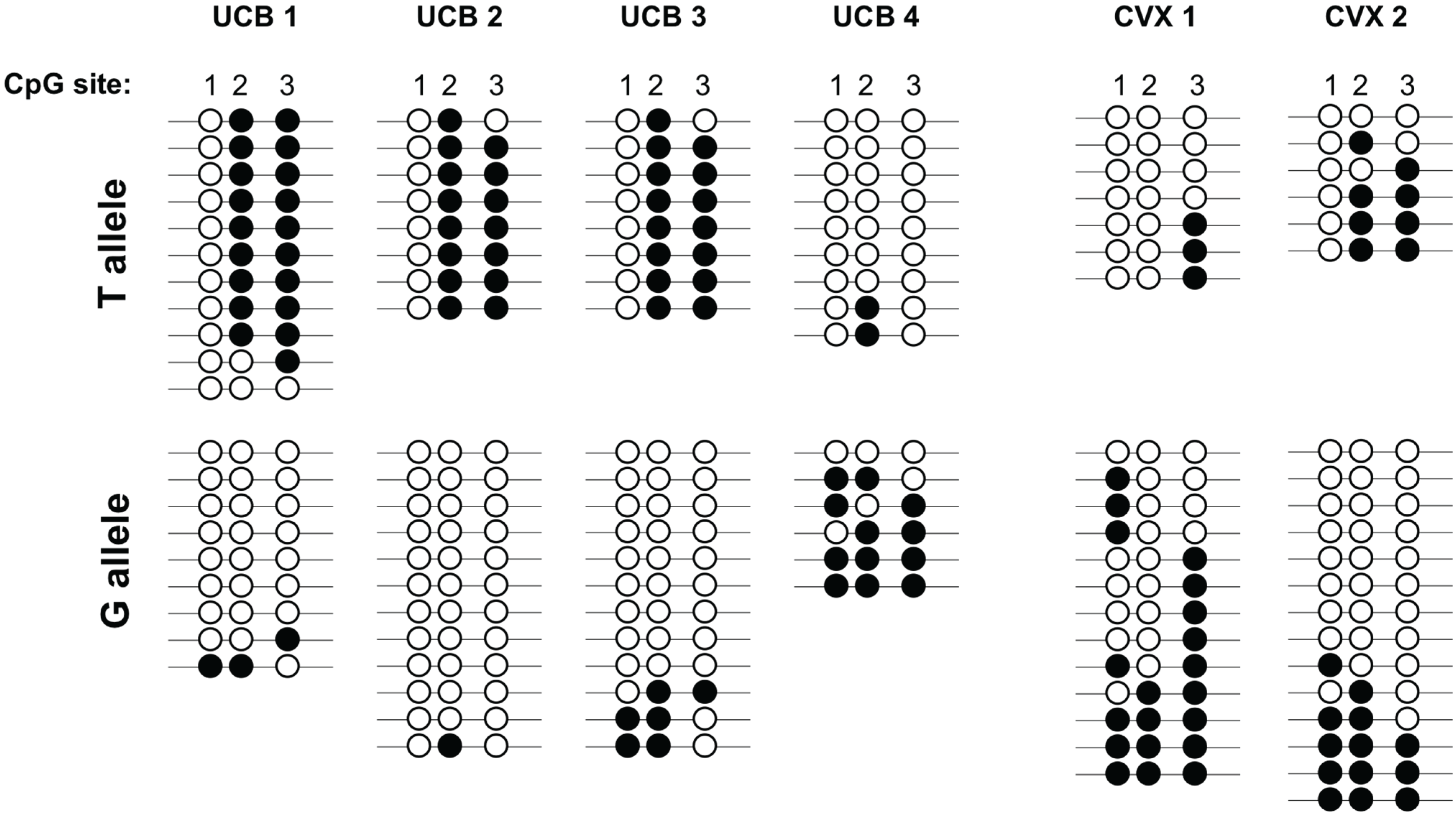
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brouwer-Visser, J.; Huang, G.S. IGF2 signaling and regulation in cancer. Cytokine Growth Factor Rev. 2015, 26, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Zhang, Q.; Liu, X.; Miao, C.; Shangguan, S.; Bao, Y.; Guo, J.; Wang, L.; Zhang, T.; Li, H.; et al. Different epigenetic alterations are associated with abnormal IGF2/IGF2 upregulation in neural tube defects. PLoS ONE 2014, 9, e113308. [Google Scholar] [CrossRef] [PubMed]
- Bracko, O.; Singer, T.; Aigner, S.; Knobloch, M.; Winner, B.; Ray, J.; Clemenson, G.D., Jr.; Suh, H.; Couillard-Despres, S.; Aigner, L.; et al. Gene expression profiling of neural stem cells and their neuronal progeny reveals IGF2 as a regulator of adult hippocampal neurogenesis. J. Neurosci. 2012, 32, 3376–3387. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Wang, L.; Shangguan, S.; Chang, S.; Wang, Z.; Lu, X.; Zhang, Q.; Wang, J.; Zhao, H.; Wang, F.; et al. Altered methylation of IGF2 DMR0 is associated with neural tube defects. Mol. Cell. Biochem. 2013, 380, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Livingstone, C. IGF2 and cancer. Endocr. Relat. Cancer 2013, 20, R321–R339. [Google Scholar] [CrossRef] [PubMed]
- Alberini, C.M.; Chen, D.Y. Memory enhancement: consolidation, reconsolidation and insulin-like growth factor 2. Trends Neurosci. 2012, 35, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, T.; Ouchi, Y. Emerging evidence of insulin-like growth factor 2 as a memory enhancer: a unique animal model of cognitive dysfunction with impaired adult neurogenesis. Rev. Neurosci. 2014, 25, 559–574. [Google Scholar] [CrossRef] [PubMed]
- Pascual-Lucas, M.; Viana da Silva, S.; di Scala, M.; Garcia-Barroso, C.; Gonzalez-Aseguinolaza, G.; Mulle, C.; Alberini, C.M.; Cuadrado-Tejedor, M.; Garcia-Osta, A. Insulin-like growth factor 2 reverses memory and synaptic deficits in APP transgenic mice. EMBO Mol. Med. 2014, 6, 1246–1262. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, L.P.; Tang, H.; Shan, W.Y.; Wang, X.; Liu, D.; Wu, Y.Y.; Tian, Q.; Wang, J.Z.; Zhu, L.Q.; et al. Acetyl-L-carnitine rescues scopolamine-induced memory deficits by restoring insulin-like growth factor II via decreasing p53 oxidation. Neuropharmacology 2014, 76, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.N. Eukaryotic DNA methylation. Hum. Genet. 1983, 64, 315–333. [Google Scholar] [CrossRef] [PubMed]
- Jurkowska, R.Z.; Jurkowski, T.P.; Jeltsch, A. Structure and function of mammalian DNA methyltransferases. Chembiochem 2011, 12, 206–222. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.K.; Huang, Z.; Wen, Y.; Spillman, M.A.; Whitaker, R.S.; Simel, L.R.; Nichols, T.D.; Marks, J.R.; Berchuck, A. Frequent IGF2/H19 domain epigenetic alterations and elevated IGF2 expression in epithelial ovarian cancer. Mol. Cancer Res. 2006, 4, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Vidal, A.C.; Henry, N.M.; Murphy, S.K.; Oneko, O.; Nye, M.; Bartlett, J.A.; Overcash, F.; Huang, Z.; Wang, F.; Mlay, P.; et al. PEG1/MEST and IGF2 DNA methylation in CIN and in cervical cancer. Clin. Transl. Oncol. 2014, 16, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Rumbajan, J.M.; Maeda, T.; Souzaki, R.; Mitsui, K.; Higashimoto, K.; Nakabayashi, K.; Yatsuki, H.; Nishioka, K.; Harada, R.; Aoki, S.; et al. Comprehensive analyses of imprinted differentially methylated regions reveal epigenetic and genetic characteristics in hepatoblastoma. BMC Cancer 2013. [Google Scholar] [CrossRef] [PubMed]
- Baba, Y.; Nosho, K.; Shima, K.; Huttenhower, C.; Tanaka, N.; Hazra, A.; Giovannucci, E.L.; Fuchs, C.S.; Ogino, S. Hypomethylation of the IGF2 DMR in colorectal tumors, detected by bisulfite pyrosequencing, is associated with poor prognosis. Gastroenterology 2010, 139, 1855–1864. [Google Scholar] [CrossRef] [PubMed]
- Perkins, E.; Murphy, S.K.; Murtha, A.P.; Schildkraut, J.; Jirtle, R.L.; Demark-Wahnefried, W.; Forman, M.R.; Kurtzberg, J.; Overcash, F.; Huang, Z.; et al. Insulin-like Growth Factor 2/H19 methylation at birth and risk of overweight and obesity in children. J. Pediatr. 2012, 161, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Murphy, S.K.; Murtha, A.P.; Fuemmeler, B.F.; Schildkraut, J.; Huang, Z.; Overcash, F.; Kurtzberg, J.; Jirtle, R.; Iversen, E.S.; et al. Depression in pregnancy, infant birth weight and DNA methylation of imprint regulatory elements. Epigenetics 2012, 7, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.K.; Adigun, A.; Huang, Z.; Overcash, F.; Wang, F.; Jirtle, R.L.; Schildkraut, J.M.; Murtha, A.P.; Iversen, E.S.; Hoyo, C. Gender-specific methylation differences in relation to prenatal exposure to cigarette smoke. Gene 2012, 494, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Hoyo, C.; Murtha, A.P.; Schildkraut, J.M.; Forman, M.R.; Calingaert, B.; Demark-Wahnefried, W.; Kurtzberg, J.; Jirtle, R.L.; Murphy, S.K. Folic acid supplementation before and during pregnancy in the Newborn Epigenetics STudy (NEST). BMC Public Health 2011. [Google Scholar] [CrossRef] [PubMed]
- Hoyo, C.; Fortner, K.; Murtha, A.P.; Schildkraut, J.M.; Soubry, A.; Demark-Wahnefried, W.; Jirtle, R.L.; Kurtzberg, J.; Forman, M.R.; Overcash, F.; et al. Association of cord blood methylation fractions at imprinted insulin-like growth factor 2 (IGF2), plasma IGF2, and birth weight. Cancer Causes Control 2012, 23, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Valero, M.A.; Rother, J.; Gorlov, I.; Frazier, M.; Gorlova, O.Y. Interplay between polymorphisms and methylation in the H19/IGF2 gene region may contribute to obesity in Mexican-American children. J. Dev. Orig. Health Dis. 2013, 4, 499–506. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Hivert, M.F.; Perron, P.; Poirier, P.; Guay, S.P.; Brisson, D.; Bouchard, L. IGF2 DNA methylation is a modulator of newborn’s fetal growth and development. Epigenetics 2012, 7, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Tobi, E.W.; Slagboom, P.E.; van Dongen, J.; Kremer, D.; Stein, A.D.; Putter, H.; Heijmans, B.T.; Lumey, L.H. Prenatal famine and genetic variation are independently and additively associated with DNA methylation at regulatory loci within IGF2/H19. PLoS ONE 2012, 7, e37933. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Onyango, P.; Brandenburg, S.; Wu, Y.; Hsieh, C.L.; Feinberg, A.P. Loss of imprinting in colorectal cancer linked to hypomethylation of H19 and IGF2. Cancer Res. 2002, 62, 6442–6446. [Google Scholar] [PubMed]
- Cui, H.; Cruz-Correa, M.; Giardiello, F.M.; Hutcheon, D.F.; Kafonek, D.R.; Brandenburg, S.; Wu, Y.; He, X.; Powe, N.R.; Feinberg, A.P. Loss of IGF2 imprinting: A potential marker of colorectal cancer risk. Science 2003, 299, 1753–1755. [Google Scholar] [CrossRef] [PubMed]
- Heijmans, B.T.; Tobi, E.W.; Stein, A.D.; Putter, H.; Blauw, G.J.; Susser, E.S.; Slagboom, P.E.; Lumey, L.H. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 17046–17049. [Google Scholar] [CrossRef] [PubMed]
- Soubry, A.; Schildkraut, J.M.; Murtha, A.; Wang, F.; Huang, Z.; Bernal, A.; Kurtzberg, J.; Jirtle, R.L.; Murphy, S.K.; Hoyo, C. Paternal obesity is associated with IGF2 hypomethylation in newborns: results from a Newborn Epigenetics Study (NEST) cohort. BMC Med. 2013. [Google Scholar] [CrossRef] [PubMed]
- Vidal, A.C.; Murphy, S.K.; Hernandez, B.Y.; Vasquez, B.; Bartlett, J.A.; Oneko, O.; Mlay, P.; Obure, J.; Overcash, F.; Smith, J.S.; et al. Distribution of HPV genotypes in cervical intraepithelial lesions and cervical cancer in Tanzanian women. Infect. Agent Cancer 2011. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hoyo, C.; Murphy, S.K.; Huang, Z.; Overcash, F.; Thompson, J.; Brown, H.; Murtha, A.P. DNA methylation at imprint regulatory regions in preterm birth and infection. Am. J. Obstet. Gynecol. 2013, 208, 395.e1–395.e7. [Google Scholar] [CrossRef] [PubMed]
- Nye, M.D.; Hoyo, C.; Huang, Z.; Vidal, A.C.; Wang, F.; Overcash, F.; Smith, J.S.; Vasquez, B.; Hernandez, B.; Swai, B.; et al. Associations between methylation of paternally expressed gene 3 (PEG3), cervical intraepithelial neoplasia and invasive cervical cancer. PLoS ONE 2013, 8, e56325. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Wen, Y.; Shandilya, R.; Marks, J.R.; Berchuck, A.; Murphy, S.K. High throughput detection of M6P/IGF2R intronic hypermethylation and LOH in ovarian cancer. Nucleic Acids Res. 2006, 34, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.L.; Byun, H.M.; Kwan, J.M.; Campan, M.; Ingles, S.A.; Laird, P.W.; Yang, A.S. Rapid and quantitative method of allele-specific DNA methylation analysis. Biotechniques 2006, 41, 734–739. [Google Scholar] [CrossRef] [PubMed]
- Genomes Project Consortium; Abecasis, G.R.; Altshuler, D.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Gibbs, R.A.; Hurles, M.E.; McVean, G.A. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [PubMed]
- Murrell, A.; Ito, Y.; Verde, G.; Huddleston, J.; Woodfine, K.; Silengo, M.C.; Spreafico, F.; Perotti, D.; de Crescenzo, A.; Sparago, A.; et al. Distinct methylation changes at the IGF2-H19 locus in congenital growth disorders and cancer. PLoS ONE 2008, 3, e1849. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Murphy, S.K. Increased Intragenic IGF2 Methylation is associated with repression of insulator activity and elevated expression in serous ovarian carcinoma. Front. Oncol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Dammann, R.H.; Kirsch, S.; Schagdarsurengin, U.; Dansranjavin, T.; Gradhand, E.; Schmitt, W.D.; Hauptmann, S. Frequent aberrant methylation of the imprinted IGF2/H19 locus and LINE1 hypomethylation in ovarian carcinoma. Int. J. Oncol. 2010, 36, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Oertel, B.G.; Doehring, A.; Roskam, B.; Kettner, M.; Hackmann, N.; Ferreiros, N.; Schmidt, P.H.; Lotsch, J. Genetic-epigenetic interaction modulates mu-opioid receptor regulation. Hum. Mol. Genet. 2012, 21, 4751–4760. [Google Scholar] [CrossRef] [PubMed]
- Cui, H. Loss of imprinting of IGF2 as an epigenetic marker for the risk of human cancer. Dis. Markers 2007, 23, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Waterland, R.A.; Lin, J.R.; Smith, C.A.; Jirtle, R.L. Post-weaning diet affects genomic imprinting at the insulin-like growth factor 2 (Igf2) locus. Hum. Mol. Genet. 2006, 15, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Boissonnas, C.C.; Abdalaoui, H.E.; Haelewyn, V.; Fauque, P.; Dupont, J.M.; Gut, I.; Vaiman, D.; Jouannet, P.; Tost, J.; Jammes, H. Specific epigenetic alterations of IGF2-H19 locus in spermatozoa from infertile men. Eur. J. Hum. Genet. 2010, 18, 73–80. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murphy, S.K.; Erginer, E.; Huang, Z.; Visco, Z.; Hoyo, C. Genotype-Epigenotype Interaction at the IGF2 DMR. Genes 2015, 6, 777-789. https://doi.org/10.3390/genes6030777
Murphy SK, Erginer E, Huang Z, Visco Z, Hoyo C. Genotype-Epigenotype Interaction at the IGF2 DMR. Genes. 2015; 6(3):777-789. https://doi.org/10.3390/genes6030777
Chicago/Turabian StyleMurphy, Susan K., Erin Erginer, Zhiqing Huang, Zachary Visco, and Cathrine Hoyo. 2015. "Genotype-Epigenotype Interaction at the IGF2 DMR" Genes 6, no. 3: 777-789. https://doi.org/10.3390/genes6030777
APA StyleMurphy, S. K., Erginer, E., Huang, Z., Visco, Z., & Hoyo, C. (2015). Genotype-Epigenotype Interaction at the IGF2 DMR. Genes, 6(3), 777-789. https://doi.org/10.3390/genes6030777