
Chromatin Remodelers: From Function to Dysfunction

Abstract
:1. Introduction
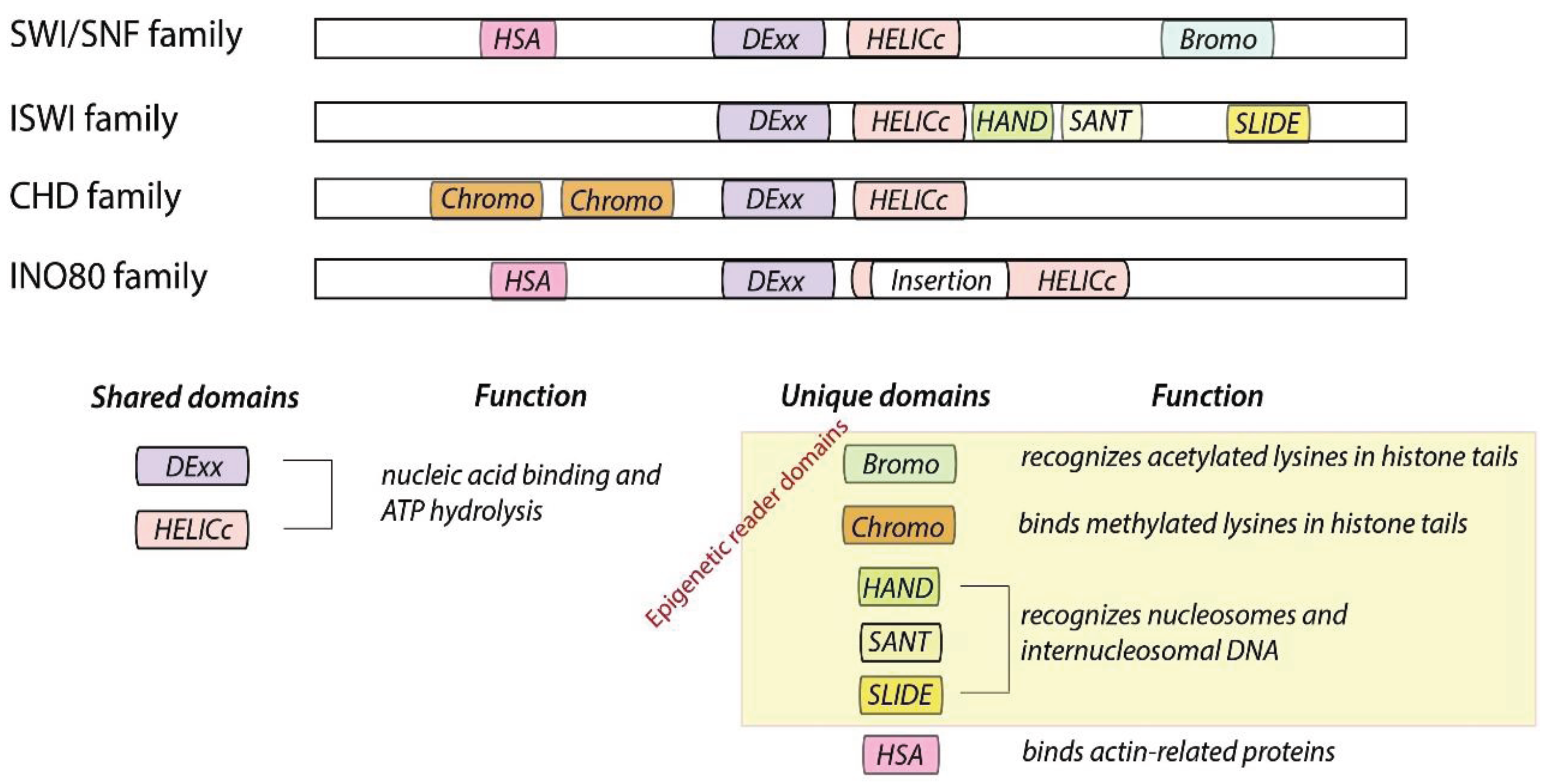
2. Chromatin Remodelers Fall into Four Families

3. Translocation Mechanism
4. Targeting Chromatin Remodeling Enzymes to Specific Genome Locations
4.1. Mechanisms
4.2. Targeting Signals Recognized by Chromatin Remodelers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular Interaction | Description Selected Examples | Refs. |
|---|---|---|
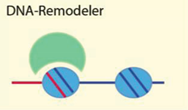 | DNA secondary structure, G-quadruplexes, DNA linker length, DNA modification | [6,51,55,59] |
| Examples: Brg1, Chd1, ISWI, ACF, NURF, Mi-2, Snf2H, Tip5/NoRC, ATRX, NURD, RSC | ||
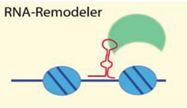 | Noncoding RNA secondary structure | [55,61,62,63,64] |
| Examples: ISWI, Tip5/NoRC, Brg1 | ||
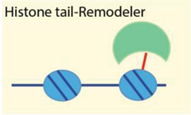 | H3 acetylation recognized by bromo domains, methylation—Chromo domains, PHD. H4 tails are required for SANT domain binding. | [65,66,67,68,69,70,71] |
| Examples: Tip5/NoRC, RSC, ISWI, Chd4 | ||
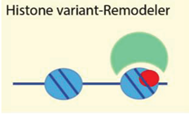 | Histone variants mH2A, H2A.Z, H2A.X | [72,73,74,75] |
| Examples: ATRX, Ino80, WICH, Lsh | ||
 | Transcription factors and other protein-protein interactions | [28,76,77,78] |
| Examples: NoRC, NuRD, Snf2H |
4.2.1. DNA Sequence, Structure and Modification
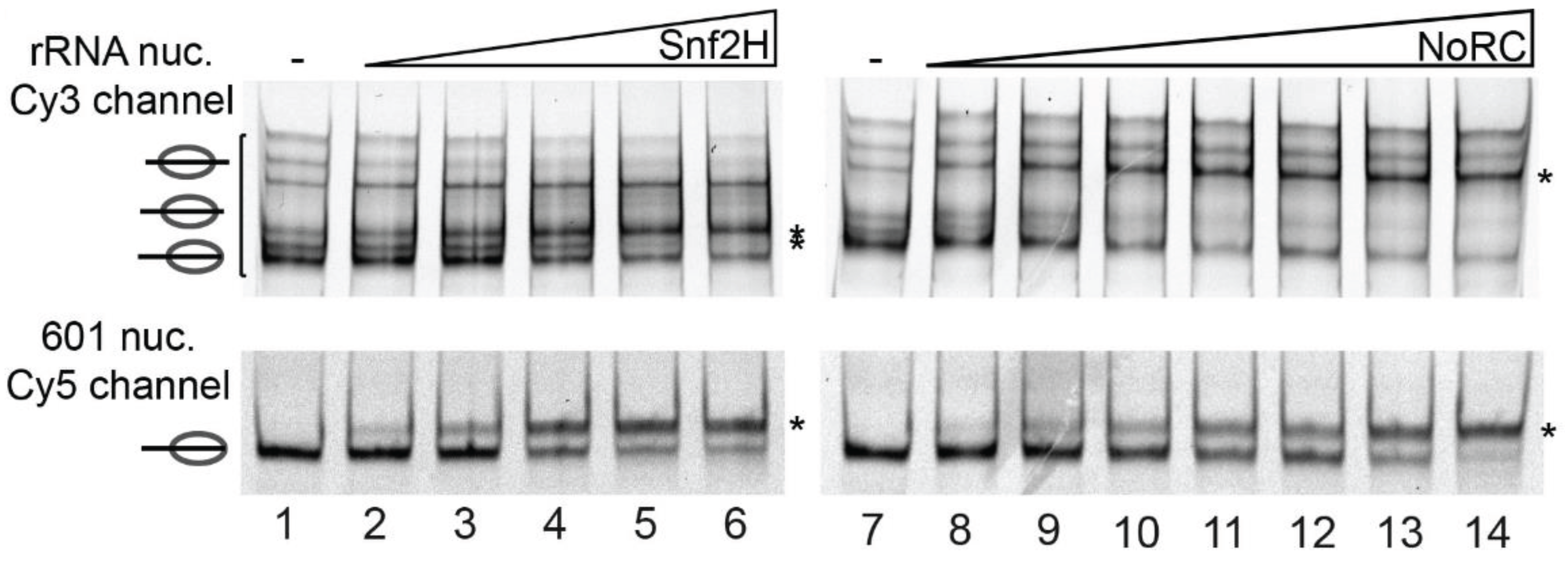
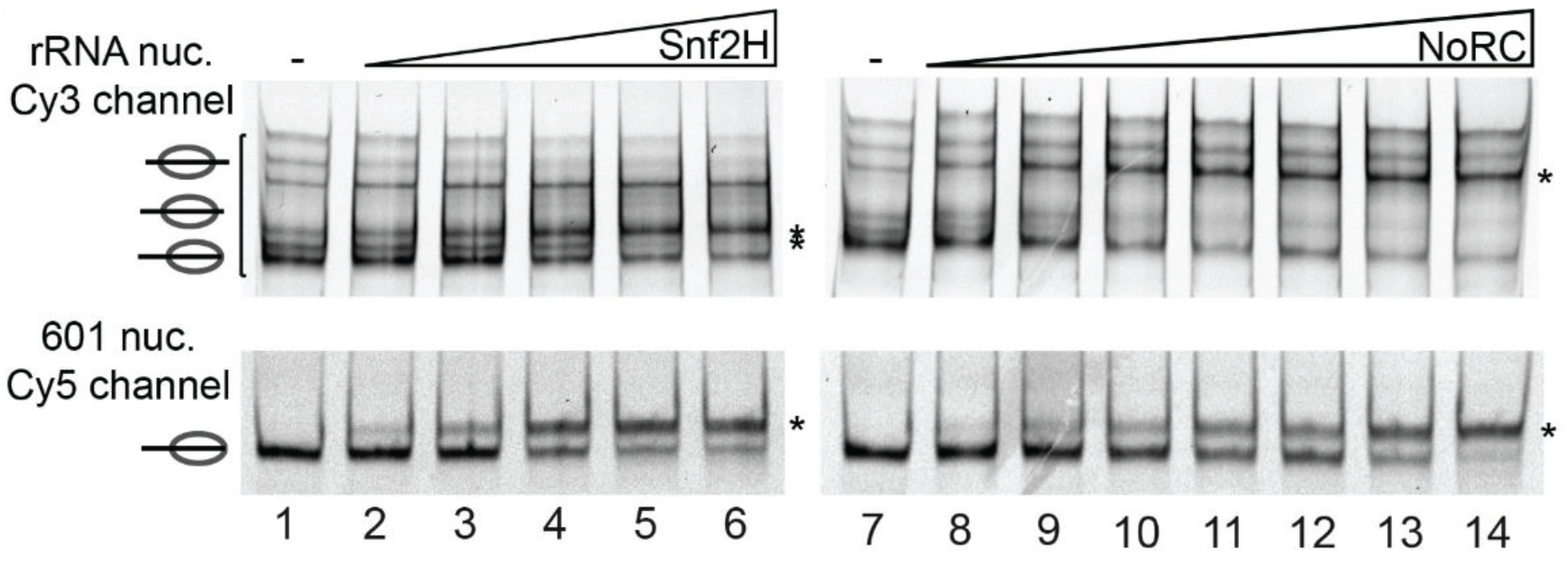
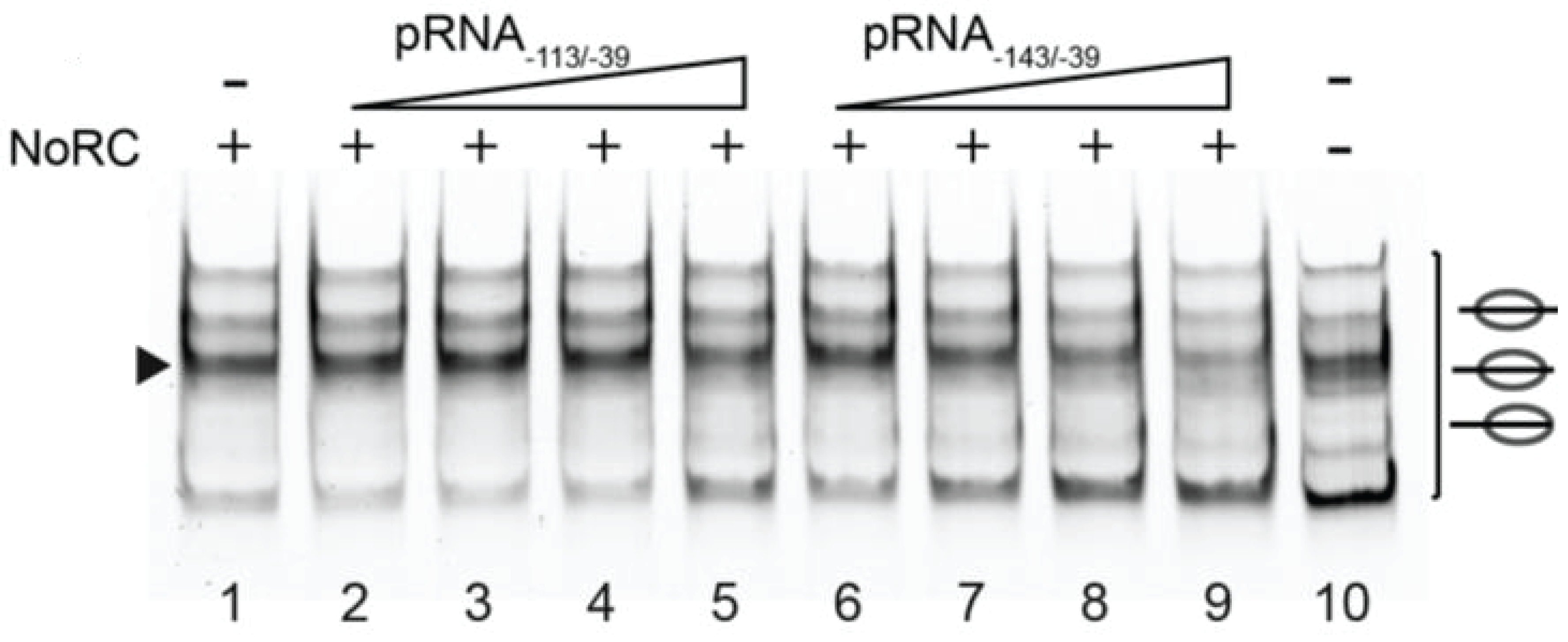
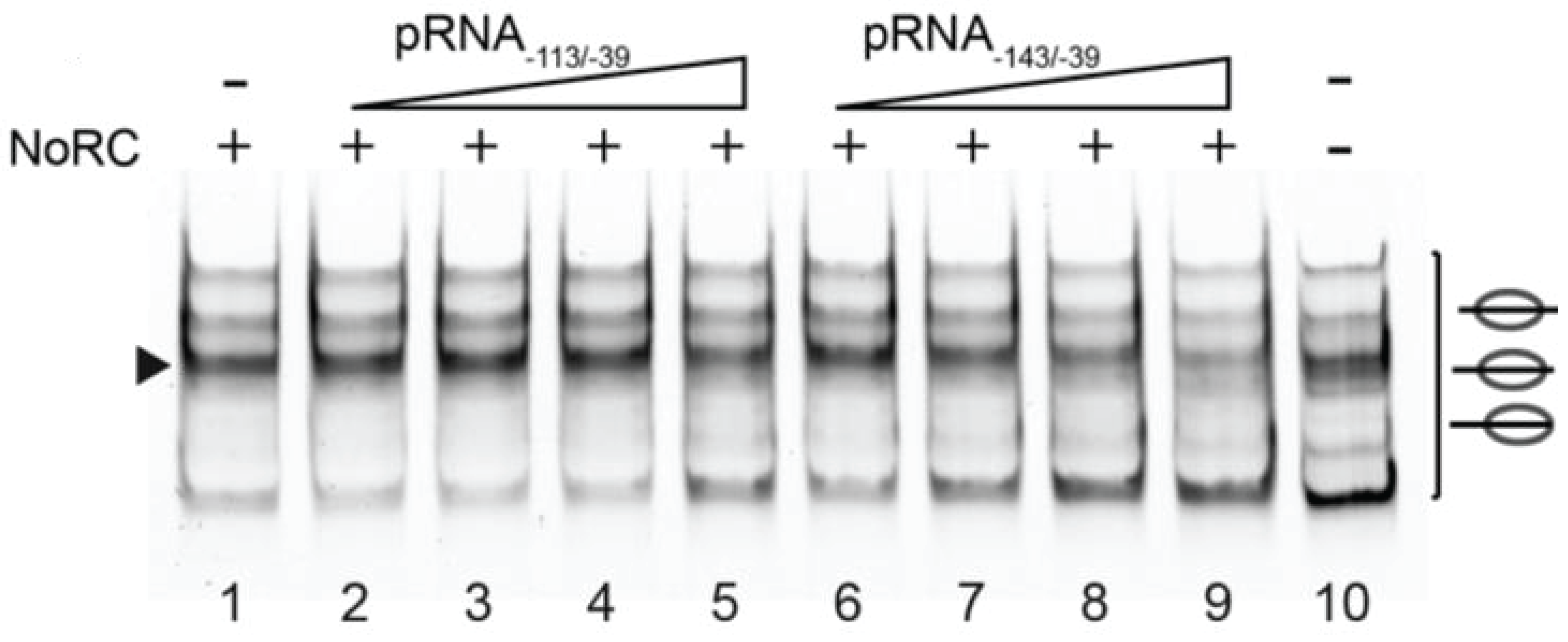
4.2.2. RNA
4.2.3. Histone Modifications
4.2.4. Histone Variants
4.2.5. Targeting to Chromatin by Sequence Specific Binding Proteins
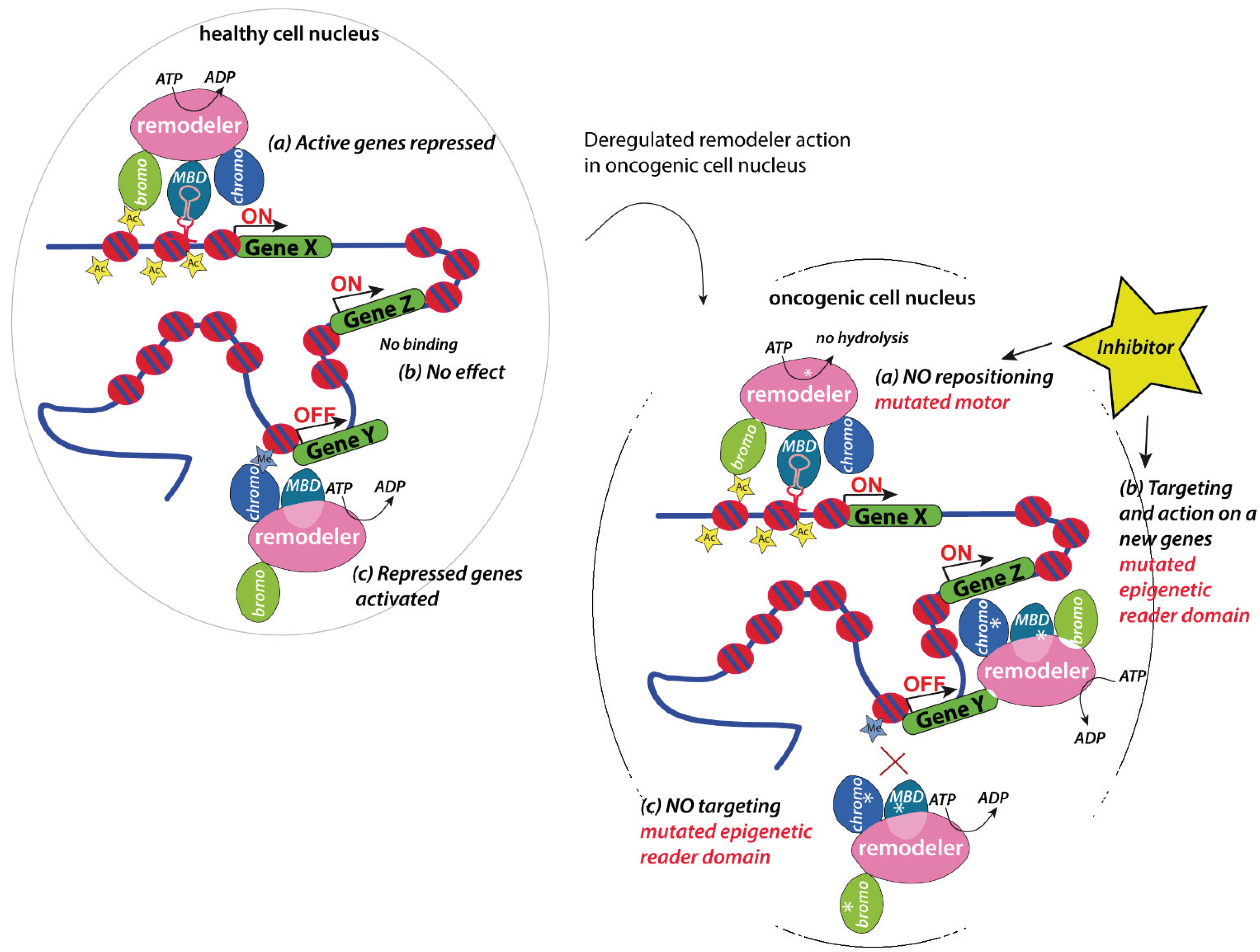
5. Implication in Cancer
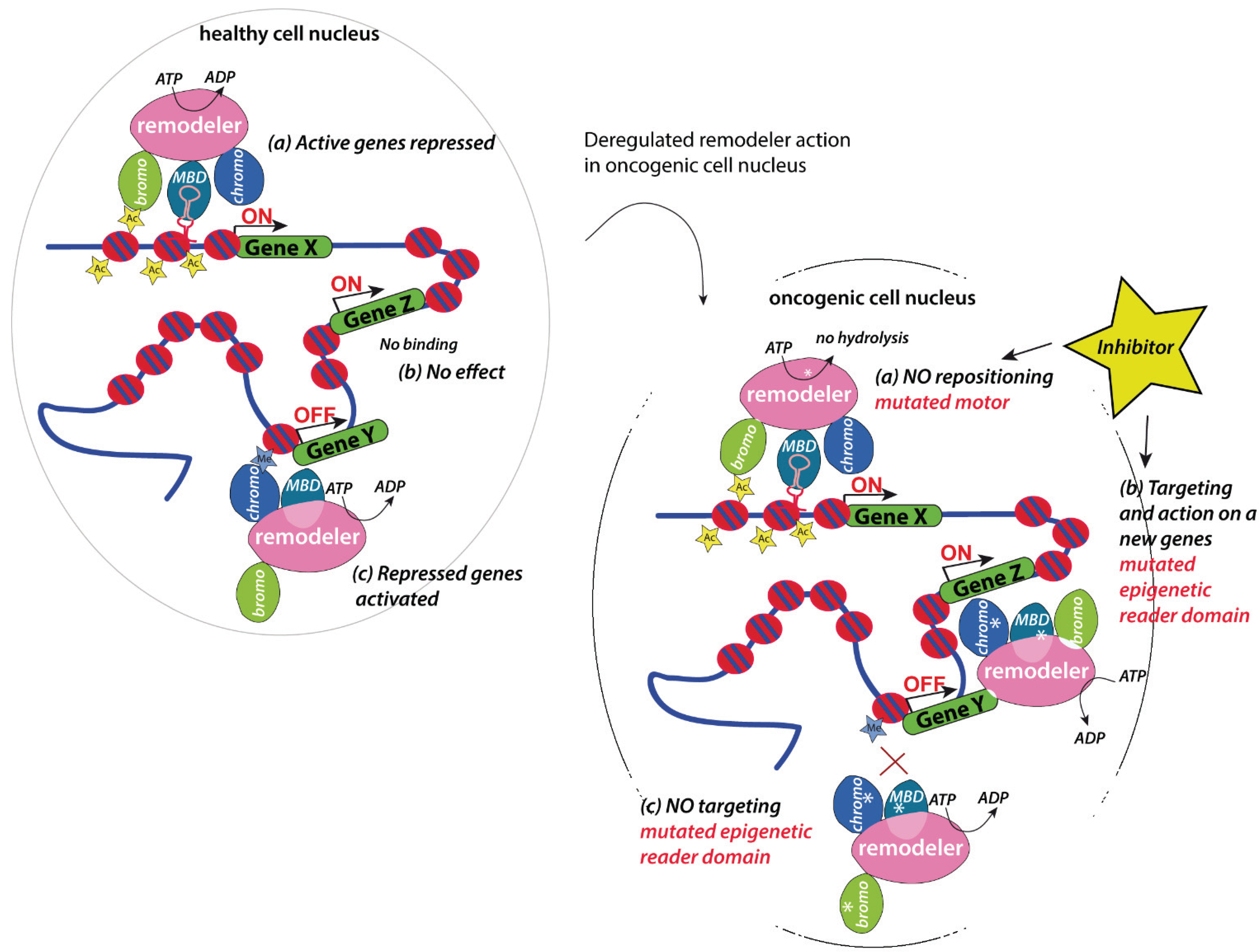
5.1. Chromatin Remodelers as Regulators of Master Regulators
5.2. Therapeutic Opportunities
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Van Holde, K.; Zlatanova, J. Chromatin fiber structure: Where is the problem now? Semin. Cell Dev. Biol. 2007, 18, 651–658. [Google Scholar]
- Zaret, K.S.; Carroll, J.S. Pioneer transcription factors: Establishing competence for gene expression. Genes Dev. 2011, 25, 2227–2241. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Clapier, C.R.; Cairns, B.R. The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 2009, 78, 273–304. [Google Scholar] [CrossRef] [PubMed]
- Erdel, F.; Rippe, K. Binding kinetics of human ISWI chromatin-remodelers to DNA repair sites elucidate their target location mechanism. Nucleus 2011, 2, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Rippe, K.; Schrader, A.; Riede, P.; Strohner, R.; Lehmann, E.; Langst, G. DNA sequence- and conformation-directed positioning of nucleosomes by chromatin-remodeling complexes. Proc. Natl. Acad. Sci. 2007, 104, 15635–15640. [Google Scholar] [CrossRef] [PubMed]
- Erdel, F.; Müller-Ott, K.; Baum, M.; Wachsmuth, M.; Rippe, K. Dissecting chromatin interactions in living cells from protein mobility maps. Chromosome Res. 2010, 19, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Eisen, J.A.; Sweder, K.S.; Hanawalt, P.C. Evolution of the SNF2 family of proteins: Subfamilies with distinct sequences and functions. Nucleic Acids Res. 1995, 23, 2715–2723. [Google Scholar] [CrossRef] [PubMed]
- Flaus, A.; Martin, D.M.A.; Barton, G.J.; Owen-Hughes, T. Identification of multiple distinct Snf2 subfamilies with conserved structural motifs. Nucleic Acids Res. 2006, 34, 2887–2905. [Google Scholar] [CrossRef] [PubMed]
- Taverna, S.D.; Li, H.; Ruthenburg, A.J.; Allis, C.D.; Patel, D.J. How chromatin-binding modules interpret histone modifications: Lessons from professional pocket pickers. Nat. Publ. Group 2007, 14, 1025–1040. [Google Scholar] [CrossRef] [PubMed]
- Simó-Riudalbas, L.; Esteller, M. Targeting the histone orthography of cancer: Drugs for writers, erasers and readers. Br. J. Pharmacol. 2014, 172, 2716–2732. [Google Scholar] [CrossRef] [PubMed]
- Gallenkamp, D.; Gelato, K.A.; Haendler, B.; Weinmann, H. Bromodomains and their pharmacological inhibitors. Chem. Med. Chem. 2014, 9, 438–464. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. At the tipping point for epigenetic therapies in cancer. J. Clin. Invest. 2014, 124, 14–16. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Knapp, S. The bromodomain interaction module. FEBS Lett. 2012, 586, 2692–2704. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.G.; Roberts, C.W.M. SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 2011, 11, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Lemon, B.; Inouye, C.; King, D.S.; Tjian, R. Selectivity of chromatin-remodelling cofactors for ligand-activated transcription. Nature 2001, 414, 924–928. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z. PBAF chromatin-remodeling complex requires a novel specificity subunit, BAF200, to regulate expression of selective interferon-responsive genes. Genes Dev. 2005, 19, 1662–1667. [Google Scholar] [CrossRef] [PubMed]
- Strohner, R.; Nemeth, A.; Nightingale, K.P.; Grummt, I.; Becker, P.B.; Längst, G. Recruitment of the nucleolar remodeling complex NoRC establishes ribosomal DNA silencing in chromatin. MCB 2004, 24, 1791–1798. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Längst, G.; Grummt, I. NoRC-dependent nucleosome positioning silences rRNA genes. EMBO J. 2006, 25, 5735–5741. [Google Scholar] [CrossRef] [PubMed]
- Santoro, R.; Li, J.; Grummt, I. The nucleolar remodeling complex NoRC mediates heterochromatin formation and silencing of ribosomal gene transcription. Nat. Genet. 2002, 32, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Alkhatib, S.G.; Landry, J.W. The nucleosome remodeling factor. FEBS Lett. 2011, 585, 3197–3207. [Google Scholar] [CrossRef] [PubMed]
- Ables, E.T.; Drummond-Barbosa, D. The steroid hormone ecdysone functions with intrinsic chromatin remodeling factors to control female germline stem cells in Drosophila. Cell Stem Cell 2010, 7, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Sims, J.K.; Wade, P.A. SnapShot: Chromatin remodeling: CHD. Cell 2011, 144. [Google Scholar] [CrossRef] [PubMed]
- Delmas, V.; Stokes, D.G.; Perry, R.P. A mammalian DNA-binding protein that contains a chromodomain and an SNF2/SWI2-like helicase domain. Proc. Natl. Acad. Sci. USA 1993, 90, 2414–2418. [Google Scholar] [CrossRef] [PubMed]
- Stokes, D.G.; Perry, R.P. DNA-binding and chromatin localization properties of CHD1. J. Mol. Biol. 1995, 15, 2745–2753. [Google Scholar]
- Ryan, D.P.; Sundaramoorthy, R.; Martin, D.; Singh, V.; Owen-Hughes, T. The DNA-binding domain of the Chd1 chromatin-remodelling enzyme contains SANT and SLIDE domains. EMBO J. 2011, 30, 2596–2609. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.A.; Mahajan, P.; Mertens, H.D.; Deery, M.J.; Zhang, W.; Pham, P.; Du, X.; Bartke, T.; Zhang, W.; Edlich, C.; et al. The PHD and chromo domains regulate the ATPase activity of the human chromatin remodeler CHD4. J. Mol. Biol. 2012, 422, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Murawska, M.; Brehm, A. CHD chromatin remodelers and the transcription cycle. Transcription 2011, 2, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Gkikopoulos, T.; Schofield, P.; Singh, V.; Pinskaya, M.; Mellor, J.; Smolle, M.; Workman, J.L.; Barton, G.J.; Owen-Hughes, T. A role for Snf2-related nucleosome-spacing enzymes in genome-wide nucleosome organization. Science 2011, 333, 1758–1760. [Google Scholar] [CrossRef] [PubMed]
- West, S.C. Processing of recombination intermediates by the RuvABC proteins. Annu. Rev. Genet. 1997, 31, 213–244. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Mizuguchi, G.; Hamiche, A.; Wu, C. A chromatin remodelling complex involved in transcription and DNA processing. Nature 2000, 406, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Shi, Y.; Mulligan, P.; Gay, F.; Landry, J.; Liu, H.; Lu, J.; Qi, H.H.; Wang, W.; Nickoloff, J.A.; et al. A YY1-INO80 complex regulates genomic stability through homologous recombination-based repair. Nat Struct Mol Biol. 2007, 14, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Shen, X. SnapShot: Chromatin remodeling: INO80 and SWR1. Cell 2011, 144. [Google Scholar] [CrossRef] [PubMed]
- Papamichos-Chronakis, M.; Watanabe, S.; Rando, O.J.; Peterson, C.L. Global regulation of H2A.Z localization by the INO80 chromatin-remodeling enzyme is essential for genome integrity. Cell 2011, 144, 200–213. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Ranallo, R.; Choi, E.; Wu, C. Involvement of actin-related proteins in ATP-dependent chromatin remodeling. Mol. Cell 2003, 12, 147–155. [Google Scholar] [CrossRef]
- Tsukuda, T.; Fleming, A.B.; Nickoloff, J.A.; Osley, M.A. Chromatin remodelling at a DNA double-strand break site in Saccharomyces cerevisiae. Nature 2005, 438, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Van Attikum, H.; Fritsch, O.; Gasser, S.M. Distinct roles for SWR1 and INO80 chromatin remodeling complexes at chromosomal double-strand breaks. EMBO J. 2007, 26, 4113–4125. [Google Scholar] [CrossRef] [PubMed]
- Langst, G.; Bonte, E.J.; Corona, D.F.; Becker, P.B. Nucleosome movement by CHRAC and ISWI without disruption or trans-displacement of the histone octamer. Cell 1999, 97, 843–852. [Google Scholar] [CrossRef]
- Tsukiyama, T.; Palmer, J.; Landel, C.C.; Shiloach, J.; Wu, C. Characterization of the imitation switch subfamily of ATP-dependent chromatin-remodeling factors in Saccharomyces cerevisiae. Genes Dev. 1999, 13, 686–697. [Google Scholar] [CrossRef] [PubMed]
- Davey, C.A.; Sargent, D.F.; Luger, K.; Maeder, A.W.; Richmond, T.J. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 a resolution. J. Mol. Biol. 2002, 319, 1097–1113. [Google Scholar] [CrossRef]
- Längst, G.; Becker, P.B. Nucleosome remodeling: one mechanism, many phenomena? Biochim. Biophys. Acta 2004, 1677, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar] [PubMed]
- Aoyagi, S.; Wade, P.A.; Hayes, J.J. Nucleosome sliding induced by the xMi-2 complex does not occur exclusively via a simple twist-diffusion mechanism. J. Biol. Chem. 2003, 278, 30562–30568. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, S.; Hayes, J.J. hSWI/SNF-catalyzed nucleosome sliding does not occur solely via a twist-diffusion mechanism. Mol. Cell. Biol. 2002, 22, 7484–7490. [Google Scholar] [CrossRef] [PubMed]
- Langst, G.; Becker, P.B. ISWI induces nucleosome sliding on nicked DNA. Mol. Cell 2001, 8, 1085–1092. [Google Scholar] [CrossRef]
- Schiessel, H.; Widom, J.; Bruinsma, R.F.; Gelbart, W.M. Polymer reptation and nucleosome repositioning. Phys. Rev. Lett. 2001, 86, 4414–4417. [Google Scholar] [CrossRef] [PubMed]
- Strohner, R.; Wachsmuth, M.; Dachauer, K.; Mazurkiewicz, J.; Hochstatter, J.; Rippe, K.; Längst, G. A “loop recapture” mechanism for ACF-dependent nucleosome remodeling. Nat. Publ. Group 2005, 12, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Peterson, C.L.; Hayes, J.J. SWI/SNF- and RSC-catalyzed nucleosome mobilization requires internal DNA loop translocation within nucleosomes. Mol. Cell Biol. 2011, 31, 4165–4175. [Google Scholar] [CrossRef] [PubMed]
- Blosser, T.R.; Yang, J.G.; Stone, M.D.; Narlikar, G.J.; Zhuang, X. Dynamics of nucleosome remodelling by individual ACF complexes. Nature 2009, 462, 1022–1027. [Google Scholar] [CrossRef] [PubMed]
- Sirinakis, G.; Clapier, C.R.; Gao, Y.; Viswanathan, R.; Cairns, B.R.; Zhang, Y. The RSC chromatin remodelling ATPase translocates DNA with high force and small step size. EMBO J. 2011, 30, 2364–2372. [Google Scholar] [CrossRef] [PubMed]
- Van Vuqt, J.J.F.A.; de Jaqer, M.; Murawska, M.; Brehm, A.; van Noort, J. Multiple aspects of ATP-dependent nucleosome translocation by RSC and Mi-2 are directed by the underlying DNA sequence. PLoS ONE 2009, 4, e6345. [Google Scholar] [CrossRef] [PubMed]
- Erdel, F.; Schubert, T.; Marth, C.; Längst, G.; Rippe, K. Human ISWI chromatin-remodeling complexes sample nucleosomes via transient binding reactions and become immobilized at active sites. Proc. Natl. Acad. Sci. 2010, 107, 19873–19878. [Google Scholar] [CrossRef] [PubMed]
- Gelbart, M.E.; Bachman, N.; Delrow, J.; Boeke, J.D.; Tsukiyama, T. Genome-wide identification of Isw2 chromatin-remodeling targets by localization of a catalytically inactive mutant. Genes Dev. 2005, 19, 942–954. [Google Scholar] [CrossRef] [PubMed]
- Erdel, F.; Krug, J.; Längst, G.; Rippe, K. Targeting chromatin remodelers: Signals and search mechanisms. Biochim. Biophys. Acta 2011, 1809, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Manelyte, L.; Strohner, R.; Gross, T.; Längst, G. Chromatin targeting signals, nucleosome positioning mechanism and non-coding RNA-mediated regulation of the chromatin remodeling complex NoRC. PLoS Genet. 2014, 10, e1004157. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.A.; Elbi, C.; Parekh, B.S.; Hager, G.L.; John, S. Chromatin remodeling complexes interact dynamically with a glucocorticoid receptor-regulated promoter. Mol. Biol. Cell 2008, 19, 3308–3322. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, A. The chromatin remodeling complex NoRC and TTF-I cooperate in the regulation of the mammalian rRNA genes in vivo. Nucleic Acids Res. 2004, 32, 4091–4099. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Navarro, S.; Reddy, S.; Comai, L. CK2-mediated stimulation of Pol I transcription by stabilization of UBF-SL1 interaction. Nucleic Acids Res. 2006, 34, 4752–4766. [Google Scholar] [CrossRef] [PubMed]
- Law, M.J.; Lower, K.M.; Voon, H.P.J.; Hughes, J.R.; Garrick, D.; Viprakasit, V.; Mitson, M.; de Gobbi, M.; Marra, M.; Morris, A.; et al. ATR-X syndrome protein targets tandem repeats and influences allele-specific expression in a size-dependent manner. Cell 2010, 143, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, O.; Li, R.; Hung, J.-H.; Chen, P.B.; Dong, X.; Ee, L.-S.; Weng, Z.; Rando, O.J.; Fazzio, T.G. Mbd3/NURD complex regulates expression of 5-hydroxymethylcytosine marked genes in embryonic stem cells. Cell 2011, 147, 1498–1510. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.; Schmitz, K.-M.; Li, J.; Grummt, I.; Santoro, R. Intergenic transcripts regulate the epigenetic state of rRNA genes. Molecular Cell 2006, 22, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.; Neubert, M.; Grummt, I. The structure of NoRC-associated RNA is crucial for targeting the chromatin remodelling complex NoRC to the nucleolus. EMBO Rep. 2008, 9, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Onorati, M.C.; Lazzaro, S.; Mallik, M.; Ingrassia, A.M.R.; Carreca, A.P.; Singh, A.K.; Chaturvedi, D.P.; Lakhotia, S.C.; Corona, D.F.V. The ISWI chromatin remodeler organizes the hsrω ncRNA-containing omega speckle nuclear compartments. PLoS Genet. 2011, 7, e1002096. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gong, Y.; Jin, B.; Wu, C.; Yang, J.; Wang, L.; Zhang, Z.; Mao, Z. Long non-coding RNA urothelial carcinoma associated 1 induces cell replication by inhibiting BRG1 in 5637 cells. Oncol. Rep. 2014, 32, 1281–1290. [Google Scholar] [PubMed]
- Clapier, C.R.; Langst, G.; Corona, D.F.; Becker, P.B.; Nightingale, K.P. Critical role for the histone H4 N terminus in nucleosome remodeling by ISWI. Mol. Cell. Biol. 2001, 21, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Grummt, I. The PHD finger/bromodomain of NoRC interacts with acetylated histone H4K16 and is sufficient for rDNA silencing. Curr. Biol. 2005, 15, 1434–1438. [Google Scholar] [CrossRef] [PubMed]
- Tallant, C.; Valentini, E.; Fedorov, O.; Overvoorde, L.; Ferguson, F.M.; Filippakopoulos, P.; Svergun, D.I.; Knapp, S.; Ciulli, A. Molecular basis of histone tail recognition by human TIP5 PHD finger and bromodomain of the chromatin remodeling complex NoRC. Structure 2015, 23, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Kasten, M.; Szerlong, H.; Erdjument-Bromage, H.; Tempst, P.; Werner, M.; Cairns, B.R. Tandem bromodomains in the chromatin remodeler RSC recognize acetylated histone H3 Lys14. EMBO J. 2004, 23, 1348–1359. [Google Scholar] [CrossRef] [PubMed]
- Musselman, C.A.; Ramírez, J.; Sims, J.K.; Mansfield, R.E.; Oliver, S.S.; Denu, J.M.; Mackay, J.P.; Wade, P.A.; Hagman, J.; Kutateladze, T.G. Bivalent recognition of nucleosomes by the tandem PHD fingers of the CHD4 ATPase is required for CHD4-mediated repression. Proc. Natl. Acad. Sci. 2012, 109, 787–792. [Google Scholar] [CrossRef] [PubMed]
- Musselman, C.A.; Mansfield, R.E.; Garske, A.L.; Davrazou, F.; Kwan, A.H.; Oliver, S.S.; O’Leary, H.; Denu, J.M.; Mackay, J.P.; Kutateladze, T.G. Binding of the CHD4 PHD2 finger to histone H3 is modulated by covalent modifications. Biochem. J. 2009, 423, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, R.E.; Musselman, C.A.; Kwan, A.H.; Oliver, S.S.; Garske, A.L.; Davrazou, F.; Denu, J.M.; Kutateladze, T.G.; Mackay, J.P. Plant homeodomain (PHD) fingers of CHD4 are histone H3-binding modules with preference for unmodified H3K4 and methylated H3K9. J. Biol. Chem. 2011, 286, 11779–11791. [Google Scholar] [CrossRef] [PubMed]
- Morrison, A.J.; Highland, J.; Krogan, N.J.; Arbel-Eden, A.; Greenblatt, J.F.; Haber, J.E.; Shen, X. INO80 and gamma-H2AX interaction links ATP-dependent chromatin remodeling to DNA damage repair. Cell 2004, 119, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Ratnakumar, K.; Duarte, L.F.; LeRoy, G.; Hasson, D.; Smeets, D.; Vardabasso, C.; Bönisch, C.; Zeng, T.; Xiang, B.; Zhang, D.Y.; et al. ATRX-mediated chromatin association of histone variant macroH2A1 regulates α-globin expression. Genes Dev. 2012, 26, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Xiao, A.; Li, H.; Shechter, D.; Ahn, S.H.; Fabrizio, L.A.; Erdjument-Bromage, H.; Ishibe-Murakami, S.; Wang, B.; Tempst, P.; Hofmann, K.; et al. WSTF regulates the H2A.X DNA damage response via a novel tyrosine kinase activity. Nature 2009, 457, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Burrage, J.; Termanis, A.; Geissner, A.; Myant, K.; Gordon, K.; Stancheva, I. The SNF2 family ATPase LSH promotes phosphorylation of H2AX and efficient repair of DNA double-strand breaks in mammalian cells. J. Cell Sci. 2012, 125, 5524–5534. [Google Scholar] [CrossRef] [PubMed]
- Strohner, R.; Nemeth, A.; Jansa, P.; Hofmann-Rohrer, U.; Santoro, R.; Langst, G.; Grummt, I. NoRC—A novel member of mammalian ISWI-containing chromatin remodeling machines. EMBO J. 2001, 20, 4892–4900. [Google Scholar] [CrossRef] [PubMed]
- Toiber, D.; Erdel, F.; Bouazoune, K.; Silberman, D.M.; Zhong, L.; Mulligan, P.; Sebastian, C.; Cosentino, C.; Martinez-Pastor, B.; Giacosa, S.; et al. SIRT6 recruits SNF2H to DNA break sites, preventing genomic instability through chromatin remodeling. Mol. Cell 2013, 51, 454–468. [Google Scholar] [CrossRef] [PubMed]
- Vidi, P.-A.; Liu, J.; Salles, D.; Jayaraman, S.; Dorfman, G.; Gray, M.; Abad, P.; Moghe, P.V.; Irudayaraj, J.M.; Wiesmüller, L.; et al. NuMA promotes homologous recombination repair by regulating the accumulation of the ISWI ATPase SNF2h at DNA breaks. Nucleic Acids Res. 2014, 42, 6365–6379. [Google Scholar] [CrossRef] [PubMed]
- Hamiche, A.; Sandaltzopoulos, R.; Gdula, D.A.; Wu, C. ATP-dependent histone octamer sliding mediated by the chromatin remodeling complex NURF. Cell 1999, 97, 833–842. [Google Scholar] [CrossRef]
- Moshkin, Y.M.; Chalkley, G.E.; Kan, T.W.; Reddy, B.A.; Ozgur, Z.; van Ijcken, W.F.J.; Dekkers, D.H.W.; Demmers, J.A.; Travers, A.A.; Verrijzer, C.P. Remodelers organize cellular chromatin by counteracting intrinsic histone-DNA sequence preferences in a class-specific manner. Mol. Cell. Biol. 2012, 32, 675–688. [Google Scholar] [CrossRef] [PubMed]
- Grüne, T.; Brzeski, J.; Eberharter, A.; Clapier, C.R.; Corona, D.F.; Becker, P.B.; Müller, C.W. Crystal structure and functional analysis of a nucleosome recognition module of the remodeling factor ISWI. Mol. Cell 2003, 12, 449–460. [Google Scholar] [CrossRef]
- Fyodorov, D.V.; Kadonaga, J.T. Binding of Acf1 to DNA involves a WAC motif and is important for ACF-mediated chromatin assembly. Mol. Cell Biol. 2002, 22, 6344–6353. [Google Scholar] [CrossRef] [PubMed]
- Poot, R.A.; Dellaire, G.; Hlsmann, B.B.; Grimaldi, M.A.; Corona, D.F.V.; Becker, P.B. HuCHRAC, a human ISWI chromatin remodelling complex contains hACF1 and two novel histone-fold proteins. EMBO J. 2000, 19, 3377–3387. [Google Scholar] [CrossRef] [PubMed]
- Jordan-Sciutto, K.L.; Dragich, J.M.; Rhodes, J.L.; Bowser, R. Fetal Alz-50 clone 1, a novel zinc finger protein, binds a specific DNA sequence and acts as a transcriptional regulator. J. Biol. Chem. 1999, 274, 35262–35268. [Google Scholar] [CrossRef] [PubMed]
- Stockdale, C.; Flaus, A.; Ferreira, H.; Owen-Hughes, T. Analysis of nucleosome repositioning by yeast ISWI and Chd1 chromatin remodeling complexes. J. Biol. Chem. 2006, 281, 16279–16288. [Google Scholar] [CrossRef] [PubMed]
- Sims, H.I.; Lane, J.M.; Ulyanova, N.P.; Schnitzler, G.R. Human SWI/SNF drives sequence-directed repositioning of nucleosomes on C-myc promoter DNA minicircles. Biochemistry 2007, 46, 11377–11388. [Google Scholar] [CrossRef] [PubMed]
- Partensky, P.D.; Narlikar, G.J. Chromatin remodelers act globally, sequence positions nucleosomes locally. J. Mol. Biol. 2009, 391, 12–25. [Google Scholar] [CrossRef] [PubMed]
- McKnight, J.N.; Jenkins, K.R.; Nodelman, I.M.; Escobar, T.; Bowman, G.D. Extranucleosomal DNA binding directs nucleosome sliding by Chd1. Mol. Cell. Biol. 2011, 31, 4746–4759. [Google Scholar] [CrossRef] [PubMed]
- Racki, L.R.; Yang, J.G.; Naber, N.; Partensky, P.D.; Acevedo, A.; Purcell, T.J.; Cooke, R.; Cheng, Y.; Narlikar, G.J. The chromatin remodeller ACF acts as a dimeric motor to space nucleosomes. Nature 2009, 462, 1016–1021. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.G.; Madrid, T.S.; Sevastopoulos, E.; Narlikar, G.J. The chromatin-remodeling enzyme ACF is an ATP-dependent DNA length sensor that regulates nucleosome spacing. Nat. Publ. Group 2006, 13, 1078–1083. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.A.; Kernohan, K.D.; Jiang, Y.; Bérubé, N.G. ATRX promotes gene expression by facilitating transcriptional elongation through guanine-rich coding regions. Hum. Mol. Genet. 2015, 24, 1824–1835. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, K.M.; Mayer, C.; Postepska, A.; Grummt, I. Interaction of noncoding RNA with the rDNA promoter mediates recruitment of DNMT3b and silencing of rRNA genes. Genes Dev. 2010, 24, 2264–2269. [Google Scholar] [CrossRef] [PubMed]
- Prensner, J.R.; Iyer, M.K.; Sahu, A.; Asangani, I.A.; Cao, Q.; Patel, L.; Vergara, I.A.; Davicioni, E.; Erho, N.; Ghadessi, M.; et al. The long noncoding RNA SChLAP1 promotes aggressive prostate cancer and antagonizes the SWI/SNF complex. Nat. Genet. 2013, 45, 1392–1398. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.S.; Roberts, C.W.M. Linking the SWI/SNF complex to prostate cancer. Nat. Genet. 2013, 45, 1268–1269. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Li, W.; Lin, C.-H.; Yang, J.; Shang, C.; Nurnberg, S.T.; Jin, K.K.; Xu, W.; Lin, C.-Y.; Lin, C.-J.; et al. A long noncoding RNA protects the heart from pathological hypertrophy. Nature 2014, 514, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Kassabov, S.R.; Zhang, B.; Persinger, J.; Bartholomew, B. SWI/SNF unwraps, slides, and rewraps the nucleosome. Mol. Cell 2003, 11, 391–403. [Google Scholar] [CrossRef]
- Duan, M.-R.; Smerdon, M.J. Histone H3 lysine 14 (H3K14) acetylation facilitates DNA repair in a positioned nucleosome by stabilizing the binding of the chromatin Remodeler RSC (Remodels Structure of Chromatin). J. Biol. Chem. 2014, 289, 8353–8363. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Radman-Livaja, M.; Rando, O.J.; Peterson, C.L. A histone acetylation switch regulates H2A.Z deposition by the SWR-C remodeling enzyme. Science 2013, 340, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Lange, M.; Kaynak, B.; Forster, U.B.; Tönjes, M.; Fischer, J.J.; Grimm, C.; Schlesinger, J.; Just, S.; Dunkel, I.; Krueger, T.; et al. Regulation of muscle development by DPF3, a novel histone acetylation and methylation reader of the BAF chromatin remodeling complex. Genes Dev. 2008, 22, 2370–2384. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Zhang, Q.; Li, S.; Plotnikov, A.N.; Walsh, M.J.; Zhou, M.-M. Mechanism and regulation of acetylated histone binding by the tandem PHD finger of DPF3b. Nature 2010, 466, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Liu, D.; Zhong, X.; Zhang, C.; Zhang, Q.; Zhou, D.-X. CHD3 protein recognizes and regulates methylated histone H3 lysines 4 and 27 over a subset of targets in the rice genome. Proc. Natl. Acad. Sci. 2012, 109, 5773–5778. [Google Scholar] [CrossRef] [PubMed]
- Volle, C.; Dalal, Y. Histone variants: The tricksters of the chromatin world. Curr. Opin. Genet. Dev. 2014, 25, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, S.; Workman, J.L. Histone exchange, chromatin structure and the regulation of transcription. Nat. Rev. Mol. Cell Biol. 2015, 16, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Voon, H.P.; Hughes, J.R.; Rode, C.; De La Rosa-Velázquez, I.A.; Jenuwein, T.; Feil, R.; Higgs, D.R.; Gibbons, R.J. ATRX plays a key role in maintaining silencing at interstitial heterochromatic loci and imprinted genes. Cell Rep. 2015, 11, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Gadue, P.; Chen, K.; Jiao, Y.; Tuteja, G.; Schug, J.; Li, W.; Kaestner, K.H. Foxa2 and H2A.Z mediate nucleosome depletion during embryonic stem cell differentiation. Cell 2012, 151, 1608–1616. [Google Scholar] [CrossRef] [PubMed]
- Weng, X.; Yu, L.; Liang, P.; Li, L.; Dai, X.; Zhou, B.; Wu, X.; Xu, H.; Fang, M.; Chen, Q.; et al. A crosstalk between chromatin remodeling and histone H3K4 methyltransferase complexes in endothelial cells regulates angiotensin II-induced cardiac hypertrophy. J. Mol. Cell. Cardiol. 2015, 82, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Garnatz, A.S.; Gao, Z.; Broman, M.; Martens, S.; Earley, J.U.; Svensson, E.C. FOG-2 mediated recruitment of the NuRD complex regulates cardiomyocyte proliferation during heart development. Dev. Biol. 2014, 395, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.S.; Kumar, R. Chromatin remodeling in cancer: a gateway to regulate gene transcription. Mol. Oncol. 2012, 6, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Perez, A.; Jene-Sanz, A.; Lopez-Bigas, N. The mutational landscape of chromatin regulatory factors across 4623 tumor samples. Genome Biol. 2013, 14, r106. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Pollack, J.R. The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLoS ONE 2013, 8, e55119. [Google Scholar] [CrossRef] [PubMed]
- Biegel, J.A.; Busse, T.M.; Weissman, B.E. SWI/SNF chromatin remodeling complexes and cancer. Am. J. Med. Genet. C Semin. Med. Genet. 2014, 166, 350–366. [Google Scholar] [CrossRef] [PubMed]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Ramos, P.; Karnezis, A.N.; Craig, D.W.; Sekulic, A.; Russell, M.L.; Hendricks, W.P.D.; Corneveaux, J.J.; Barrett, M.T.; Shumansky, K.; Yang, Y.; et al. Small cell carcinoma of the ovary, hypercalcemic type, displays frequent inactivating germline and somatic mutations in SMARCA4. Nat. Genet. 2014, 46, 427–429. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, L.; Carrot-Zhang, J.; Albrecht, S.; Fahiminiya, S.; Hamel, N.; Tomiak, E.; Grynspan, D.; Saloustros, E.; Nadaf, J.; Rivera, B.; et al. Germline and somatic SMARCA4 mutations characterize small cell carcinoma of the ovary, hypercalcemic type. Nat. Genet. 2014, 46, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Li, M.; Zhang, X.; Jones, S.; Leary, R.J.; Lin, J.C.-H.; Boca, S.M.; Carter, H.; Samayoa, J.; Bettegowda, C.; et al. The genetic landscape of the childhood cancer medulloblastoma. Science 2011, 331, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Helming, K.C.; Wang, X.; Roberts, C.W.M. Vulnerabilities of mutant SWI/SNF complexes in cancer. Cancer Cell 2014, 26, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Whyte, W.A.; Zepeda-Mendoza, C.J.; Milazzo, J.P.; Shen, C.; Roe, J.-S.; Minder, J.L.; Mercan, F.; Wang, E.; Eckersley-Maslin, M.A.; et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev. 2013, 27, 2648–2662. [Google Scholar] [CrossRef] [PubMed]
- Singhal, N.; Graumann, J.; Wu, G.; Araúzo-Bravo, M.J.; Han, D.W.; Greber, B.; Gentile, L.; Mann, M.; Schöler, H.R. Chromatin-remodeling components of the BAF complex facilitate reprogramming. Cell 2010, 141, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.G.; Helming, K.C.; Wang, X.; Kim, Y.; Vazquez, F.; Jagani, Z.; Hahn, W.C.; Roberts, C.W.M. Residual complexes containing SMARCA2 (BRM) underlie the oncogenic drive of SMARCA4 (BRG1) mutation. Mol. Cell. Biol. 2014, 34, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Workman, P.; Al-Lazikani, B. Drugging cancer genomes. Nat. Rev. Drug Discov. 2013, 12, 889–890. [Google Scholar] [CrossRef] [PubMed]
- Weissman, B.; Knudsen, K.E. Hijacking the chromatin remodeling machinery: Impact of SWI/SNF perturbations in cancer. Cancer Res. 2009, 69, 8223–8230. [Google Scholar]
- Jones, S.; Wang, T.-L.; Shih, I.-M.; Mao, T.-L.; Nakayama, K.; Roden, R.; Glas, R.; Slamon, D.; Diaz, L.A.; Vogelstein, B.; et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 2010, 330, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.S.; Kalloger, S.E.; et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N. Engl. J. Med. 2010, 363, 1532–1543. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Kan, J.; Yuen, S.T.; Shi, S.T.; Chu, K.M.; Law, S.; Chan, T.L.; Kan, Z.; Chan, A.S.Y.; Tsui, W.Y.; et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat. Genet. 2011, 43, 1219–1223. [Google Scholar] [CrossRef] [PubMed]
- Gui, Y.; Guo, G.; Huang, Y.; Hu, X.; Tang, A.; Gao, S.; Wu, R.; Chen, C.; Li, X.; Zhou, L.; et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat. Genet. 2011, 43, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Guichard, C.; Amaddeo, G.; Imbeaud, S.; Ladeiro, Y.; Pelletier, L.; Maad, I.B.; Calderaro, J.; Bioulac-Sage, P.; Letexier, M.; Degos, F.; et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 2012, 44, 694–698. [Google Scholar] [CrossRef] [PubMed]
- Nagymanyoki, Z.; Mutter, G.L.; Hornick, J.L.; Cibas, E.S. ARID1A is a useful marker of malignancy in peritoneal washings for endometrial carcinoma. Cancer Cytopathol. 2015, 123, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Itamochi, H.; Oumi, N.; Oishi, T.; Shoji, T.; Fujiwara, H.; Sugiyama, T.; Suzuki, M.; Kigawa, J.; Harada, T. Loss of ARID1A expression is associated with poor prognosis in patients with stage I/II clear cell carcinoma of the ovary. Int. J. Clin. Oncol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Gu, M.J.; Chang, H.-K.; Yu, E. Loss of ARID1A expression is associated with poor prognosis in small intestinal carcinoma. Histopathology 2015, 66, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Lee, C.; Suh, J.H.; Chae, J.Y.; Kim, H.W.; Moon, K.C. Decreased ARID1A expression correlates with poor prognosis of clear cell renal cell carcinoma. Hum. Pathol. 2015, 46, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Basta, J.; Rauchman, M. The nucleosome remodeling and deacetylase complex in development and disease. Transl. Res. 2015, 165, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Nicolson, G.L.; Nawa, A.; Toh, Y.; Taniguchi, S.; Nishimori, K.; Moustafa, A. Tumor metastasis-associated human MTA1 gene and its MTA1 protein product: Role in epithelial cancer cell invasion, proliferation and nuclear regulation. Clin. Exp. Metastasis 2003, 20, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-Y.; DeSalle, L.M.; Patel, J.H.; Capobianco, A.J.; Yu, D.; Thomas-Tikhonenko, A.; McMahon, S.B. Metastasis-associated protein 1 (MTA1) is an essential downstream effector of the c-MYC oncoprotein. Proc. Natl. Acad. Sci. USA 2005, 102, 13968–13973. [Google Scholar] [CrossRef] [PubMed]
- Le Gallo, M.; O’Hara, A.J.; Rudd, M.L.; Urick, M.E.; Hansen, N.F.; O’Neil, N.J.; Price, J.C.; Zhang, S.; England, B.M.; Godwin, A.K.; et al. Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nat. Genet. 2012, 44, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Geutjes, E.-J.; de Lint, K.; Roepman, P.; Bruurs, L.; Yu, L.-R.; Wang, W.; van Blijswijk, J.; Mohammad, H.; de Rink, I.; et al. The NuRD complex cooperates with DNMTs to maintain silencing of key colorectal tumor suppressor genes. Oncogene 2013, 33, 2157–2168. [Google Scholar] [CrossRef] [PubMed]
- Marfella, C.G.A.; Imbalzano, A.N. The Chd family of chromatin remodelers. Mutat. Res. 2007, 618, 30–40. [Google Scholar]
- Ansari, D.; Andersson, R.; Bauden, M.P.; Andersson, B.; Connolly, J.B.; Welinder, C.; Sasor, A.; Marko-Varga, G. Protein deep sequencing applied to biobank samples from patients with pancreatic cancer. J. Cancer Res. Clin. Oncol. 2014, 141, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Frommel, S.C.; Oakes, C.C.; Simon, R.; Grupp, K.; Gerig, C.Y.; Bär, D.; Robinson, M.D.; Baer, C.; Weiss, M.; et al. BAZ2A (TIP5) is involved in epigenetic alterations in prostate cancer and its overexpression predicts disease recurrence. Nat. Genet. 2015, 47, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, A.; Totoki, Y.; Abe, T.; Boroevich, K.A.; Hosoda, F.; Nguyen, H.H.; Aoki, M.; Hosono, N.; Kubo, M.; Miya, F.; et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat. Genet. 2012, 44, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Leite, K.R.M.; Morais, D.R.; Reis, S.T.; Viana, N.; Moura, C.; Florez, M.G.; Silva, I.A.; Dip, N.; Srougi, M. MicroRNA 100: A context dependent miRNA in prostate cancer. Clinics 2013, 68, 797–802. [Google Scholar] [CrossRef]
- Tolstorukov, M.Y.; Sansam, C.G.; Lu, P.; Koellhoffer, E.C.; Helming, K.C.; Alver, B.H.; Tillman, E.J.; Evans, J.A.; Wilson, B.G.; Park, P.J.; et al. Swi/Snf chromatin remodeling/tumor suppressor complex establishes nucleosome occupancy at target promoters. Proc. Natl. Acad. Sci. 2013, 110, 10165–10170. [Google Scholar] [CrossRef] [PubMed]
- Isakoff, M.S.; Sansam, C.G.; Tamayo, P.; Subramanian, A.; Evans, J.A.; Fillmore, C.M.; Wang, X.; Biegel, J.A.; Pomeroy, S.L.; Mesirov, J.P.; et al. Inactivation of the Snf5 tumor suppressor stimulates cell cycle progression and cooperates with p53 loss in oncogenic transformation. Proc. Natl. Acad. Sci. USA 2005, 102, 17745–17750. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Yan, W.; Chen, X. SNF5, a core component of the SWI/SNF complex, is necessary for p53 expression and cell survival, in part through eIF4E. Oncogene 2010, 29, 4090–4100. [Google Scholar] [CrossRef] [PubMed]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.-P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Müller, S.; Pawson, T.; et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Chaikuad, A.; Bamborough, P.; Bantscheff, M.; Bountra, C.; Chung, C.-W.; Fedorov, O.; Grandi, P.; Jung, D.; Lesniak, R.; et al. Discovery and characterization of GSK2801, a selective chemical probe for the bromodomains BAZ2A and BAZ2B. J. Med. Chem. 2015. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Längst, G.; Manelyte, L. Chromatin Remodelers: From Function to Dysfunction. Genes 2015, 6, 299-324. https://doi.org/10.3390/genes6020299
Längst G, Manelyte L. Chromatin Remodelers: From Function to Dysfunction. Genes. 2015; 6(2):299-324. https://doi.org/10.3390/genes6020299
Chicago/Turabian StyleLängst, Gernot, and Laura Manelyte. 2015. "Chromatin Remodelers: From Function to Dysfunction" Genes 6, no. 2: 299-324. https://doi.org/10.3390/genes6020299
APA StyleLängst, G., & Manelyte, L. (2015). Chromatin Remodelers: From Function to Dysfunction. Genes, 6(2), 299-324. https://doi.org/10.3390/genes6020299