
Replication Stress in Mammalian Cells and Its Consequences for Mitosis

Abstract
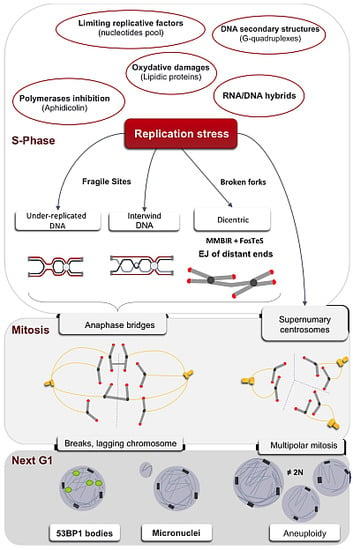
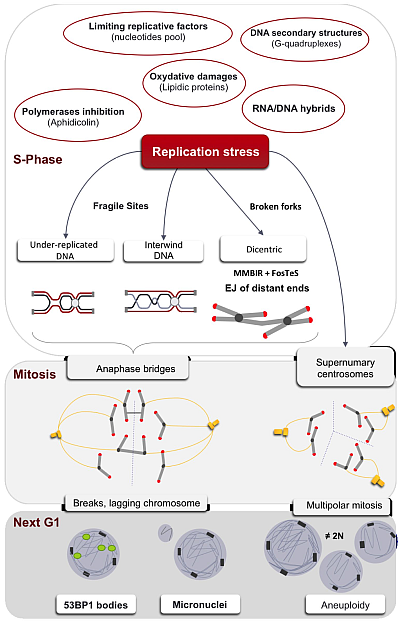
:
{kind=link}
{kind=link}
{kind=link}
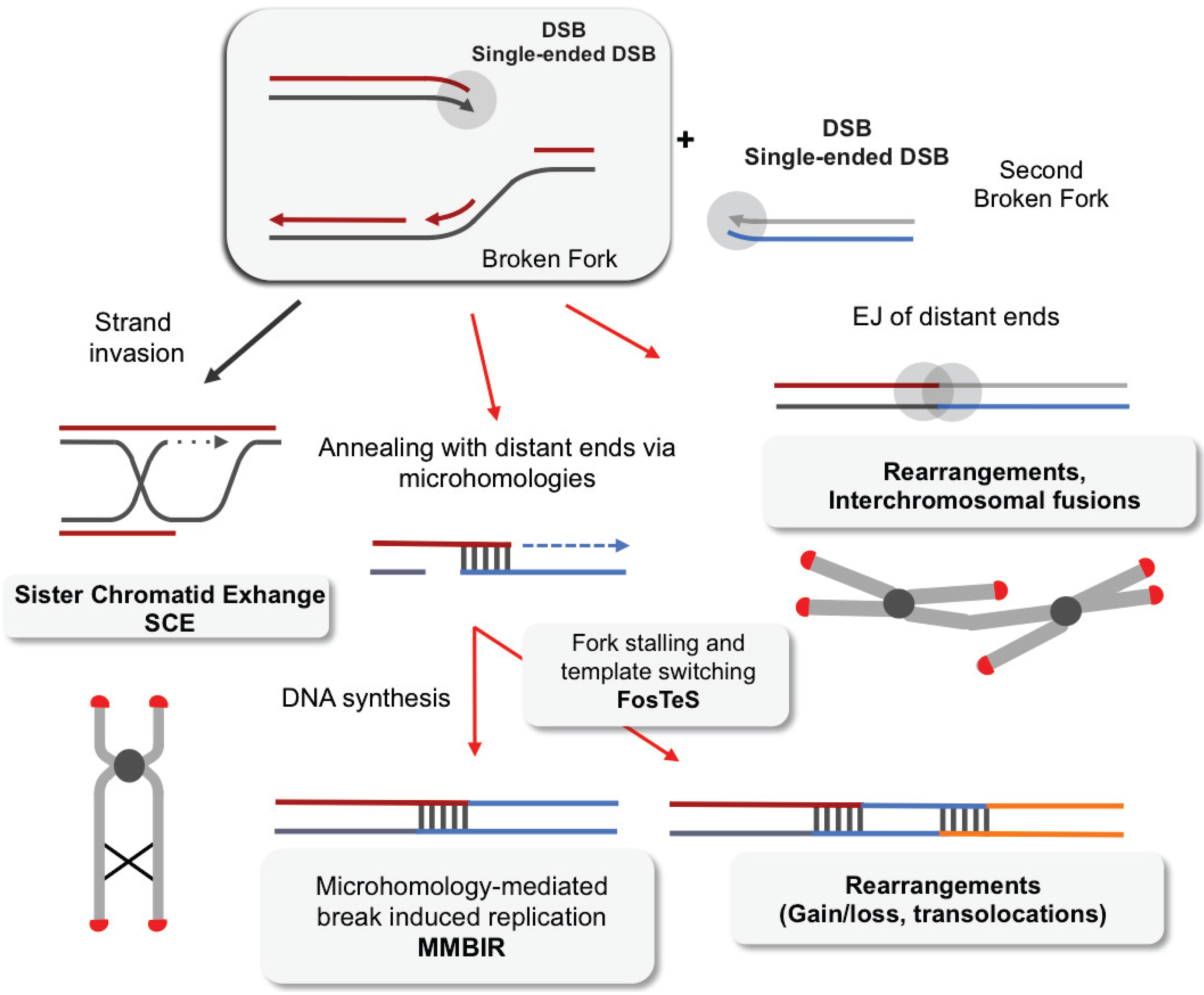{kind=link}
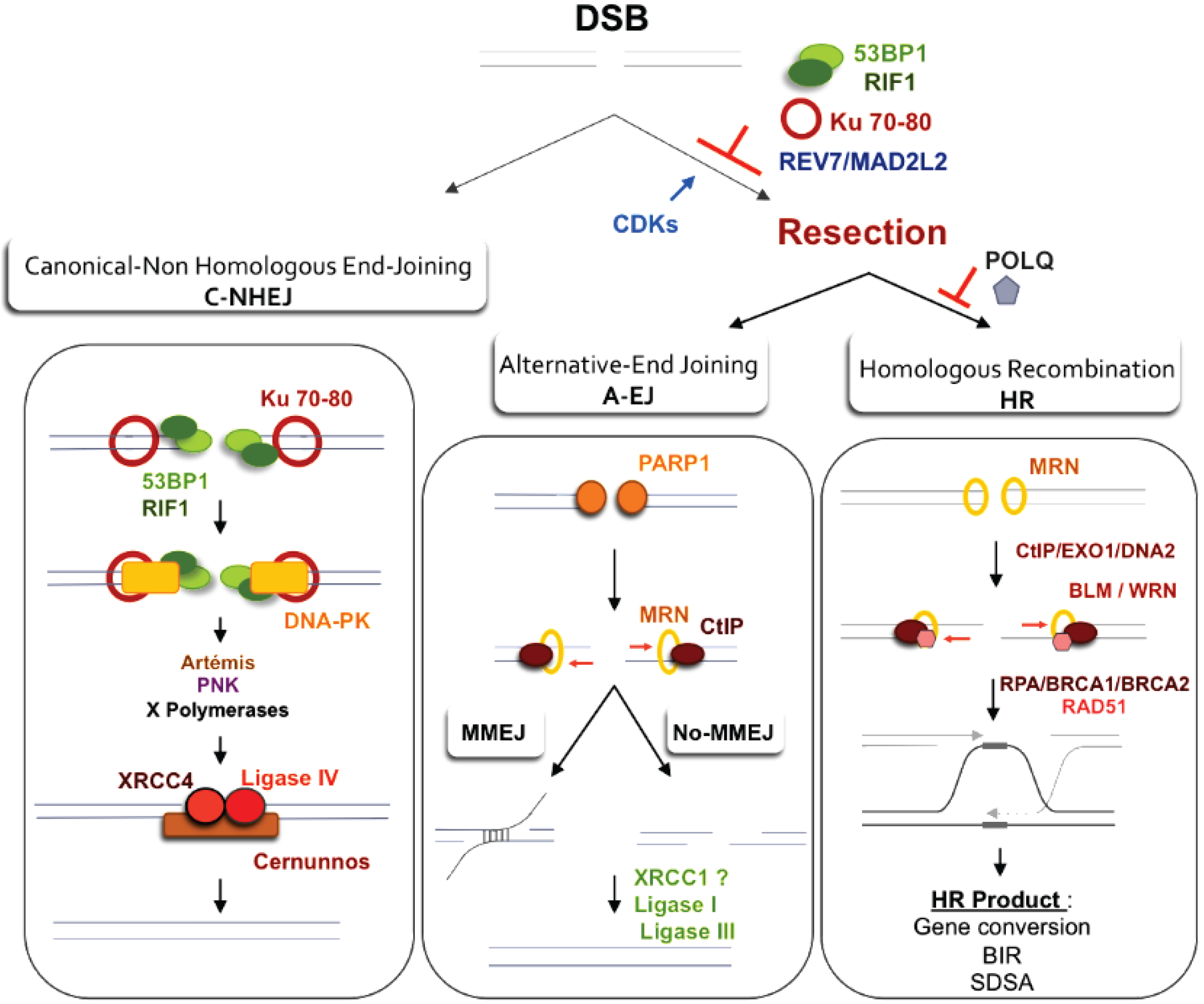{kind=link}
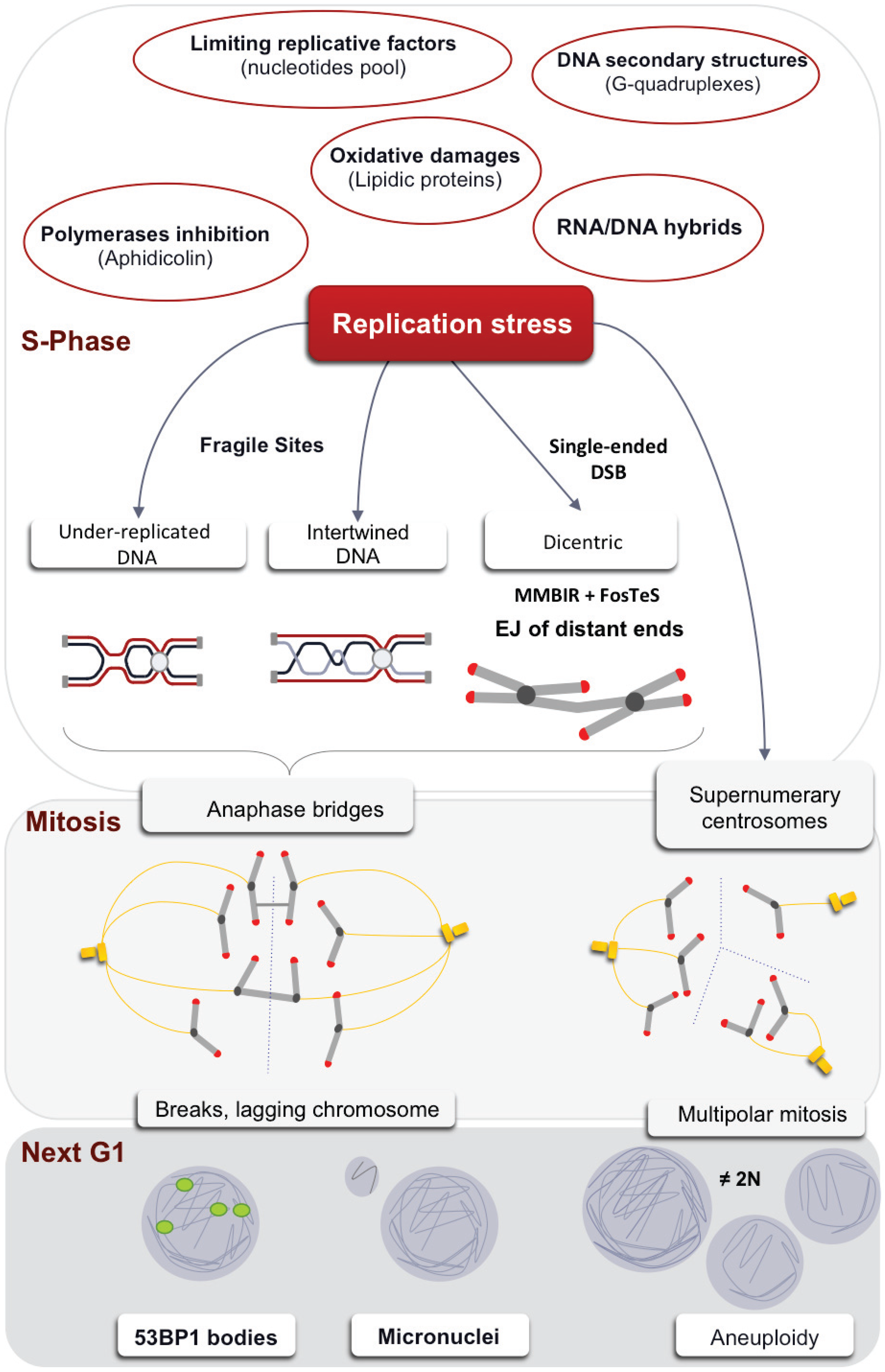{kind=link}
1. The Multiple Causes of the Replication Stress
1.1. Down-Regulation of Limiting Factors of Replication
1.2. Obstacles to Replication Fork Progression
1.3. Interference between Replication and Transcription Machineries/Programs
2. Replication of Particular Regions of the Genome
2.1. Under-Replicated DNA and Common Fragile Sites
2.2. Telomeric Sequences
2.3. Centromeric Sequences
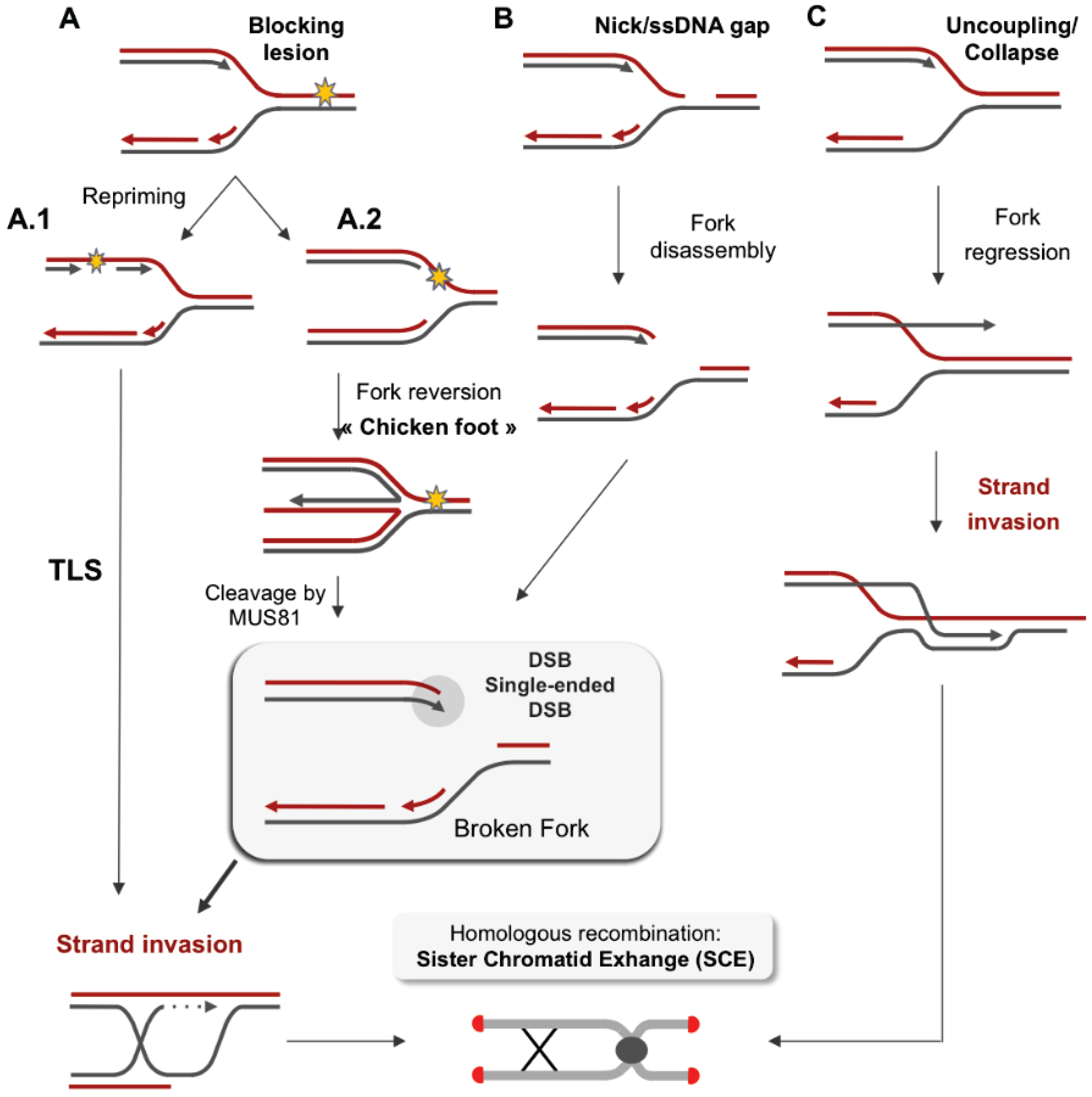
3. Stalled Forks and Replication Restart
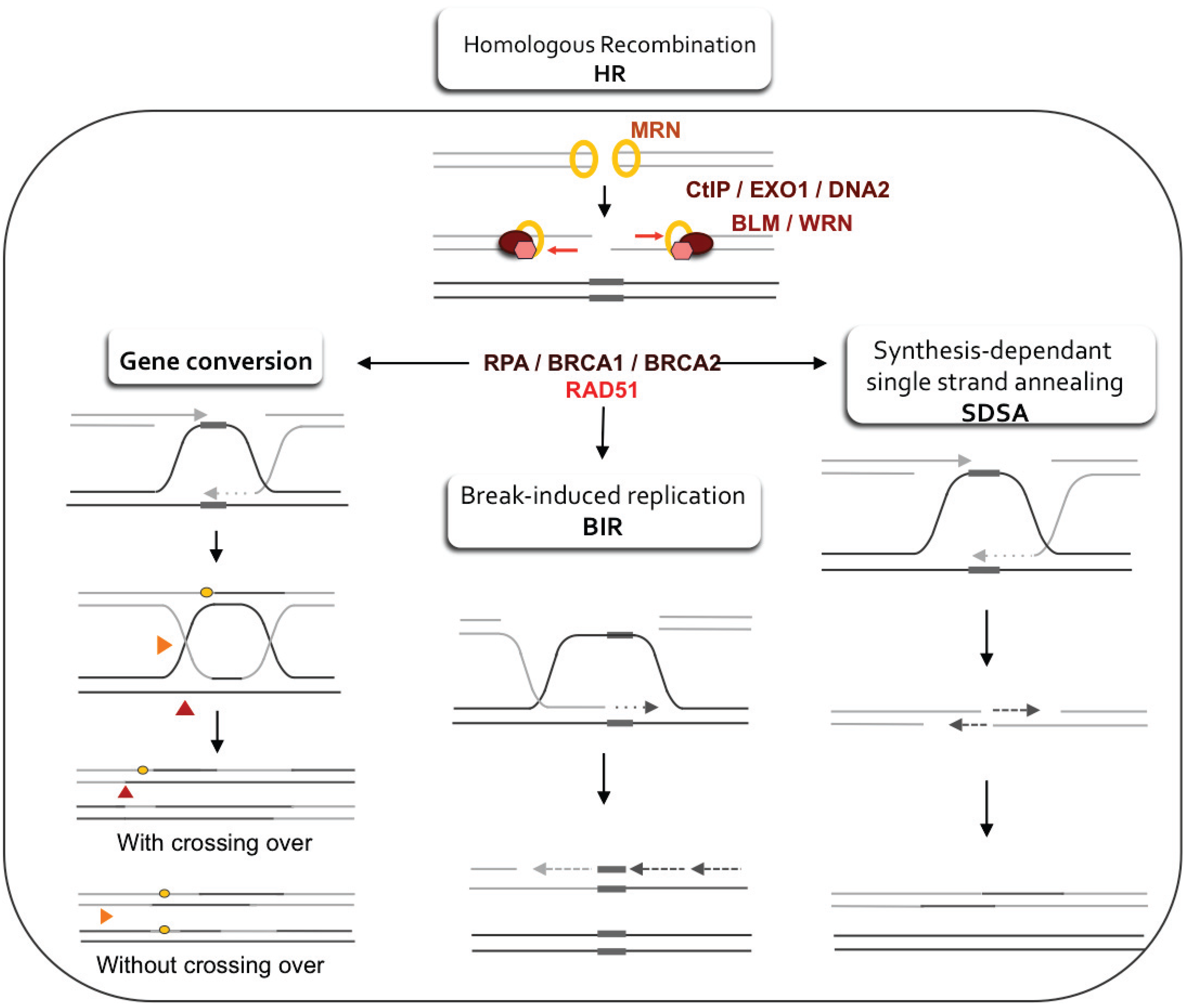
3.1. Regulation of Resection and Homologous Recombination (HR)
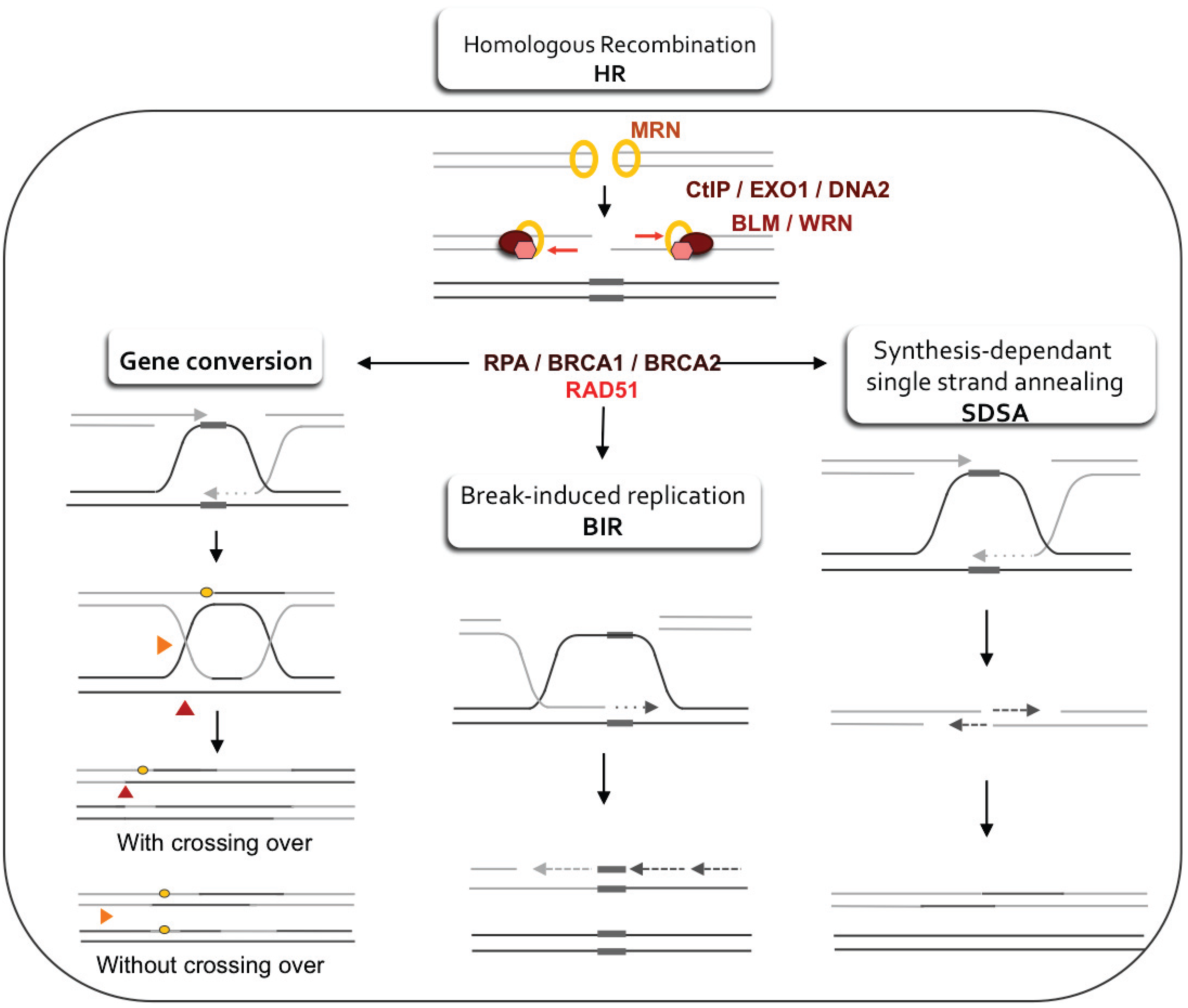
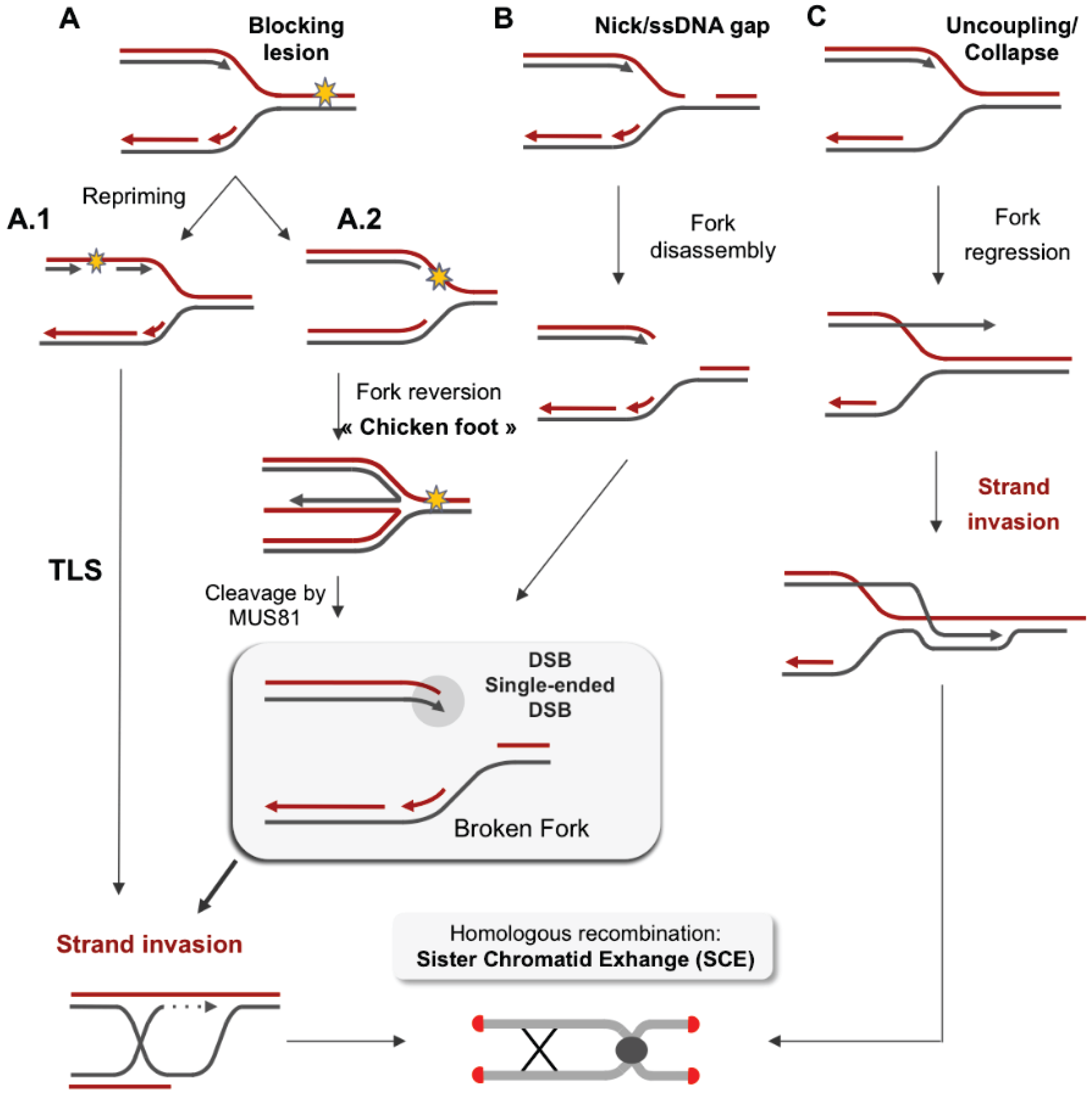
3.2. Stabilization of the Arrested Replication Fork
3.3. Restart of the Arrested Replication Fork
3.4. Error-Prone Replication Forks Restart
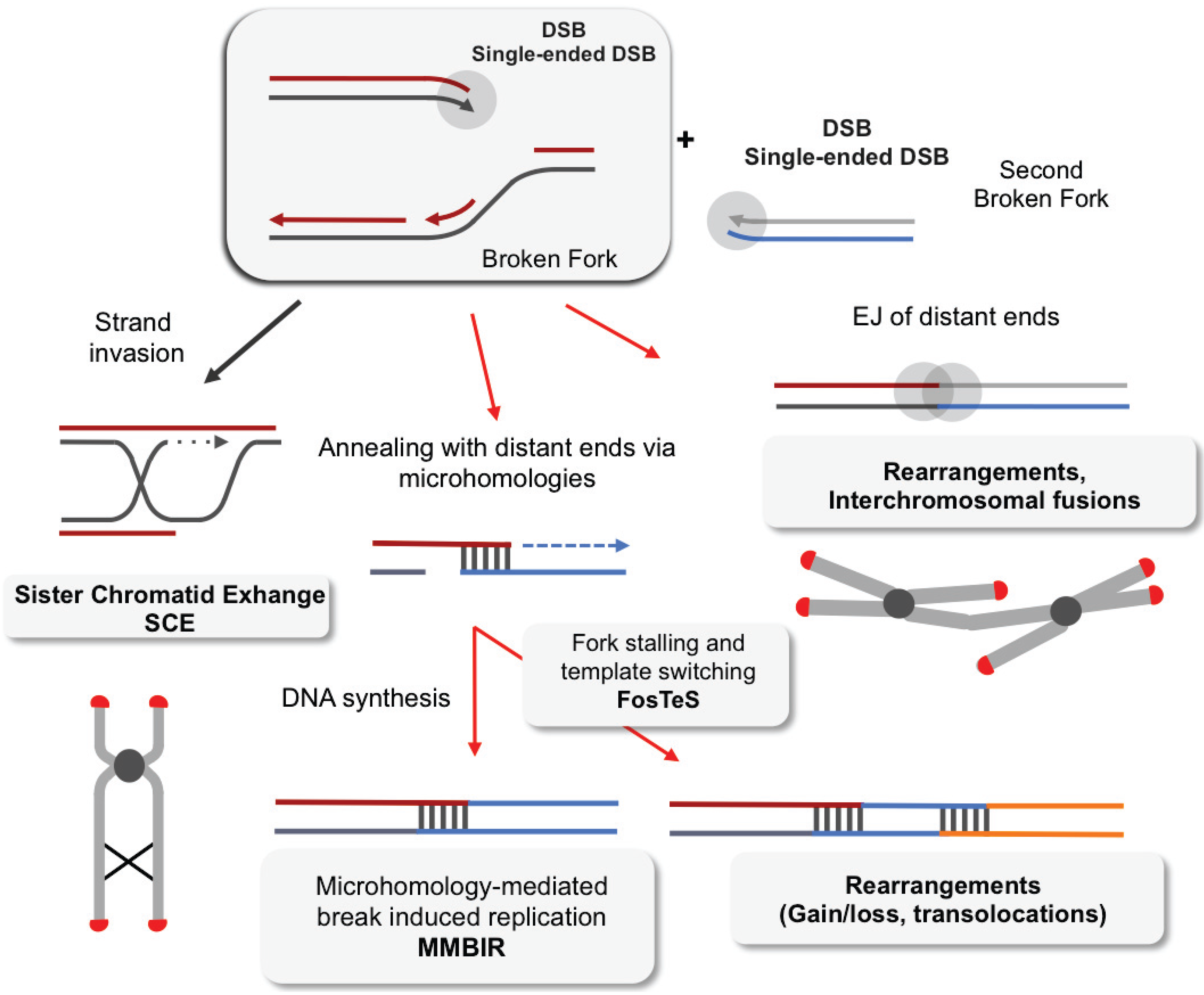
4. Impact of Single-Ended DSBs Formed by Replication Stress on Chromosome Instability
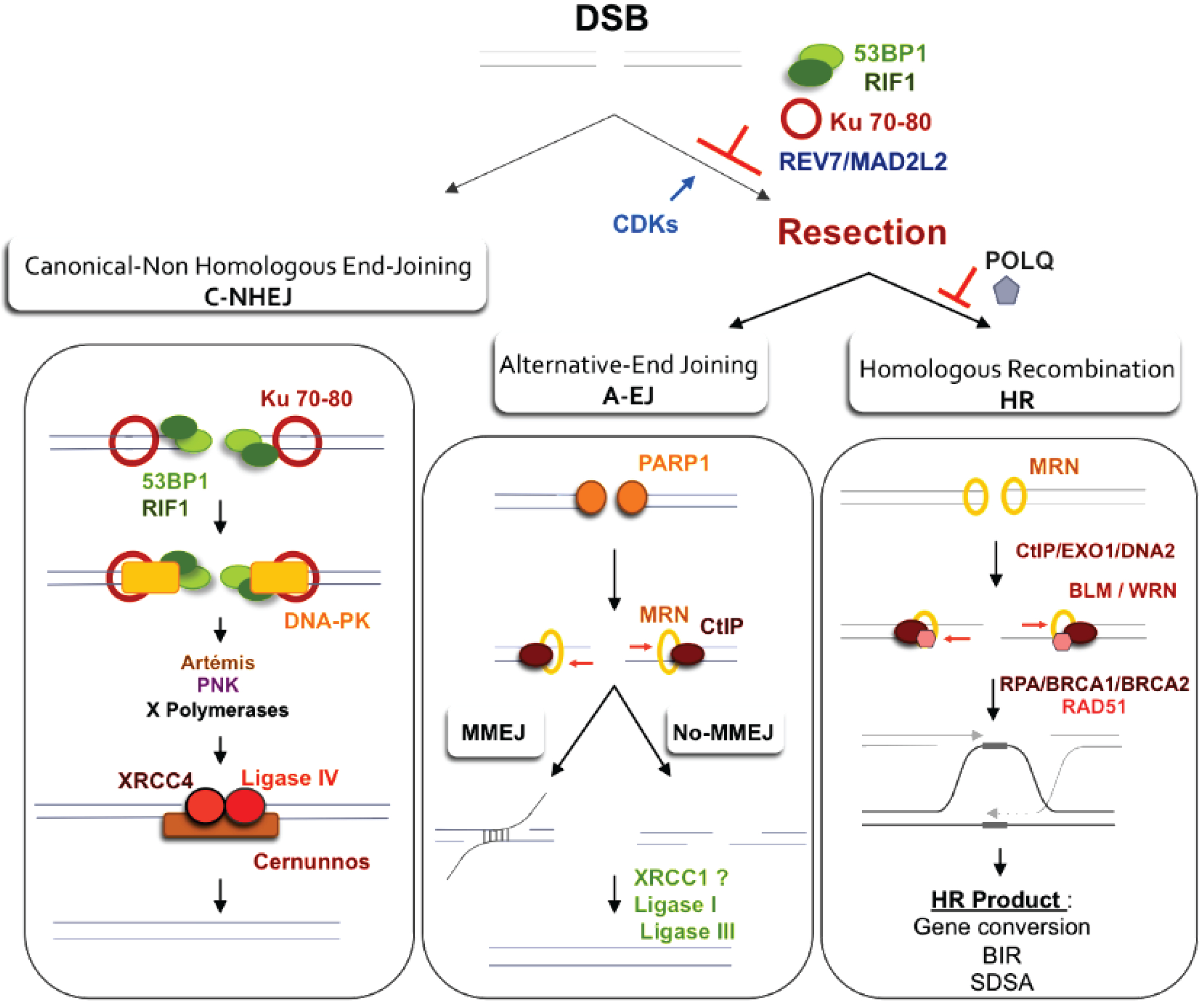
4.1. DSB Repair
4.2. EJ of Distant DBS Ends Leads to Radial Structure Formation and Dicentric Chromosomes
5. Consequences of Replication Stress on Mitosis
5.1. A Threshold of Stress for S and G2/M Arrests
5.2. Anaphases Bridges
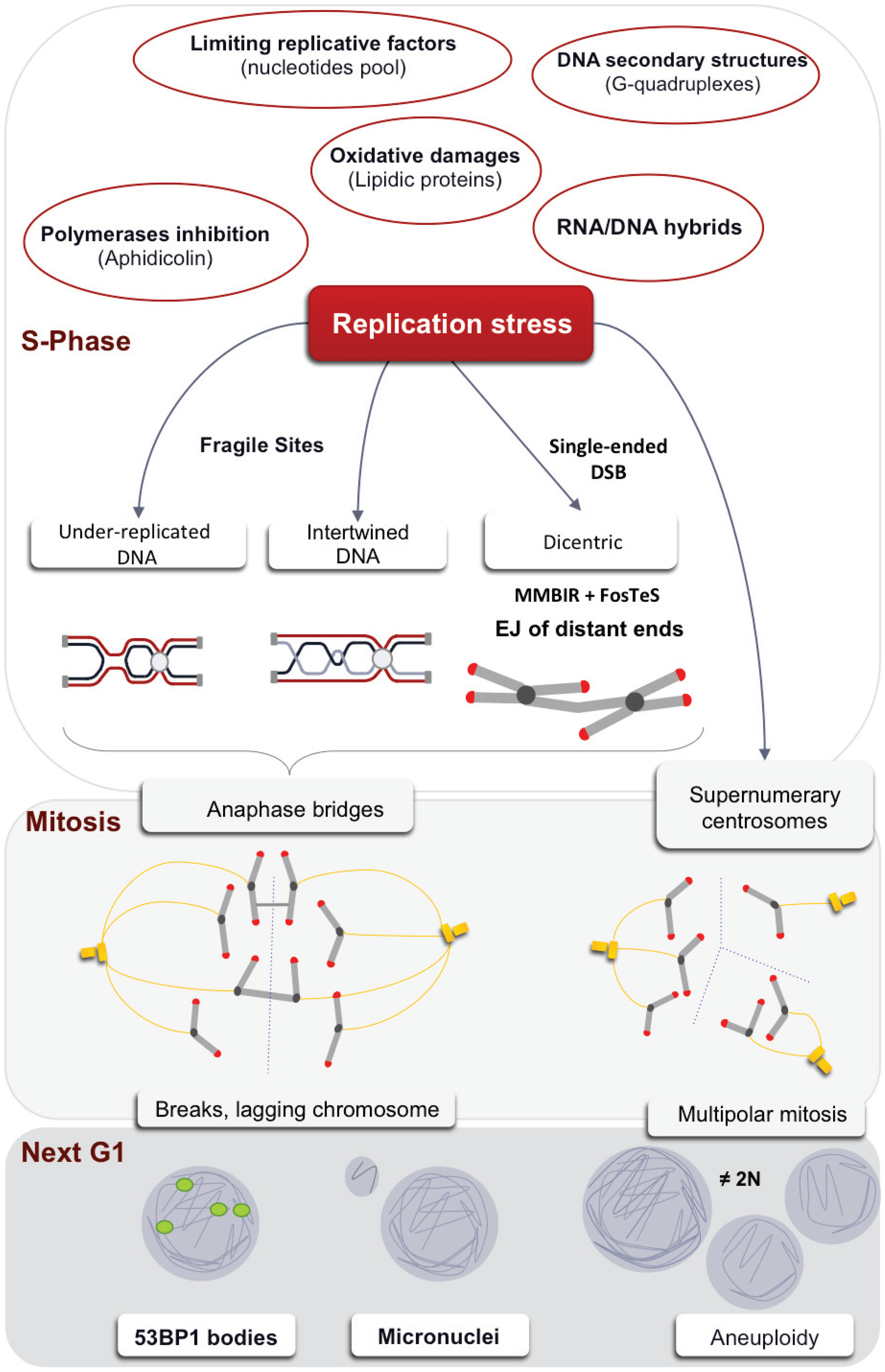
5.3. Centrosome Defects
6. Transmission of the Stress Signal to the Daughter Cell
6.1. Micronuclei
6.2. 53BP1 Bodies
7. Conclusions
Acknowledgments
Author Contributions
Abbreviations
| 53BP1 | P53 Binding Protein 1 |
| A-EJ | Alternative End-Joining |
| APH | Aphidicolin |
| BIR | Break-induced replication |
| C-NHEJ | Canonical Non Homologous End-Joining |
| CENP | Centromer protein |
| CDK | Cyclin Dependant kinase |
| CFSs | Common fragile sites |
| CIN | Chromosomal instability |
| DDR | DNA damage response |
| D-loop | Displacement loop |
| dNTPs | Deoxynucleotides |
| DSBs | Double-strand breaks |
| DSE | Double-strand end |
| EJ | End-Joining |
| ERFS | Early replicating fragile sites |
| FANC | Fanconi |
| FoSTeS | Fork Stalling and Template Switching |
| GC | Gene conversion |
| HC | Heterochromatin |
| HP | Heterochromatin Protein |
| HR | Homologous Recombination |
| HORs | Higher-Order Repeats |
| HJ | Holliday junctions |
| IR | Ionizing Radiation |
| MH | Microhomology |
| MMBIR | Microhomology-Mediated Break Induced Replication |
| MMEJ | Microhomology-Mediated End Joining |
| MN | MicroNuclei |
| MRN | MRE11/RAD50/NBS1 complex |
| OPT | Oct-1, PTF, transcription |
| ROS | Reactive Oxygen Species |
| SCE | Sister chromatid exchange |
| ssDNA | Single-Strand DNA |
| SDSA | Synthesis-Dependent Strand Annealing |
| T-SCE | Telomere Sister Chromatid Exchange |
| TLS | Translesion Synthesis |
| UFB | Ultra-Fine Bridge |
Conflicts of Interest
References
- Gorgoulis, V.G.; Vassiliou, L.F.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; Ditullio, R.A., Jr.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Horejsí, Z.; Koed, K.; Krämer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.-C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Flach, J.; Bakker, S.T.; Mohrin, M.; Conroy, P.C.; Pietras, E.M.; Reynaud, D.; Alvarez, S.; Diolaiti, M.E.; Ugarte, F.; Forsberg, E.C.; et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature 2014, 512, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Anglana, M.; Apiou, F.; Bensimon, A.; Debatisse, M. Dynamics of DNA replication in mammalian somatic cells: Nucleotide pool modulates origin choice and interorigin spacing. Cell 2003, 114, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.; García-Muse, T. Causes of genome instability. Annu. Rev. Genet. 2013, 47, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Bruschi, C.V.; McMillan, J.N.; Coglievina, M.; Esposito, M.S. The genomic instability of yeast cdc6-1/cdc6-1 mutants involves chromosome structure and recombination. Mol. Gen. Genet. 1995, 249, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Loupart, M.L.; Krause, S.A.; Heck, M.S. Aberrant replication timing induces defective chromosome condensation in Drosophila ORC2 mutants. Curr. Biol. 2000, 10, 1547–1556. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Diffley, J.F.X. Deregulated G1-cyclin expression induces genomic instability by preventing efficient pre-RC formation. Genes Dev. 2002, 16, 2639–2649. [Google Scholar] [CrossRef] [PubMed]
- Shima, N.; Alcaraz, A.; Liachko, I.; Buske, T.R.; Andrews, C.A.; Munroe, R.J.; Hartford, S.A.; Tye, B.K.; Schimenti, J.C. A viable allele of Mcm4 causes chromosome instability and mammary adenocarcinomas in mice. Nat. Genet. 2007, 39, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Wold, M.S. Replication protein A: A heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem. 1997, 66, 61–92. [Google Scholar] [CrossRef] [PubMed]
- Fanning, E.; Klimovich, V.; Nager, A.R. A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic Acids Res. 2006, 34, 4126–4137. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.I.I.; Altmeyer, M.; Rask, M.-B.; Lukas, C.; Larsen, D.H.H.; Povlsen, L.K.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef] [PubMed]
- Poli, J.; Tsaponina, O.; Crabbé, L.; Keszthelyi, A.; Pantesco, V.; Chabes, A.; Lengronne, A.; Pasero, P. dNTP pools determine fork progression and origin usage under replication stress. EMBO J. 2012, 31, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, T.; Magdalou, I.; Barascu, A.; Técher, H.; Debatisse, M.; Lopez, B.S. Spontaneous slow replication fork progression elicits mitosis alterations in homologous recombination-deficient mammalian cells. Proc. Natl. Acad. Sci. USA 2014, 111, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Gad, H.; Koolmeister, T.; Jemth, A.-S.; Eshtad, S.; Jacques, S.A.; Ström, C.E.; Svensson, L.M.; Schultz, N.; Lundbäck, T.; Einarsdottir, B.O.; et al. MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature 2014, 508, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Saldivar, J.C.; Miuma, S.; Bene, J.; Hosseini, S.A.; Shibata, H.; Sun, J.; Wheeler, L.J.; Mathews, C.K.; Huebner, K. Initiation of genome instability and preneoplastic processes through loss of Fhit expression. PLoS Genet. 2012, 8, e1003077. [Google Scholar] [CrossRef] [PubMed]
- Petermann, E.; Orta, M.L.; Issaeva, N.; Schultz, N.; Helleday, T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol. Cell 2010, 37, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Groth, A.; Rocha, W.; Verreault, A.; Almouzni, G. Chromatin challenges during DNA replication and repair. Cell 2007, 128, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Clemente-Ruiz, M.; González-Prieto, R.; Prado, F. Histone H3K56 acetylation, CAF1, and Rtt106 coordinate nucleosome assembly and stability of advancing replication forks. PLoS Genet. 2011, 7, e1002376. [Google Scholar] [CrossRef] [PubMed]
- Gunjan, A.; Verreault, A. A Rad53 kinase-dependent surveillance mechanism that regulates histone protein levels in S. cerevisiae. Cell 2003, 115, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Mejlvang, J.; Feng, Y.; Alabert, C.; Neelsen, K.J.; Jasencakova, Z.; Zhao, X.; Lees, M.; Sandelin, A.; Pasero, P.; Lopes, M.; Groth, A. New histone supply regulates replication fork speed and PCNA unloading. J. Cell Biol. 2014, 204, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Choi, T.-Y.; Park, S.Y.; Kang, H.-S.; Cheong, J.-H.; Kim, H.-D.; Lee, B.L.; Hirose, F.; Yamaguchi, M.; Yoo, M.-A. Redox regulation of DNA binding activity of DREF (DNA replication-related element binding factor) in Drosophila. Biochem. J. 2004, 378, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Girard, P.-M.; Pozzebon, M.; Delacôte, F.; Douki, T.; Smirnova, V.; Sage, E. Inhibition of S-phase progression triggered by UVA-induced ROS does not require a functional DNA damage checkpoint response in mammalian cells. DNA Repair 2008, 7, 1500–1516. [Google Scholar] [CrossRef] [PubMed]
- Montaner, B.; O’Donovan, P.; Reelfs, O.; Perrett, C.M.; Zhang, X.; Xu, Y.-Z.; Ren, X.; Macpherson, P.; Frith, D.; Karran, P. Reactive oxygen-mediated damage to a human DNA replication and repair protein. EMBO Rep. 2007, 8, 1074–1079. [Google Scholar] [CrossRef] [PubMed]
- Onn, I.; Milman-Shtepel, N.; Shlomai, J. Redox potential regulates binding of universal minicircle sequence binding protein at the kinetoplast DNA replication origin. Eukaryot. Cell 2004, 3, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Sanders, C.M.; Sizov, D.; Seavers, P.R.; Ortiz-Lombardía, M.; Antson, A.A. Transcription activator structure reveals redox control of a replication initiation reaction. Nucleic Acids Res. 2007, 35, 3504–3515. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; You, J.S.; Lee, S.H. Role of zinc-finger motif in redox regulation of human replication protein A. Antioxid. Redox Signal. 2001, 3, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Weiner, B.E.; Huang, H.; Dattilo, B.M.; Nilges, M.J.; Fanning, E.; Chazin, W.J. An iron-sulfur cluster in the C-terminal domain of the p58 subunit of human DNA primase. J. Biol. Chem. 2007, 282, 33444–33451. [Google Scholar] [CrossRef] [PubMed]
- Wallace, S.S. Biological consequences of free radical-damaged DNA bases. Free Radic. Biol. Med. 2002, 33, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-L.; Pasero, P. Interference between DNA replication and transcription as a cause of genomic instability. Curr. Genomics 2012, 13, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Wei, X. Segregation of transcription and replication sites into higher order domains. Science 1998, 281, 1502–1505. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Huertas, P.; Aguilera, A. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol. Cell 2003, 12, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Ginno, P.A.; Lim, Y.W.; Lott, P.L.; Korf, I.; Chédin, F. GC skew at the 5' and 3' ends of human genes links R-loop formation to epigenetic regulation and transcription termination. Genome Res. 2013, 23, 1590–1600. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Lima, W.F.; Crooke, S.T. Properties of cloned and expressed human RNase H1. J. Biol. Chem. 1999, 274, 28270–28278. [Google Scholar] [CrossRef] [PubMed]
- Alzu, A.; Bermejo, R.; Begnis, M.; Lucca, C.; Piccini, D.; Carotenuto, W.; Saponaro, M.; Brambati, A.; Cocito, A.; Foiani, M.; et al. Senataxin associates with replication forks to protect fork integrity across RNA-polymerase-II-transcribed genes. Cell 2012, 151, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Baker, T.A.; Sekimizu, K.; Funnell, B.E.; Kornberg, A. Extensive unwinding of the plasmid template during staged enzymatic initiation of DNA replication from the origin of the Escherichia coli chromosome. Cell 1986, 45, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Kouzine, F.; Gupta, A.; Baranello, L.; Wojtowicz, D.; Ben-Aissa, K.; Liu, J.; Przytycka, T.M.; Levens, D. Transcription-dependent dynamic supercoiling is a short-range genomic force. Nat. Struct. Mol. Biol. 2013, 20, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Bermejo, R.; Doksani, Y.; Capra, T.; Katou, Y.-M.; Tanaka, H.; Shirahige, K.; Foiani, M. Top1- and Top2-mediated topological transitions at replication forks ensure fork progression and stability and prevent DNA damage checkpoint activation. Genes Dev. 2007, 21, 1921–1936. [Google Scholar] [CrossRef] [PubMed]
- Olavarrieta, L.; Hernández, P.; Krimer, D.B.; Schvartzman, J.B. DNA knotting caused by head-on collision of transcription and replication. J. Mol. Biol. 2002, 322, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Tuduri, S.; Crabbé, L.; Conti, C.; Tourrière, H.; Holtgreve-Grez, H.; Jauch, A.; Pantesco, V.; de Vos, J.; Thomas, A.; Theillet, C.; et al. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat. Cell Biol. 2009, 11, 1315–1324. [Google Scholar] [CrossRef] [PubMed]
- Bermejo, R.; Capra, T.; Gonzalez-Huici, V.; Fachinetti, D.; Cocito, A.; Natoli, G.; Katou, Y.; Mori, H.; Kurokawa, K.; Shirahige, K.; et al. Genome-organizing factors Top2 and Hmo1 prevent chromosome fragility at sites of S phase transcription. Cell 2009, 138, 870–884. [Google Scholar] [CrossRef] [PubMed]
- Helmrich, A.; Ballarino, M.; Tora, L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 2011, 44, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Barlow, J.H.; Faryabi, R.B.; Callén, E.; Wong, N.; Malhowski, A.; Chen, H.T.; Gutierrez-Cruz, G.; Sun, H.-W.; McKinnon, P.; Wright, G.; et al. Identification of early replicating fragile sites that contribute to genome instability. Cell 2013, 152, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Glover, T.W.; Berger, C.; Coyle, J.; Echo, B. DNA polymerase alpha inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum. Genet. 1984, 67, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Durkin, S.G.; Glover, T.W. Chromosome fragile sites. Annu. Rev. Genet. 2007, 41, 169–192. [Google Scholar] [CrossRef] [PubMed]
- Elder, F.F.; Robinson, T.J. Rodent common fragile sites: Are they conserved? Evidence from mouse and rat. Chromosoma 1989, 97, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Raveendranathan, M.; Chattopadhyay, S.; Bolon, Y.-T.; Haworth, J.; Clarke, D.J.; Bielinsky, A.-K. Genome-wide replication profiles of S-phase checkpoint mutants reveal fragile sites in yeast. EMBO J. 2006, 25, 3627–3639. [Google Scholar] [CrossRef] [PubMed]
- Helmrich, A.; Stout-Weider, K.; Hermann, K.; Schrock, E.; Heiden, T. Common fragile sites are conserved features of human and mouse chromosomes and relate to large active genes. Genome Res. 2006, 16, 1222–1230. [Google Scholar] [CrossRef] [PubMed]
- Burrow, A.A.; Williams, L.E.; Pierce, L.C.T.; Wang, Y.-H. Over half of breakpoints in gene pairs involved in cancer-specific recurrent translocations are mapped to human chromosomal fragile sites. BMC Genomics 2009, 10, 59. [Google Scholar] [CrossRef] [PubMed]
- Fungtammasan, A.; Walsh, E.; Chiaromonte, F.; Eckert, K.A.; Makova, K.D. A genome-wide analysis of common fragile sites: What features determine chromosomal instability in the human genome? Genome Res. 2012, 22, 993–1005. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Freudenreich, C.H. An AT-rich sequence in human common fragile site FRA16D causes fork stalling and chromosome breakage in S. cerevisiae. Mol. Cell 2007, 27, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Dillon, L.W.; Pierce, L.C.T.; Ng, M.C.Y.; Wang, Y.-H. Role of DNA secondary structures in fragile site breakage along human chromosome 10. Hum. Mol. Genet. 2013, 22, 1443–1456. [Google Scholar] [CrossRef] [PubMed]
- Corbin, S.; Neilly, M.E.; Espinosa, R.; Davis, E.M.; McKeithan, T.W.; Le Beau, M.M. Identification of unstable sequences within the common fragile site at 3p14.2: Implications for the mechanism of deletions within fragile histidine triad gene/common fragile site at 3p14.2 in tumors. Cancer Res. 2002, 62, 3477–3484. [Google Scholar] [PubMed]
- Finnis, M.; Dayan, S.; Hobson, L.; Chenevix-Trench, G.; Friend, K.; Ried, K.; Venter, D.; Woollatt, E.; Baker, E.; Richards, R.I. Common chromosomal fragile site FRA16D mutation in cancer cells. Hum. Mol. Genet. 2005, 14, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Debatisse, M.; Le Tallec, B.; Letessier, A.; Dutrillaux, B.; Brison, O. Common fragile sites: Mechanisms of instability revisited. Trends Genet. 2012, 28, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, E.; Matricardi, L.; Tosoni, E.; Bensimon, A.; Russo, A. Replication dynamics at common fragile site FRA6E. Chromosoma 2010, 119, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Letessier, A.; Millot, G.A.; Koundrioukoff, S.; Lachagès, A.-M.; Vogt, N.; Hansen, R.S.; Malfoy, B.; Brison, O.; Debatisse, M. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 2011, 470, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Le Tallec, B.; Dutrillaux, B.; Lachages, A.-M.; Millot, G.A.; Brison, O.; Debatisse, M. Molecular profiling of common fragile sites in human fibroblasts. Nat. Struct. Mol. Biol. 2011, 18, 1421–1423. [Google Scholar] [CrossRef] [PubMed]
- Cayrou, C.; Coulombe, P.; Méchali, M. Programming DNA replication origins and chromosome organization. Chromosome Res. 2010, 18, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Dazy, S.; Gandrillon, O.; Hyrien, O.; Prioleau, M.-N. Broadening of DNA replication origin usage during metazoan cell differentiation. EMBO Rep. 2006, 7, 806–811. [Google Scholar] [PubMed]
- Weddington, N.; Stuy, A.; Hiratani, I.; Ryba, T.; Yokochi, T.; Gilbert, D.M. ReplicationDomain: A visualization tool and comparative database for genome-wide replication timing data. BMC Bioinform. 2008, 9, 530. [Google Scholar] [CrossRef]
- Hansen, R.S.; Thomas, S.; Sandstrom, R.; Canfield, T.K.; Thurman, R.E.; Weaver, M.; Dorschner, M.O.; Gartler, S.M.; Stamatoyannopoulos, J.A. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc. Natl. Acad. Sci. USA 2010, 107, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Le Tallec, B.; Millot, G.A.; Blin, M.E.; Brison, O.; Dutrillaux, B.; Debatisse, M. Common fragile site profiling in epithelial and erythroid cells reveals that most recurrent cancer deletions lie in fragile sites hosting large genes. Cell Rep. 2013, 4, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Le Tallec, B.; Koundrioukoff, S.; Wilhelm, T.; Letessier, A.; Brison, O.; Debatisse, M. Updating the mechanisms of common fragile site instability: How to reconcile the different views? Cell. Mol. Life Sci. 2014, 71, 4489–4494. [Google Scholar] [CrossRef] [PubMed]
- Le Beau, M. Replication of a common fragile site, FRA3B, occurs late in S phase and is delayed further upon induction: Implications for the mechanism of fragile site induction. Hum. Mol. Genet. 1998, 7, 755–761. [Google Scholar] [CrossRef] [PubMed]
- El Achkar, E.; Gerbault-Seureau, M.; Muleris, M.; Dutrillaux, B.; Debatisse, M. Premature condensation induces breaks at the interface of early and late replicating chromosome bands bearing common fragile sites. Proc. Natl. Acad. Sci. USA 2005, 102, 18069–18074. [Google Scholar] [CrossRef] [PubMed]
- Naim, V.; Rosselli, F. The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat. Cell Biol. 2009, 11, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Bergoglio, V.; Boyer, A.-S.; Walsh, E.; Naim, V.; Legube, G.; Lee, M.Y.; Rey, L.; Rosselli, F.; Cazaux, C.; Eckert, K.A.; et al. DNA synthesis by Pol η promotes fragile site stability by preventing under-replicated DNA in mitosis. J. Cell Biol. 2013, 201, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Koundrioukoff, S.; Carignon, S.; Técher, H.; Letessier, A.; Brison, O.; Debatisse, M. Stepwise activation of the ATR signaling pathway upon increasing replication stress impacts fragile site integrity. PLoS Genet. 2013, 9, e1003643. [Google Scholar] [CrossRef] [PubMed]
- Sfeir, A.; Kosiyatrakul, S.T.; Hockemeyer, D.; MacRae, S.L.; Karlseder, J.; Schildkraut, C.L.; de Lange, T. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 2009, 138, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Vannier, J.-B.; Pavicic-Kaltenbrunner, V.; Petalcorin, M.I.R.; Ding, H.; Boulton, S.J. RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell 2012, 149, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Martínez, P.; Thanasoula, M.; Muñoz, P.; Liao, C.; Tejera, A.; McNees, C.; Flores, J.M.; Fernández-Capetillo, O.; Tarsounas, M.; Blasco, M.A. Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev. 2009, 23, 2060–2075. [Google Scholar] [CrossRef] [PubMed]
- Martínez, P.; Flores, J.M.; Blasco, M. A 53BP1 deficiency combined with telomere dysfunction activates ATR-dependent DNA damage response. J. Cell Biol. 2012, 197, 283–300. [Google Scholar] [CrossRef] [PubMed]
- Westhorpe, F.G.; Straight, A.F. Functions of the centromere and kinetochore in chromosome segregation. Curr. Opin. Cell Biol. 2013, 25, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Willard, H.F. Evolution of alpha satellite. Curr. Opin. Genet. Dev. 1991, 1, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Waye, J.S.; Willard, H.F. Nucleotide sequence heterogeneity of alpha satellite repetitive DNA: A survey of alphoid sequences from different human chromosomes. Nucleic Acids Res. 1987, 15, 7549–7569. [Google Scholar] [CrossRef] [PubMed]
- Vissel, B.; Choo, K.H. Human alpha satellite DNA—Consensus sequence and conserved regions. Nucleic Acids Res. 1987, 15, 6751–6752. [Google Scholar] [CrossRef] [PubMed]
- Greenfeder, S.A.; Newlon, C.S. Replication forks pause at yeast centromeres. Mol. Cell. Biol. 1992, 12, 4056–4066. [Google Scholar] [PubMed]
- Simi, S.; Simili, M.; Bonatti, S.; Campagna, M.; Abbondandolo, A. Fragile sites at the centromere of Chinese hamster chromosomes: A possible mechanism of chromosome loss. Mutat. Res. 1998, 397, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Sundin, O.; Varshavsky, A. Terminal stages of SV40 DNA replication proceed via multiply intertwined catenated dimers. Cell 1980, 21, 103–114. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, R.J.; Humphrey, T.C. A role for recombination in centromere function. Trends Genet. 2010, 26, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-H.; Jones, M.J.K.; Yin, Y.; Crist, S.B.; Colnaghi, L.; Sims, R.J.; Rothenberg, E.; Jallepalli, P.V.; Huang, T.T. ATR-mediated phosphorylation of FANCI regulates dormant origin firing in response to replication stress. Mol. Cell 2015, 58, 323–338. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, D.; Blow, J.J. Dormant origins, the licensing checkpoint, and the response to replicative stresses. Cold Spring Harb. Perspect. Biol. 2012, 4, 1–10. [Google Scholar] [CrossRef]
- Haber, J.E. Genome Stability: DNA Repair and Recombinaition; Garland Science: New York, NY, USA, 2014; p. 399. [Google Scholar]
- Saintigny, Y. Characterization of homologous recombination induced by replication inhibition in mammalian cells. EMBO J. 2001, 20, 3861–3870. [Google Scholar] [CrossRef] [PubMed]
- Grabarz, A.; Guirouilh-Barbat, J.; Barascu, A.; Pennarun, G.; Genet, D.; Rass, E.; Germann, S.M.; Bertrand, P.; Hickson, I.D.; Lopez, B.S. A role for BLM in double-strand break repair pathway choice: Prevention of CtIP/Mre11-mediated alternative nonhomologous end-joining. Cell Rep. 2013, 5, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Guirouilh-Barbat, J.; Huck, S.; Bertrand, P.; Pirzio, L.; Desmaze, C.; Sabatier, L.; Lopez, B.S. Impact of the KU80 pathway on NHEJ-induced genome rearrangements in mammalian cells. Mol. Cell 2004, 14, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Di Virgilio, M.; Callen, E.; Yamane, A.; Zhang, W.; Jankovic, M.; Gitlin, A.D.; Feldhahn, N.; Resch, W.; Oliveira, T.Y.; Chait, B.T.; et al. Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science 2013, 339, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Lottersberger, F.; Buonomo, S.B.; Sfeir, A.; de Lange, T. 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science 2013, 339, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Bunting, S.F.; Callén, E.; Wong, N.; Chen, H.T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 inhibits homologous recombination in brca1-deficient cells by blocking resection of DNA breaks. Cell 2010, 141, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Reina-San-Martin, B.; Chen, J.; Nussenzweig, A.A.; Nussenzweig, M.C. Enhanced intra-switch region recombination during immunoglobulin class switch recombination in 53BP1−/− B cells. Eur. J. Immunol. 2007, 37, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Huertas, P.; Jackson, S.P. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J. Biol. Chem. 2009, 284, 9558–9565. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Chen, J. DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol. Cell. Biol. 2004, 24, 9478–9486. [Google Scholar] [CrossRef] [PubMed]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Langerak, P.; Mejia-Ramirez, E.; Limbo, O.; Russell, P. Release of Ku and MRN from DNA ends by Mre11 nuclease activity and Ctp1 is required for homologous recombination repair of double-strand breaks. PLoS Genet. 2011, 7, e1002271. [Google Scholar] [CrossRef] [PubMed]
- Rass, E.; Grabarz, A.; Plo, I.; Gautier, J.; Bertrand, P.; Lopez, B.S. Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat. Struct. Mol. Biol. 2009, 16, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Escribano-Díaz, C.; Orthwein, A.; Fradet-Turcotte, A.; Xing, M.; Young, J.T.F.; Tkáč, J.; Cook, M.A.; Rosebrock, A.P.; Munro, M.; Canny, M.D.; et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. Cell 2013, 49, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Fong, K.W.; Wang, J.; Wang, W.; Chen, J. RIF1 counteracts BRCA1-mediated end resection during DNA repair. J. Biol. Chem. 2013, 288, 11135–11143. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.R.; Sossick, A.J.; Boulton, S.J.; Jackson, S.P. BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. J. Cell Sci. 2012, 125, 3529–3534. [Google Scholar] [CrossRef] [PubMed]
- Tomimatsu, N.; Mukherjee, B.; Hardebeck, M.C.; Ilcheva, M.; Camacho, C.V.; Harris, J.L.; Porteus, M.; Llorente, B. Phosphorylation of EXO1 by CDKs 1 and 2 regulates DNA end resection and repair pathway choice Nozomi. Nat. Commun. 2014, 5, 3561. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Chapman, J.R.; Brandsma, I.; Yuan, J.; Mistrik, M.; Bouwman, P.; Bartkova, J.; Gogola, E.; Warmerdam, D.; Barazas, M.; et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 2015. [Google Scholar] [CrossRef]
- Boersma, V.; Moatti, N.; Segura-Bayona, S.; Peuscher, M.H.; van der Torre, J.; Wevers, B.A.; Orthwein, A.; Durocher, D.; Jacobs, J.J.L. MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5′ end resection. Nature 2015. [Google Scholar] [CrossRef]
- Potts, P.R.; Porteus, M.H.; Yu, H. Human SMC5/6 complex promotes sister chromatid homologous recombination by recruiting the SMC1/3 cohesin complex to double-strand breaks. EMBO J. 2006, 25, 3377–3388. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Balakrishnan, K.; Malaterre, J.; Beasley, M.; Yan, Y.; Essers, J.; Appeldoorn, E.; Tomaszewski, J.M.; Thomaszewski, J.M.; Vazquez, M.; et al. Rad21-cohesin haploinsufficiency impedes DNA repair and enhances gastrointestinal radiosensitivity in mice. PLoS ONE 2010, 5, e12112. [Google Scholar] [CrossRef] [PubMed]
- De Piccoli, G.; Cortes-Ledesma, F.; Ira, G.; Torres-Rosell, J.; Uhle, S.; Farmer, S.; Hwang, J.-Y.; Machin, F.; Ceschia, A.; McAleenan, A.; et al. Smc5-Smc6 mediate DNA double-strand-break repair by promoting sister-chromatid recombination. Nat. Cell Biol. 2006, 8, 1032–1034. [Google Scholar] [CrossRef] [PubMed]
- Nickoloff, J.A. Transcription enhances intrachromosomal homologous recombination in mammalian cells. Mol. Cell. Biol. 1992, 12, 5311–5318. [Google Scholar] [PubMed]
- Aymard, F.; Bugler, B.; Schmidt, C.K.; Guillou, E.; Caron, P.; Briois, S.; Iacovoni, J.S.; Daburon, V.; Miller, K.M.; Jackson, S.P.; et al. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat. Struct. Mol. Biol. 2014, 21, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.; Cotta-Ramusino, C.; Pellicioli, A.; Liberi, G.; Plevani, P.; Muzi-Falconi, M.; Newlon, C.S.; Foiani, M. The DNA replication checkpoint response stabilizes stalled replication forks. Nature 2001, 412, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Cobb, J.A.; Bjergbaek, L.; Shimada, K.; Frei, C.; Gasser, S.M. DNA polymerase stabilization at stalled replication forks requires Mec1 and the RecQ helicase Sgs. EMBO J. 2003, 22, 4325–4336. [Google Scholar] [CrossRef] [PubMed]
- Lucca, C.; Vanoli, F.; Cotta-Ramusino, C.; Pellicioli, A.; Liberi, G.; Haber, J.; Foiani, M. Checkpoint-mediated control of replisome-fork association and signalling in response to replication pausing. Oncogene 2004, 23, 1206–1213. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Ray Chaudhuri, A.; Lopes, M.; Costanzo, V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat. Struct. Mol. Biol. 2010, 17, 1305–1311. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Puddu, F.; Costanzo, V. RAD51- and MRE11-dependent reassembly of uncoupled CMG helicase complex at collapsed replication forks. Nat. Struct. Mol. Biol. 2011, 19, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Schlacher, K.; Wu, H.; Jasin, M. A distinct replication fork protection pathway connects fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.; Hamdy, F.C.; Helleday, T. Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res. 2012, 72, 2814–2821. [Google Scholar] [CrossRef] [PubMed]
- Pefani, D.-E.; Latusek, R.; Pires, I.; Grawenda, A.M.; Yee, K.S.; Hamilton, G.; van der Weyden, L.; Esashi, F.; Hammond, E.M.; O’Neill, E. RASSF1A-LATS1 signalling stabilizes replication forks by restricting CDK2-mediated phosphorylation of BRCA2. Nat. Cell Biol. 2014, 16, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Higgins, N.P.; Kato, K.; Strauss, B. A model for replication repair in mammalian cells. J. Mol. Biol. 1976, 101, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Seigneur, M.; Bidnenko, V.; Ehrlich, S.D.; Michel, B. RuvAB acts at arrested replication forks. Cell 1998, 95, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Sogo, J.M.; Lopes, M.; Foiani, M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 2002, 297, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Ray Chaudhuri, A.; Hashimoto, Y.; Herrador, R.; Neelsen, K.J.; Fachinetti, D.; Bermejo, R.; Cocito, A.; Costanzo, V.; Lopes, M. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat. Struct. Mol. Biol. 2012, 19, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Bansbach, C.E.; Bétous, R.; Lovejoy, C.A.; Glick, G.G.; Cortez, D. The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes Dev. 2009, 23, 2405–2414. [Google Scholar] [CrossRef] [PubMed]
- Zellweger, R.; Dalcher, D.; Mutreja, K.; Berti, M.; Schmid, J.A.; Herrador, R.; Vindigni, A.; Lopes, M. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol. 2015, 208, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Ragland, R.L.; Patel, S.; Rivard, R.S.; Smith, K.; Peters, A.A.; Bielinsky, A.-K.; Brown, E.J. RNF4 and PLK1 are required for replication fork collapse in ATR-deficient cells. Genes Dev. 2013, 27, 2259–2273. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.I.; Murga, M.; Fernandez-Capetillo, O. Targeting ATR and Chk1 kinases for cancer treatment: A new model for new (and old) drugs. Mol. Oncol. 2011, 5, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Nam, E.A.; Cortez, D. ATR signalling: More than meeting at the fork. Biochem. J. 2011, 436, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Labib, K.; de Piccoli, G. Surviving chromosome replication: The many roles of the S-phase checkpoint pathway. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 3554–3561. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, K.; Lambert, S.; Baldacci, G.; Murray, J.M.; Carr, A.M. Nearby inverted repeats fuse to generate acentric and dicentric palindromic chromosomes by a replication template exchange mechanism. Genes Dev. 2009, 23, 2876–2886. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.; Mizuno, K.; Blaisonneau, J.; Martineau, S.; Chanet, R.; Fréon, K.; Murray, J.M.; Carr, A.M.; Baldacci, G. Homologous recombination restarts blocked replication forks at the expense of genome rearrangements by template exchange. Mol. Cell 2010, 39, 346–359. [Google Scholar] [CrossRef] [PubMed]
- Neelsen, K.J.; Zanini, I.M.Y.; Herrador, R.; Lopes, M. Oncogenes induce genotoxic stress by mitotic processing of unusual replication intermediates. J. Cell Biol. 2013, 200, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Matos, J.; Blanco, M.G.; Maslen, S.; Skehel, J.M.; West, S.C. Regulatory control of the resolution of DNA recombination intermediates during meiosis and mitosis. Cell 2011, 147, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Pageau, G.J.; Lawrence, J.B. BRCA1 foci in normal S-phase nuclei are linked to interphase centromeres and replication of pericentric heterochromatin. J. Cell Biol. 2006, 175, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Buonomo, S.B.C.; Wu, Y.; Ferguson, D.; de Lange, T. Mammalian Rif1 contributes to replication stress survival and homology-directed repair. J. Cell Biol. 2009, 187, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Okamoto, A.; Katou, Y.; Yadani, C.; Shitanda, T.; Kaweeteerawat, C.; Takahashi, T.S.; Itoh, T.; Shirahige, K.; Masukata, H.; et al. Rad51 suppresses gross chromosomal rearrangement at centromere in Schizosaccharomyces pombe. EMBO J. 2008, 27, 3036–3046. [Google Scholar] [CrossRef] [PubMed]
- Jaco, I.; Canela, A.; Vera, E.; Blasco, M.A. Centromere mitotic recombination in mammalian cells. J. Cell Biol. 2008, 181, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Daboussi, F.; Courbet, S.; Benhamou, S.; Kannouche, P.; Zdzienicka, M.Z.; Debatisse, M.; Lopez, B.S. A homologous recombination defect affects replication-fork progression in mammalian cells. J. Cell Sci. 2008, 121, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Guirouilh-Barbat, J.; Lambert, S.; Bertrand, P.; Lopez, B.S. Is homologous recombination really an error-free process? Front. Genet. 2014, 5, 175. [Google Scholar] [CrossRef] [PubMed]
- Crasta, K.; Ganem, N.J.; Dagher, R.; Lantermann, A.B.; Ivanova, E.V.; Pan, Y.; Nezi, L.; Protopopov, A.; Chowdhury, D.; Pellman, D. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012, 482, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.J.K.; Jallepalli, P.V. Chromothripsis: Chromosomes in crisis. Dev. Cell 2012, 23, 908–917. [Google Scholar] [CrossRef] [PubMed]
- Forment, J.V.; Kaidi, A.; Jackson, S.P. Chromothripsis and cancer: Causes and consequences of chromosome shattering. Nat. Rev. Cancer 2012, 12, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Erez, A.; Nagamani, S.C.S.; Dhar, S.U.; Ko, K.E.; Dharmadhikari, A.V.; Cooper, M.L.; Zhang, F.; Withers, M.A.; Bacino, C.A.; et al. Chromosome catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell 2012, 146, 889–903. [Google Scholar] [CrossRef]
- Lee, J.A.; Carvalho, C.M.B.; Lupski, J.R. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 2007, 131, 1235–1247. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Khajavi, M.; Connolly, A.M.; Towne, C.F.; Batish, S.D.; Lupski, J.R. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat. Genet. 2009, 41, 849–853. [Google Scholar] [CrossRef] [PubMed]
- Roseaulin, L.; Yamada, Y.; Tsutsui, Y.; Russell, P.; Iwasaki, H.; Arcangioli, B. Mus81 is essential for sister chromatid recombination at broken replication forks. EMBO J. 2008, 27, 1378–1387. [Google Scholar] [CrossRef] [PubMed]
- Hanada, K.; Budzowska, M.; Davies, S.L.; van Drunen, E.; Onizawa, H.; Beverloo, H.B.; Maas, A.; Essers, J.; Hickson, I.D.; Kanaar, R. The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat. Struct. Mol. Biol. 2007, 14, 1096–1104. [Google Scholar] [CrossRef] [PubMed]
- Bétermier, M.; Bertrand, P.; Lopez, B.S. Is non-homologous end-joining really an inherently error-prone process? PLoS Genet. 2014, 10, e1004086. [Google Scholar] [CrossRef] [PubMed]
- Bothmer, A.; Robbiani, D.F.; di Virgilio, M.; Bunting, S.F.; Klein, I.A.; Feldhahn, N.; Barlow, J.; Chen, H.-T.T.; Bosque, D.; Callen, E.; et al. Regulation of DNA end joining, resection, and immunoglobulin class switch recombination by 53BP1. Mol. Cell 2011, 42, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Bunting, S.F.; Nussenzweig, A. End-joining, translocations and cancer. Nat. Rev. Cancer 2013, 13, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Deriano, L.; Roth, D.B. Modernizing the nonhomologous end-joining repertoire: Alternative and classical NHEJ share the stage. Annu. Rev. Genet. 2013, 47, 433–455. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Niu, H.; Chung, W.-H.; Zhu, Z.; Papusha, A.; Shim, E.Y.; Lee, S.E.; Sung, P.; Ira, G. Cell cycle regulation of DNA double-strand break end resection by Cdk1-dependent Dna2 phosphorylation. Nat. Struct. Mol. Biol. 2011, 18, 1015–1019. [Google Scholar] [CrossRef] [PubMed]
- Yousefzadeh, M.J.; Wyatt, D.W.; Takata, K.-I.; Mu, Y.; Hensley, S.C.; Tomida, J.; Bylund, G.O.; Doublié, S.; Johansson, E.; Ramsden, D.A.; et al. Mechanism of suppression of chromosomal instability by DNA polymerase POLQ. PLoS Genet. 2014, 10, e1004654. [Google Scholar] [CrossRef] [PubMed]
- Mateos-gomez, P.A.; Gong, F.; Nair, N.; Miller, K.M.; Lazzerini-denchi, E.; Sfeir, A. Mammalian polymerase θ promotes alternative NHEJ and suppresses recombination. Nature 2015, 518, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Sturzenegger, A.; Burdova, K.; Kanagaraj, R.; Levikova, M.; Pinto, C.; Cejka, P.; Janscak, P. DNA2 cooperates with the WRN and BLM RecQ helicases to mediate long-range DNA-end resection in human cells. J. Biol. Chem. 2014, 289, 27314–27326. [Google Scholar] [CrossRef] [PubMed]
- Couëdel, C.; Mills, K.D.; Barchi, M.; Shen, L.; Olshen, A.; Johnson, R.D.; Nussenzweig, A.; Essers, J.; Kanaar, R.; Li, G.C.; et al. Collaboration of homologous recombination and nonhomologous end-joining factors for the survival and integrity of mice and cells. Genes Dev. 2004, 18, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.D.; Ferguson, D.O.; Essers, J.; Eckersdorff, M.; Kanaar, R.; Alt, F.W. Rad54 and DNA Ligase IV cooperate to maintain mammalian chromatid stability. Genes Dev. 2004, 18, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Adamo, A.; Collis, S.J.; Adelman, C.A.; Silva, N.; Horejsi, Z.; Ward, J.D.; Martinez-Perez, E.; Boulton, S.J.; La Volpe, A. Preventing nonhomologous end joining suppresses DNA repair defects of fanconi anemia. Mol. Cell 2010, 39, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Pace, P.; Mosedale, G.; Hodskinson, M.R.; Rosado, I.V.; Sivasubramaniam, M.; Patel, K.J. Ku70 corrupts DNA repair in the absence of the Fanconi anemia pathway. Science 2010, 329, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Smogorzewska, A.; Karlseder, J.; Holtgreve-Grez, H.; Jauch, A.; de Lange, T. DNA ligase IV-dependent NHEJ of deprotected mammalian telomeres in G1 and G2. Curr. Biol. 2002, 12, 1635–1644. [Google Scholar] [CrossRef] [PubMed]
- Celli, G.B.; de Lange, T. DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat. Cell Biol. 2005, 7, 712–718. [Google Scholar] [CrossRef] [PubMed]
- Celli, G.B.; Denchi, E.L.; de Lange, T. Ku70 stimulates fusion of dysfunctional telomeres yet protects chromosome ends from homologous recombination. Nat. Cell Biol. 2006, 8, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Aten, J.A.; Stap, J.; Krawczyk, P.M.; van Oven, C.H.; Hoebe, R.A.; Essers, J.; Kanaar, R. Dynamics of DNA double-strand breaks revealed by clustering of damaged chromosome domains. Science 2004, 303, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Kruhlak, M.J.; Celeste, A.; Dellaire, G.; Fernandez-Capetillo, O.; Müller, W.G.; McNally, J.G.; Bazett-Jones, D.P.; Nussenzweig, A. Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J. Cell Biol. 2006, 172, 823–834. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, P.M.; Borovski, T.; Stap, J.; Cijsouw, T.; ten Cate, R.; Medema, J.P.; Kanaar, R.; Franken, N.A.P.; Aten, J.A. Chromatin mobility is increased at sites of DNA double-strand breaks. J. Cell Sci. 2012, 125, 2127–2133. [Google Scholar] [CrossRef] [PubMed]
- Soutoglou, E.; Dorn, J.F.; Sengupta, K.; Jasin, M.; Nussenzweig, A.; Ried, T.; Danuser, G.; Misteli, T. Positional stability of single double-strand breaks in mammalian cells. Nat. Cell Biol. 2007, 9, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Roukos, V.; Voss, T.C.; Schmidt, C.K.; Lee, S.; Wangsa, D.; Misteli, T. Spatial dynamics of chromosome translocations in living cells. Science 2013, 341, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Girst, S.; Hable, V.; Drexler, G.A.; Greubel, C.; Siebenwirth, C.; Haum, M.; Friedl, A.A.; Dollinger, G. Subdiffusion supports joining of correct ends during repair of DNA double-strand breaks. Sci. Rep. 2013, 3, 2511. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.; Durante, M.; Taucher-Scholz, G.; Jakob, B. ATM alters the otherwise robust chromatin mobility at sites of DNA double-strand breaks (DSBs) in human cells. PLoS ONE 2014, 9, e92640. [Google Scholar] [CrossRef] [PubMed]
- Krenning, L.; Feringa, F.M.; Shaltiel, I.A.; vanden Berg, J.; Medema, R.H. Transient activation of p53 in G2 phase is sufficient to induce senescence. Mol. Cell 2014, 55, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Müllers, E.; Cascales, H.S.; Jaiswal, H.; Saurin, A.T.; Lindqvist, A. Translocation of Cyclin B1 marks the restriction point for terminal cell cycle exit in G2 phase. Cell Cycle 2014, 13, 2733–2743. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.L.; Palmai-Pallag, T.; Ying, S.; Hickson, I.D. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell Biol. 2009, 11, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Baumann, C.; Körner, R.; Hofmann, K.; Nigg, E.A. PICH, a centromere-associated SNF2 family atpase, is regulated by plk1 and required for the spindle checkpoint. Cell 2007, 128, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Vinciguerra, P.; Godinho, S.A.; Parmar, K.; Pellman, D.; D’Andrea, A.D. Cytokinesis failure occurs in Fanconi anemia pathway-deficient murine and human bone marrow hematopoietic cells. J. Clin. Investig. 2010, 120, 3834–3842. [Google Scholar] [CrossRef] [PubMed]
- Naim, V.; Wilhelm, T.; Debatisse, M.; Rosselli, F. ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat. Cell Biol. 2013, 15, 1008–1015. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Nielsen, C.F.; Yao, Q.; Hickson, I.D. The origins and processing of ultra fine anaphase DNA bridges. Curr. Opin. Genet. Dev. 2014, 26, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Guervilly, J.-H.; Takedachi, A.; Naim, V.; Scaglione, S.; Chawhan, C.; Lovera, Y.; Despras, E.; Kuraoka, I.; Kannouche, P.; Rosselli, F.; et al. The SLX4 Complex Is a SUMO E3 Ligase that Impacts on Replication Stress Outcome and Genome Stability. Mol. Cell 2015, 57, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.K.; Hickson, I.D. RecQ helicases: Multifunctional genome caretakers. Nat. Rev. Cancer 2009, 9, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Barefield, C.; Karlseder, J. The BLM helicase contributes to telomere maintenance through processing of late-replicating intermediate structures. Nucleic Acids Res. 2012, 40, 7358–7367. [Google Scholar] [CrossRef] [PubMed]
- Lahkim Bennani-Belhaj, K.; Rouzeau, S.; Buhagiar-Labarchède, G.; Chabosseau, P.; Onclercq-Delic, R.; Bayart, E.; Cordelières, F.; Couturier, J.; Amor-Guéret, M. The Bloom syndrome protein limits the lethality associated with RAD51 deficiency. Mol. Cancer Res. 2010, 8, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Laulier, C.; Cheng, A.; Stark, J.M. The relative efficiency of homology-directed repair has distinct effects on proper anaphase chromosome separation. Nucleic Acids Res. 2011, 39, 5935–5944. [Google Scholar] [CrossRef] [PubMed]
- Rodrigue, A.; Coulombe, Y.; Jacquet, K.; Gagné, J.-P.; Roques, C.; Gobeil, S.; Poirier, G.; Masson, J.-Y. The RAD51 paralogs ensure cellular protection against mitotic defects and aneuploidy. J. Cell Sci. 2013, 126, 348–359. [Google Scholar] [CrossRef] [PubMed]
- Tahara, H.; Shin-Ya, K.; Seimiya, H.; Yamada, H.; Tsuruo, T.; Ide, T. G-Quadruplex stabilization by telomestatin induces TRF2 protein dissociation from telomeres and anaphase bridge formation accompanied by loss of the 3′ telomeric overhang in cancer cells. Oncogene 2006, 25, 1955–1966. [Google Scholar] [CrossRef] [PubMed]
- Van Steensel, B.; Smogorzewska, A.; de Lange, T. TRF2 protects human telomeres from end-to-end fusions. Cell 1998, 92, 401–413. [Google Scholar] [CrossRef] [PubMed]
- Skoufias, D.A.; Andreassen, P.R.; Lacroix, F.B.; Wilson, L.; Margolis, R.L. Mammalian mad2 and bub1/bubR1 recognize distinct spindle-attachment and kinetochore-tension checkpoints. Proc. Natl. Acad. Sci. USA 2001, 98, 4492–4497. [Google Scholar] [CrossRef] [PubMed]
- Cimini, D.; Tanzarella, C.; Degrassi, F. Differences in malsegregation rates obtained by scoring ana-telophases or binucleate cells. Mutagenesis 1999, 14, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Griffin, C.S.; Simpson, P.J.; Wilson, C.R.; Thacker, J. Mammalian recombination-repair genes XRCC2 and XRCC3 promote correct chromosome segregation. Nat. Cell Biol. 2000, 2, 757–761. [Google Scholar] [CrossRef] [PubMed]
- Kraakman-van der Zwet, M.; Overkamp, W.J.I.; van Lange, R.E.E.; Essers, J.; van Duijn-Goedhart, A.; Wiggers, I.; Swaminathan, S.; van Buul, P.P.W.; Errami, A.; Tan, R.T.L.; et al. Brca2 (XRCC11) deficiency results in radioresistant DNA synthesis and a higher frequency of spontaneous deletions. Mol. Cell. Biol. 2002, 22, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, P.; Lambert, S.; Joubert, C.; Lopez, B.S. Overexpression of mammalian Rad51 does not stimulate tumorigenesis while a dominant-negative Rad51 affects centrosome fragmentation, ploidy and stimulates tumorigenesis, in p53-defective CHO cells. Oncogene 2003, 22, 7587–7592. [Google Scholar] [CrossRef] [PubMed]
- Daboussi, F.; Thacker, J.; Lopez, B.S. Genetic interactions between RAD51 and its paralogues for centrosome fragmentation and ploidy control, independently of the sensitivity to genotoxic stresses. Oncogene 2005, 24, 3691–3696. [Google Scholar] [CrossRef] [PubMed]
- Rattner, J.B.; Phillips, S.G. Independence of centriole formation and DNA synthesis. J. Cell Biol. 1973, 57, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Balczon, R.; Bao, L.; Zimmer, W.E.; Brown, K.; Zinkowski, R.P.; Brinkley, B.R. Dissociation of centrosome replication events from cycles of DNA synthesis and mitotic division in hydroxyurea-arrested Chinese hamster ovary cells. J. Cell Biol. 1995, 130, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.; Stearns, T. Centrosome number is controlled by a centrosome-intrinsic block to reduplication. Nat. Cell Biol. 2003, 5, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Katsura, M.; Tsuruga, T.; Date, O.; Yoshihara, T.; Ishida, M.; Tomoda, Y.; Okajima, M.; Takaku, M.; Kurumizaka, H.; Kinomura, A.; et al. The ATR-Chk1 pathway plays a role in the generation of centrosome aberrations induced by Rad51C dysfunction. Nucleic Acids Res. 2009, 37, 3959–3968. [Google Scholar] [CrossRef] [PubMed]
- Goepfert, T.M.; Adigun, Y.E.; Zhong, L.; Gay, J.; Medina, D.; Brinkley, W.R. Centrosome amplification and overexpression of aurora a are early events in rat mammary carcinogenesis. Cancer Res. 2002, 62, 4115–4122. [Google Scholar] [PubMed]
- Pihan, G.A.; Wallace, J.; Zhou, Y.; Doxsey, S.J. Centrosome abnormalities and chromosome instability occur together in pre-invasive carcinomas. Cancer Res. 2003, 63, 1398–1404. [Google Scholar] [PubMed]
- Liontos, M.; Koutsami, M.; Sideridou, M.; Evangelou, K.; Kletsas, D.; Levy, B.; Kotsinas, A.; Nahum, O.; Zoumpourlis, V.; Kouloukoussa, M.; et al. Deregulated overexpression of hCdt1 and hCdc6 promotes malignant behavior. Cancer Res. 2007, 67, 10899–10909. [Google Scholar] [CrossRef] [PubMed]
- Hontz, A.E.; Li, S.A.; Lingle, W.L.; Negron, V.; Bruzek, A.; Salisbury, J.L.; Li, J.J. Aurora A and B overexpression and centrosome amplification in early estrogen-induced tumor foci in the Syrian hamster kidney: Implications for chromosomal instability, aneuploidy, and neoplasia. Cancer Res. 2007, 67, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Duensing, A.; Chin, A.; Wang, L.; Kuan, S.-F.; Duensing, S. Analysis of centrosome overduplication in correlation to cell division errors in high-risk human papillomavirus (HPV)-associated anal neoplasms. Virology 2008, 372, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Howell, W.H. The life-history of the formed elements of the blood, especially the red blood corpuscles. J. Morphol. 1890, 4, 57–116. [Google Scholar] [CrossRef]
- Jolly, M. Recherches sur la formation des globules rouges des mammiferes. Arch. D’anatomie Microsc. 1907, 9, 133–314. [Google Scholar]
- Dawson, D.W.; Bury, H.P. The significance of Howell-Jolly bodies and giant metamyelocytes in marrow smears. J. Clin. Pathol. 1961, 14, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.L.; Compton, D.A. Chromosome missegregation in human cells arises through specific types of kinetochore-microtubule attachment errors. Proc. Natl. Acad. Sci. USA 2011, 108, 17974–17978. [Google Scholar] [CrossRef] [PubMed]
- Gisselsson, D.; Björk, J.; Höglund, M.; Mertens, F.; dal Cin, P.; Akerman, M.; Mandahl, N. Abnormal nuclear shape in solid tumors reflects mitotic instability. Am. J. Pathol. 2001, 158, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Terradas, M.; Martín, M.; Tusell, L.; Genescà, A. Genetic activities in micronuclei: Is the DNA entrapped in micronuclei lost for the cell? Mutat. Res. Rev. Mutat. Res. 2010, 705, 60–67. [Google Scholar] [CrossRef]
- Savage, J.R. A comment on the quantitative relationship between micronuclei and chromosomal aberrations. Mutat. Res. 1988, 207, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Fenech, M. Cytokinesis-block micronucleus cytome assay. Nat. Protoc. 2007, 2, 1084–1104. [Google Scholar] [CrossRef] [PubMed]
- Fenech, M.; Kirsch-Volders, M.; Natarajan, A.T.; Surralles, J.; Crott, J.W.; Parry, J.; Norppa, H.; Eastmond, D.A.; Tucker, J.D.; Thomas, P. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis 2011, 26, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Sun, Z.; Liu, Z.; Guo, H.; Liu, Q.; Jiang, H.; Zou, Y.; Gong, Y.; Tischfield, J.A.; Shao, C. Replication stress induces micronuclei comprising of aggregated DNA double-strand breaks. PLoS ONE 2011, 6, e18618. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Fang, X.; Baker, D.J.; Guo, L.; Gao, X.; Wei, Z.; Han, S.; van Deursen, J.M.; Zhang, P. The ATM-p53 pathway suppresses aneuploidy-induced tumorigenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 14188–14193. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.L.; Compton, D.A. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J. Cell Biol. 2010, 188, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Harrigan, J.A.; Belotserkovskaya, R.; Coates, J.; Dimitrova, D.S.; Polo, S.E.; Bradshaw, C.R.; Fraser, P.; Jackson, S.P. Replication stress induces 53BP1-containing OPT domains in G1 cells. J. Cell Biol. 2011, 193, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Lukas, C.; Savic, V.; Bekker-Jensen, S.; Doil, C.; Neumann, B.; Pedersen, R.S.; Grøfte, M.; Chan, K.L.; Hickson, I.D.; Bartek, J.; et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat. Cell Biol. 2011, 13, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Benada, J.; Burdová, K.; Lidak, T.; von Morgen, P.; Macurek, L.; Benada, J.; Burdov, K. Polo-like kinase 1 inhibits DNA damage response during mitosis. Cell Cycle 2015, 14, 37–41. [Google Scholar] [CrossRef]
- Orthwein, A.; Fradet-Turcotte, A.; Noordermeer, S.M.; Canny, M.D.; Brun, C.M.; Strecker, J.; Escribano-Diaz, C.; Durocher, D. Mitosis inhibits DNA double-strand break repair to guard against telomere fusions. Science 2014, 344, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-H.; Acharya, S.S.; Kwon, M.; Drane, P.; Guan, Y.; Adelmant, G.; Kalev, P.; Shah, J.; Pellman, D.; Marto, J.A.; et al. Dephosphorylation enables the recruitment of 53BP1 to double-strand DNA breaks. Mol. Cell 2014, 54, 515–525. [Google Scholar]
- Boveri, T.; Gustav Fischer, J. Zur frage der entstehung maligner tumoren. Science 1914, 2, 676–679. [Google Scholar]
- Boveri, T. Concerning the origin of malignant tumours by Theodor Boveri. Translated and annotated by Henry Harris. J. Cell Sci. 2008, 121, 1–84. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gelot, C.; Magdalou, I.; Lopez, B.S. Replication Stress in Mammalian Cells and Its Consequences for Mitosis. Genes 2015, 6, 267-298. https://doi.org/10.3390/genes6020267
Gelot C, Magdalou I, Lopez BS. Replication Stress in Mammalian Cells and Its Consequences for Mitosis. Genes. 2015; 6(2):267-298. https://doi.org/10.3390/genes6020267
Chicago/Turabian StyleGelot, Camille, Indiana Magdalou, and Bernard S. Lopez. 2015. "Replication Stress in Mammalian Cells and Its Consequences for Mitosis" Genes 6, no. 2: 267-298. https://doi.org/10.3390/genes6020267
APA StyleGelot, C., Magdalou, I., & Lopez, B. S. (2015). Replication Stress in Mammalian Cells and Its Consequences for Mitosis. Genes, 6(2), 267-298. https://doi.org/10.3390/genes6020267