Altered Actions of Memantine and NMDA-Induced Currents in a New Grid2-Deleted Mouse Line

Abstract
:1. Introduction
2. Results
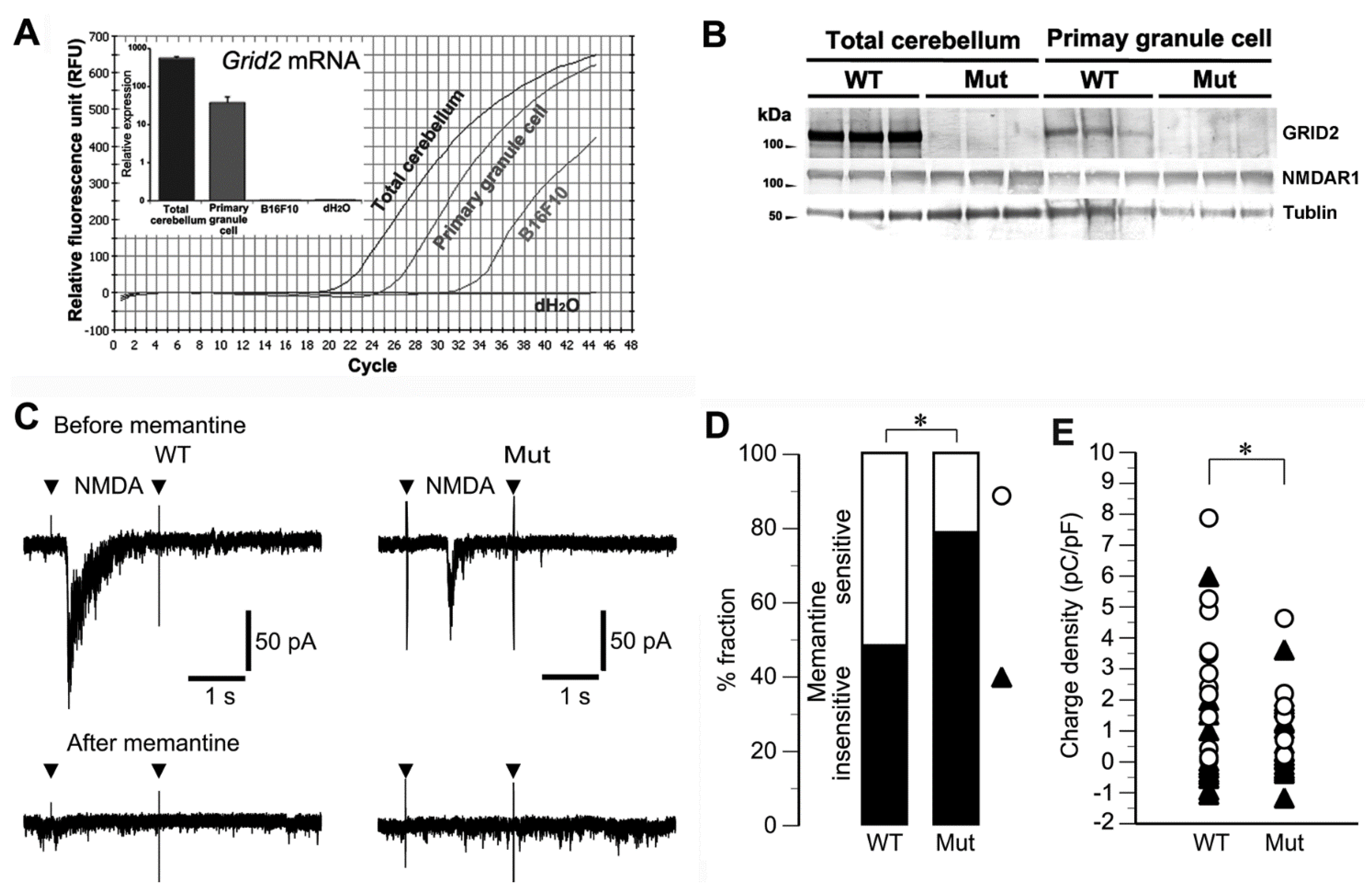
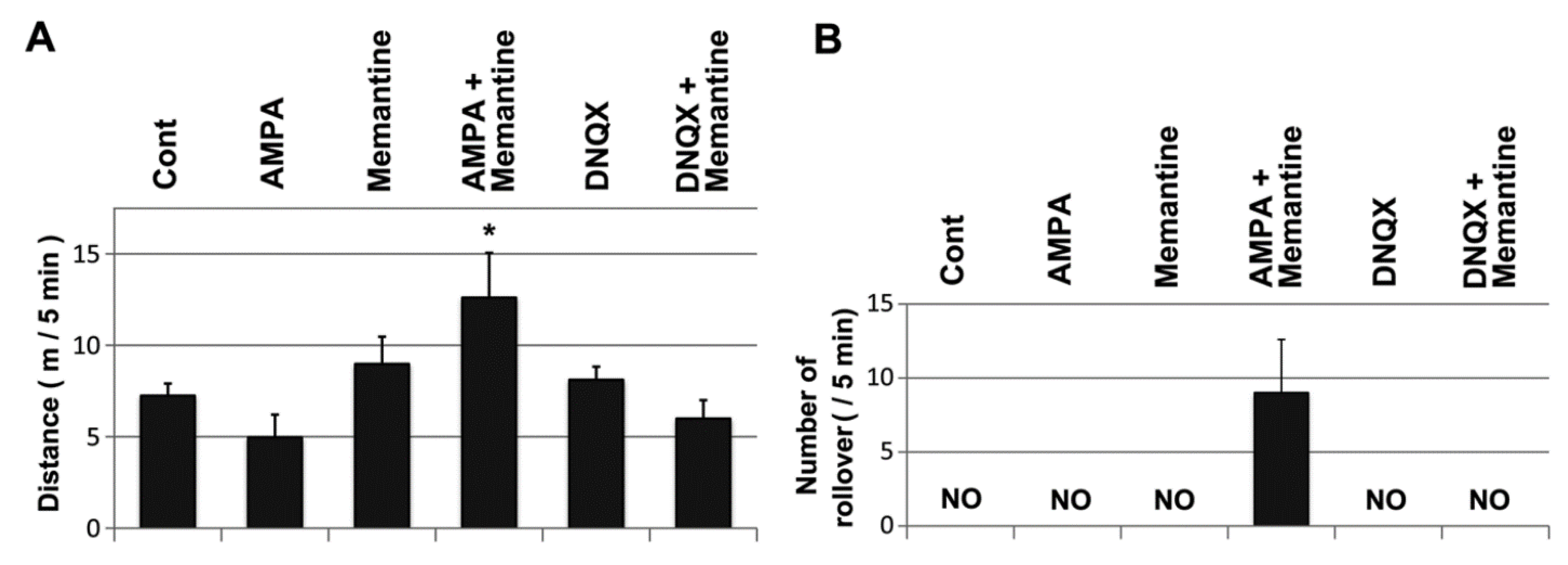
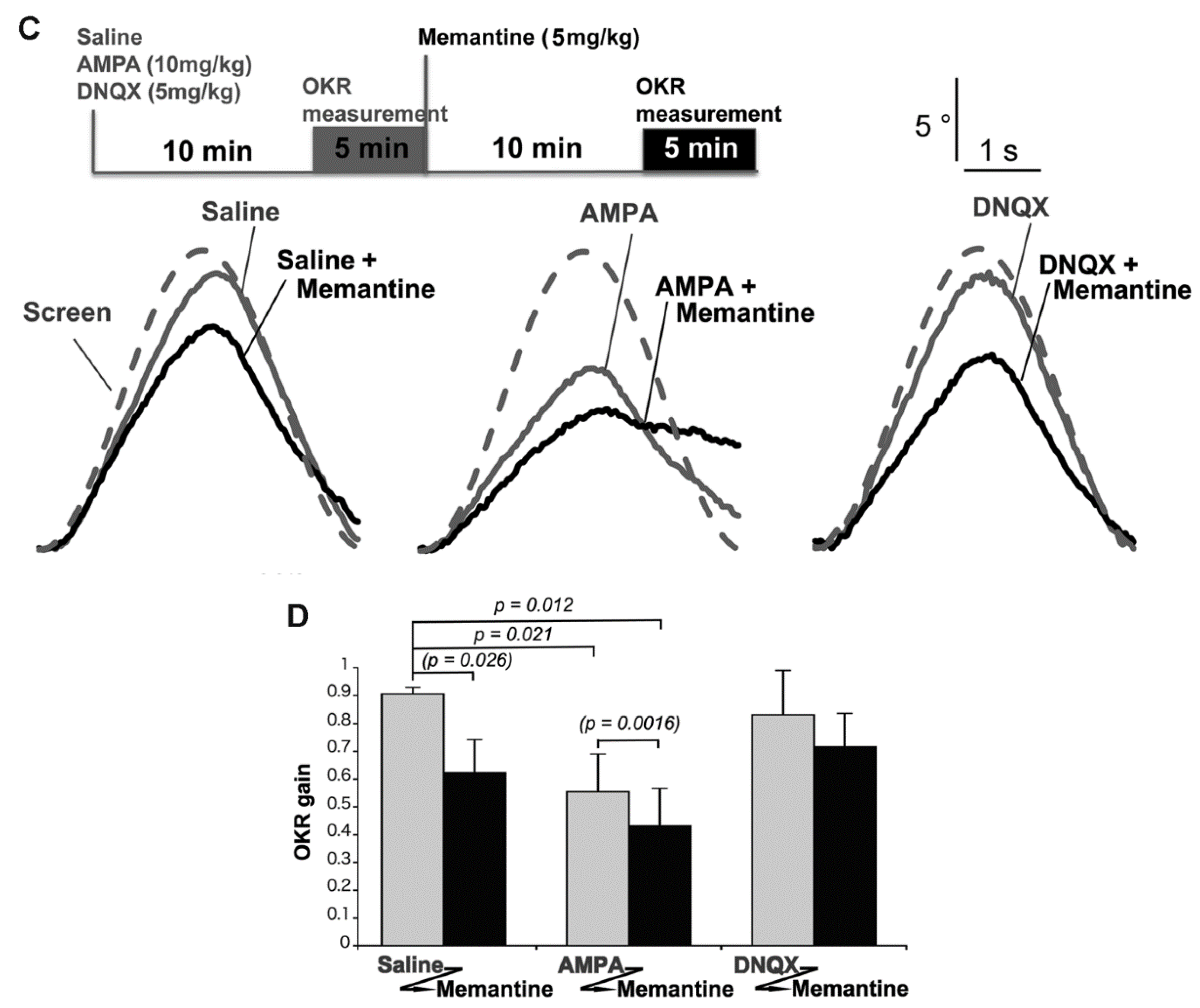
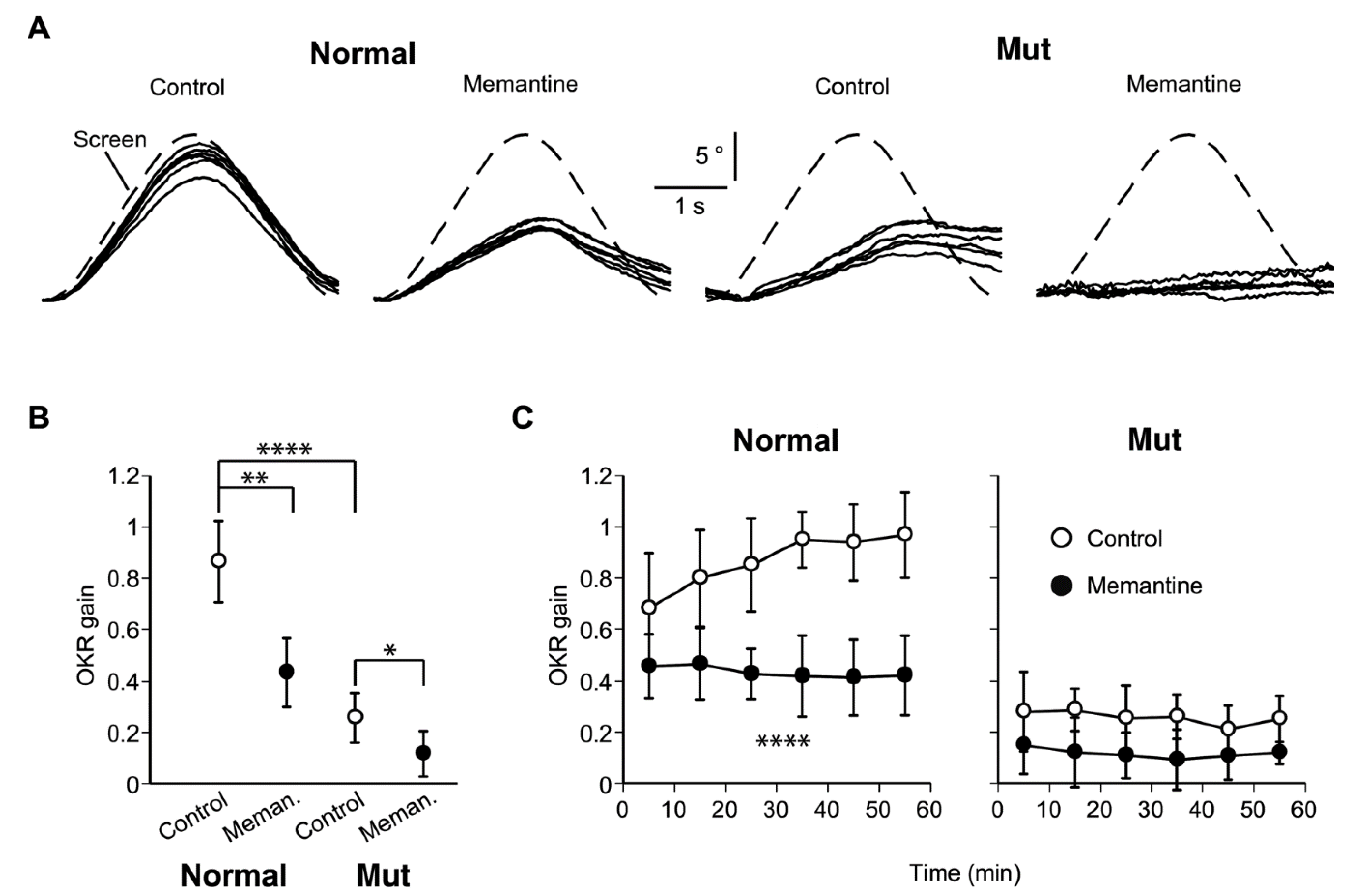
2.1. The Hereditary Ataxic Mouse is Sensitive to Memantine
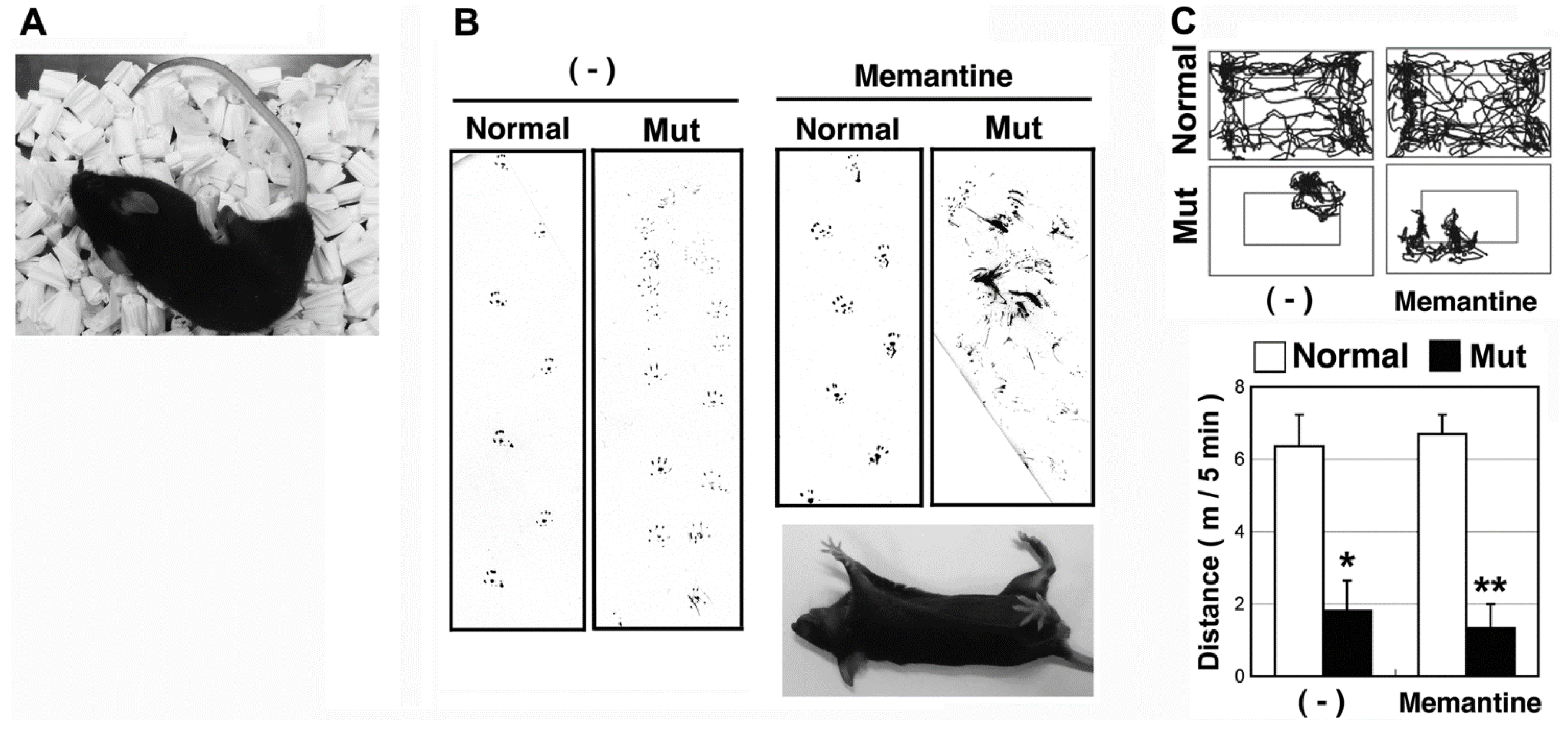

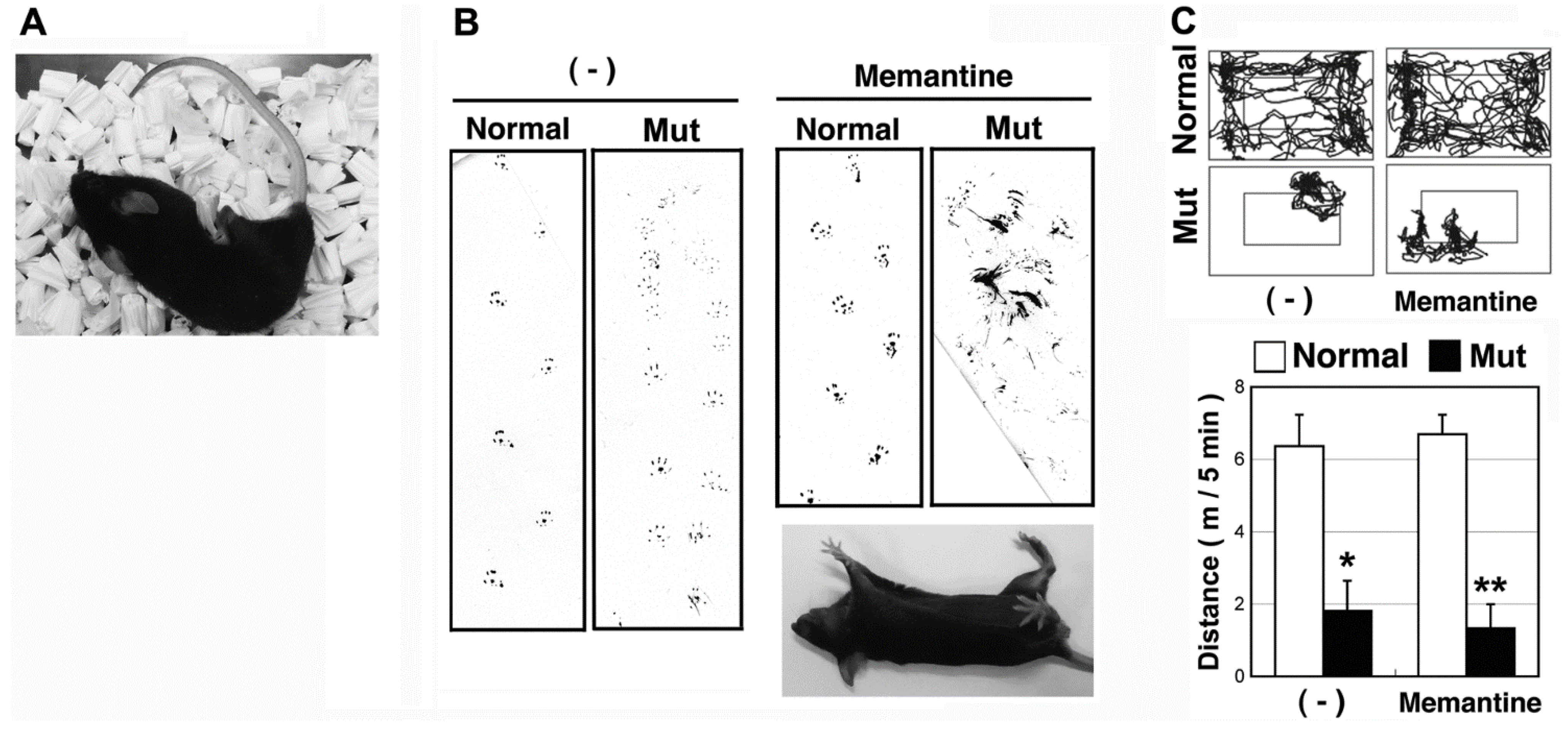{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical | Category | Dose (mg/kg) | Number of Mice | Movement of Normal Mice | Movement of Mutant Mice |
|---|---|---|---|---|---|
| Memantine | NMDA-R antagonist | 2 10 | 2 2 | NS NS | NS Balance loss |
| MK-801 | NMDA-R antagonist | 2 10 | 2 3 | NS Balance loss | NS Balance loss |
| DL-AP7 | NMDA-R antagonist | 30 | 2 | NS | Slow movement |
| (R)-CPP | NMDA-R antagonist (NR2A antagonist) | 10 20 | 4 2 | NS NS | Balance loss * Balance loss |
| Ro25-6981 | NMDA-R antagonist | 2 10 | 3 3 | NS NS | NS NS |
| Ifenprodil | NMDA-R antagonist (NR2B antagonist) | 2 10 | 3 3 | NS NS | NS NS |
| Felbamate | NR2B antagonist GABA-R activator | 30 | 2 | NS | NS |
| Nitrazepam | GABA-R activator | 30 | 2 | NS | NS |
| Isoflurane | GABA-R activator | 1% in air | 2 | Slip | Slip |
| DNQX | AMPA/Kainate receptor antagonist | 2 10 | 3 3 | NS NS | NS NS |
| Ondansetron | 5-HT3 antagonist | 5 10 | 2 2 | NS Not move | NS Not move |
| Donepezil | acetylcholinesterase inhibitor | 10 | 3 | NS | NS |
| l-Dopa | Dopamine precursor | 20 | 3 | NS | NS |
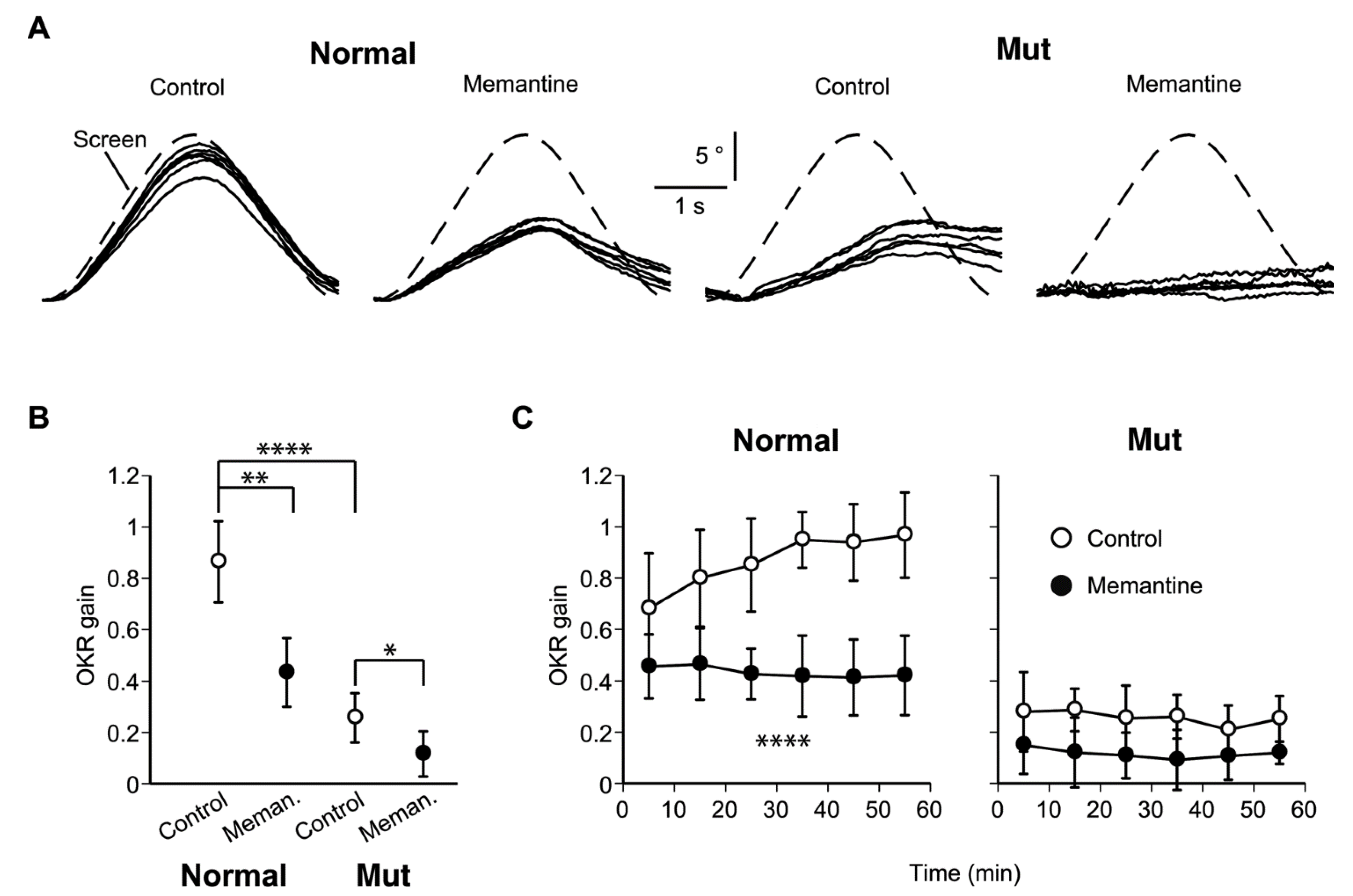
2.2. Impaired OKR and Learning in the Ataxic Mice
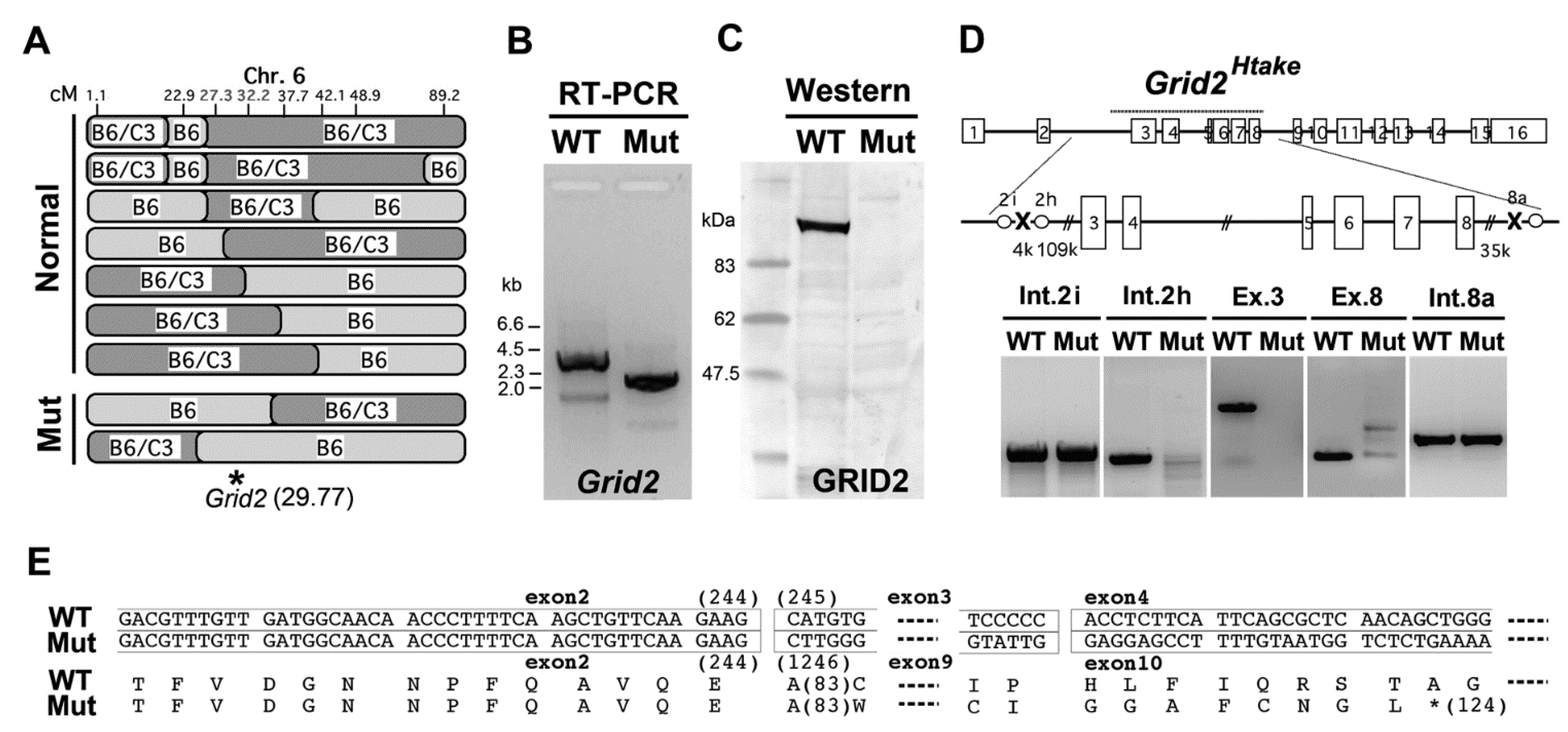
2.3. Microsatellite Analysis in the Ataxic Mice
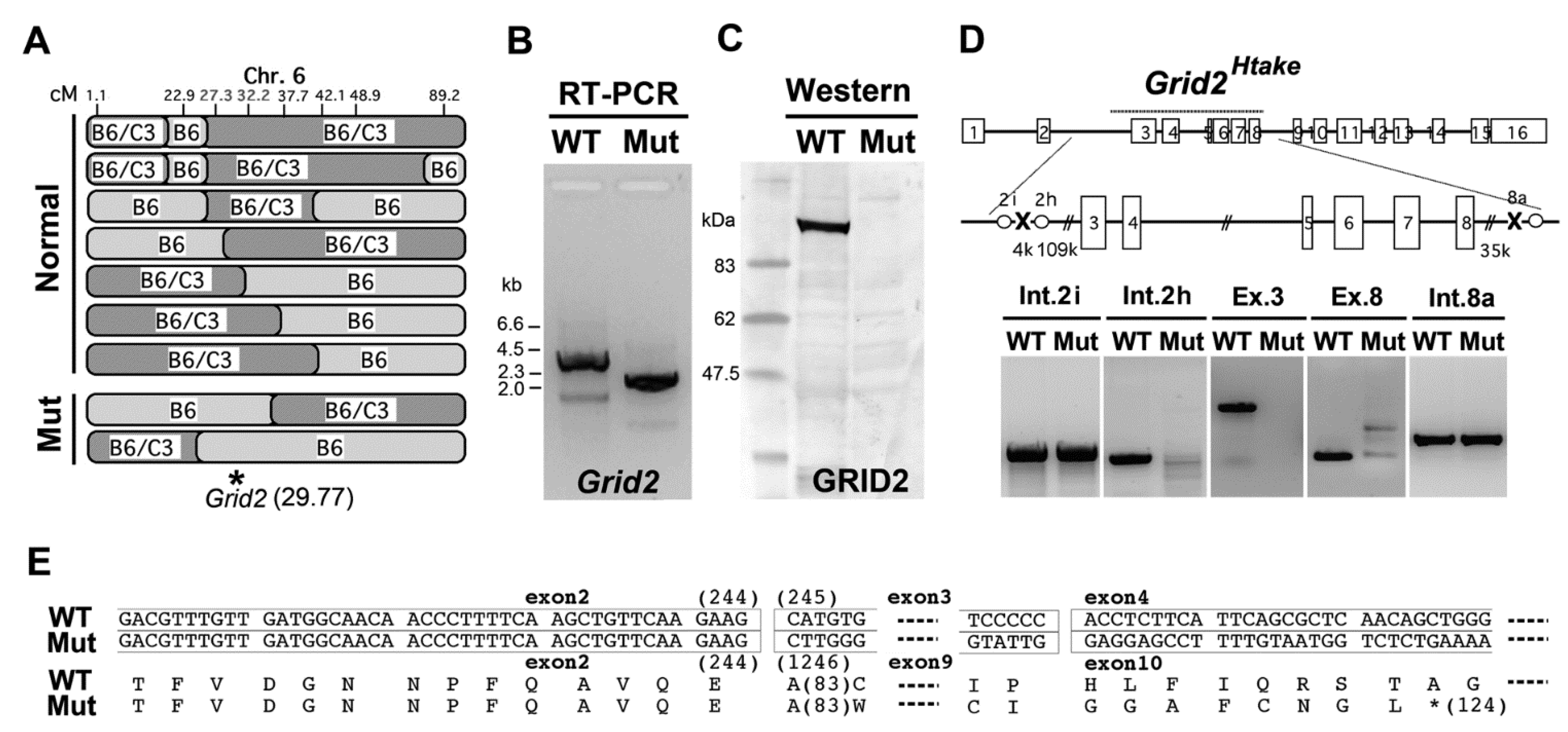
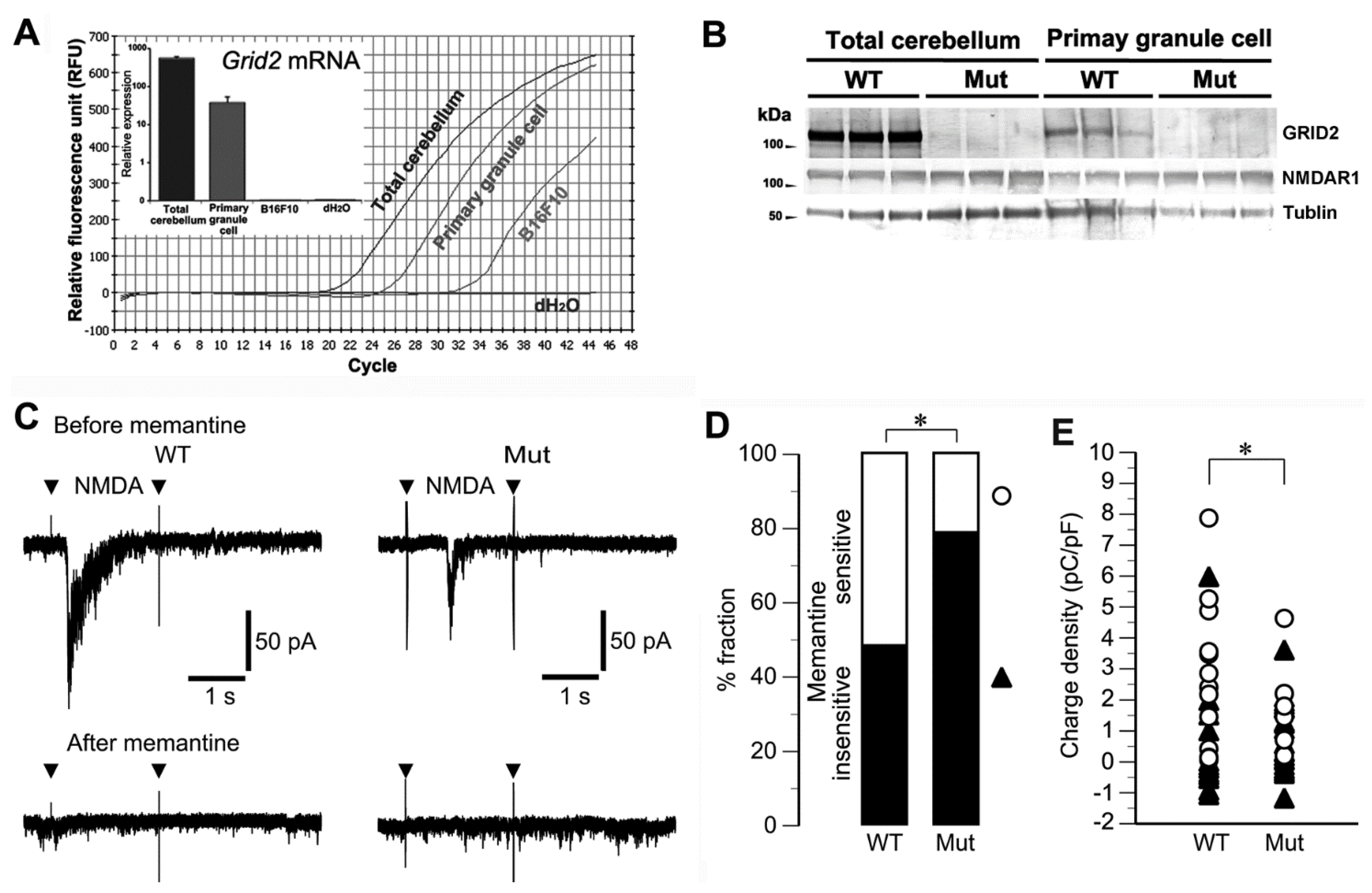
2.4. Altered Sensitivity to NMDA in Cultured Granule Cells of Grid2Htake/Htake Mice
2.5. Mice Treated with Memantine and AMPA Were Unable to Walk Smoothly
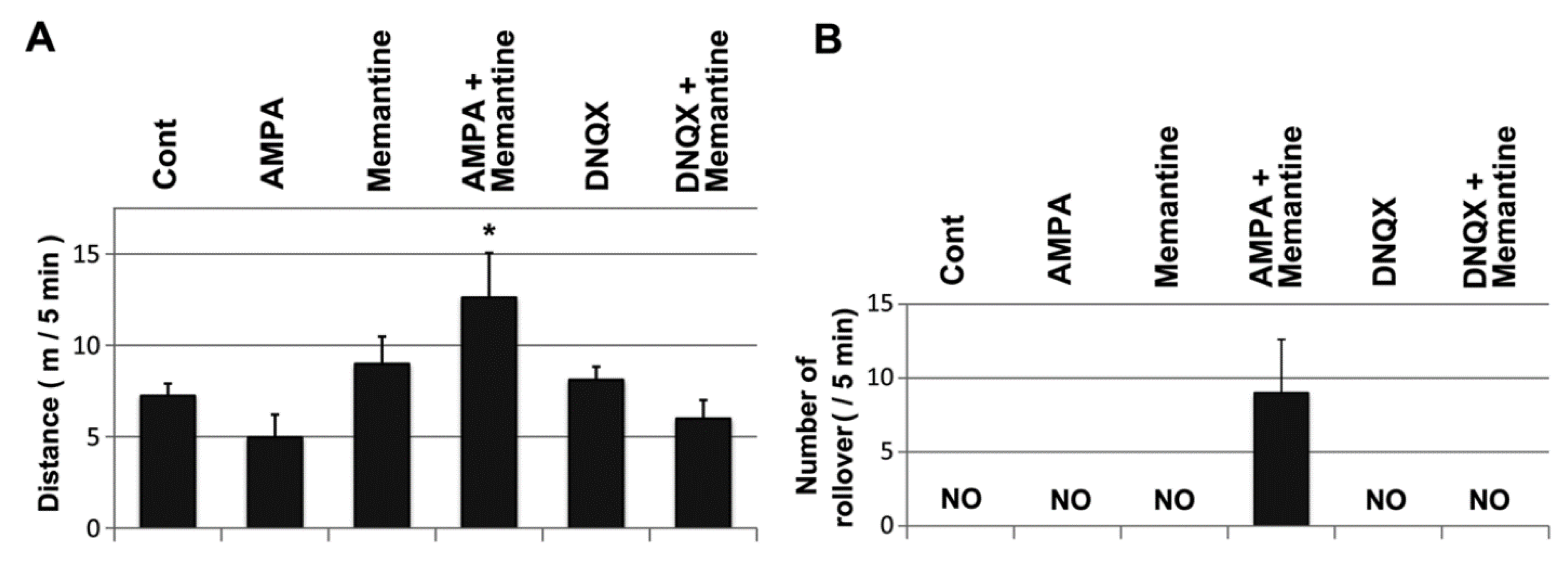
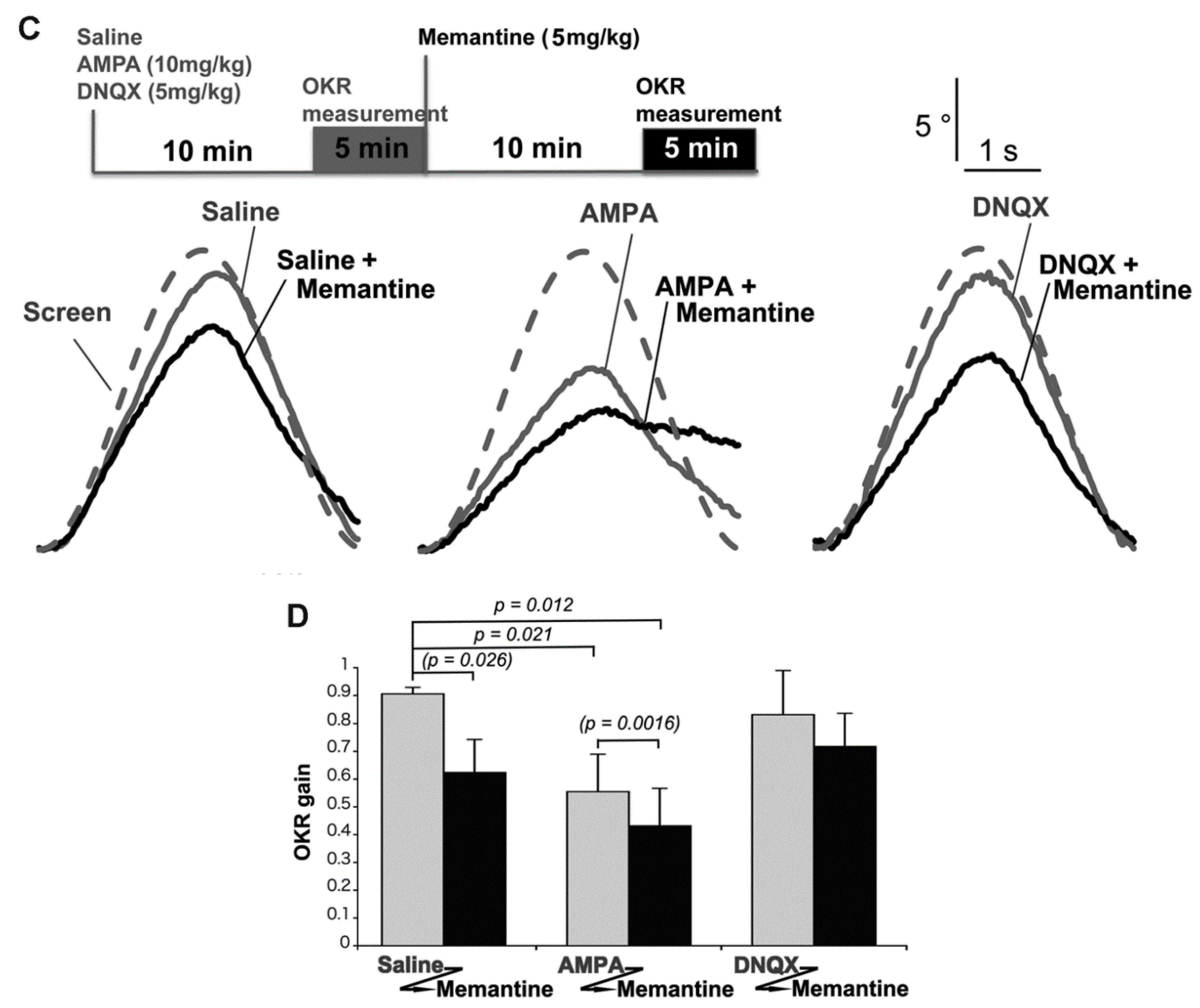
3. Discussion
4. Experimental Section
4.1. Animals
4.2. Reagents
4.3. mRNA Analyses
4.4. Measurements of Walking Distance and the OKR
4.5. Cell Culture and Electrophysiology
4.6. Statistical Analyses
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sonkusare, S.K.; Kaul, C.L.; Ramarao, P. Dementia of Alzheimer’s disease and other neurodegenerative disorders—Memantine, a new hope. Pharmacol. Res. 2005, 51, 1–17. [Google Scholar] [CrossRef]
- Lipton, S.A. Paradigm shift in neuroprotection by NMDA receptor blockade: Memantine and beyond. Nat. Rev. Drug Discov. 2006, 5, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Szczurowska, E.; Mares, P. NMDA and AMPA receptors: Development and status epilepticus. Physiol. Res. 2013, 62 (Suppl. 1), S21–S38. [Google Scholar] [PubMed]
- Kupper, J.; Ascher, P.; Neyton, J. Probing the pore region of recombinant N-methyl-d-aspartate channels using external and internal magnesium block. Proc. Natl. Acad. Sci. USA 1996, 93, 8648–8653. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.H.; Kemp, J.A.; Priestley, T.; Knight, A.R.; Woodruff, G.N.; Iversen, L.L. The anticonvulsant MK-801 is a potent N-methyl-d-aspartate antagonist. Proc. Natl. Acad. Sci. USA 1986, 83, 7104–7108. [Google Scholar] [CrossRef] [PubMed]
- Hansen, R.A.; Gartlehner, G.; Lohr, K.N.; Kaufer, D.I. Functional outcomes of drug treatment in Alzheimer’s disease: A systematic review and meta-analysis. Drugs Aging 2007, 24, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Uemura, T.; Lee, S.J.; Yasumura, M.; Takeuchi, T.; Yoshida, T.; Ra, M.; Taguchi, R.; Sakimura, K.; Mishina, M. Trans-synaptic interaction of GluRdelta2 and Neurexin through Cbln1 mediates synapse formation in the cerebellum. Cell 2010, 141, 1068–1079. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, K.; Miura, E.; Miyazaki, T.; Kakegawa, W.; Emi, K.; Narumi, S.; Fukazawa, Y.; Ito-Ishida, A.; Kondo, T.; Shigemoto, R.; et al. Cbln1 is a ligand for an orphan glutamate receptor delta2, a bidirectional synapse organizer. Science 2010, 328, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Kakegawa, W.; Miyoshi, Y.; Hamase, K.; Matsuda, S.; Matsuda, K.; Kohda, K.; Emi, K.; Motohashi, J.; Konno, R.; Zaitsu, K.; et al. d-Serine regulates cerebellar LTD and motor coordination through the delta2 glutamate receptor. Nat. Neurosci. 2011, 14, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Jardon, B.; Bonaventure, N. N-Methyl-d-aspartate antagonists suppress the development of frog symmetric monocular optokynetic nystagmus observed after unilateral visual deprivation. Brain Res. Dev. Brain Res. 1992, 67, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Godaux, E.; Cheron, G.; Mettens, P. Ketamine induces failure of the oculomotor neural integrator in the cat. Neurosci. Lett. 1990, 116, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Mettens, P.; Cheron, G.; Godaux, E. NMDA receptors are involved in temporal integration in the oculomotor system of the cat. Neuroreport 1994, 5, 1333–1336. [Google Scholar] [PubMed]
- Huang, Y.J.; Lin, C.H.; Lane, H.Y.; Tsai, G.E. NMDA Neurotransmission Dysfunction in Behavioral and Psychological Symptoms of Alzheimer’s Disease. Curr. Neuropharmacol. 2012, 10, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Puangthong, U.; Hsiung, G.Y. Critical appraisal of the long-term impact of memantine in treatment of moderate to severe Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2009, 5, 553–561. [Google Scholar] [PubMed]
- Utine, G.E.; Haliloglu, G.; Salanci, B.; Cetinkaya, A.; Kiper, P.O.; Alanay, Y.; Aktas, D.; Boduroglu, K.; Alikasifoglu, M. A Homozygous Deletion in GRID2 Causes a Human Phenotype With Cerebellar Ataxia and Atrophy. J. Child Neurol. 2013, 28, 926–932. [Google Scholar] [CrossRef] [PubMed]
- Maier, A.; Klopocki, E.; Horn, D.; Tzschach, A.; Holm, T.; Meyer, R.; Meyer, T. De novo partial deletion in GRID2 presenting with complicated spastic paraplegia. Muscle Nerve 2014, 49, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Hills, L.B.; Masri, A.; Konno, K.; Kakegawa, W.; Lam, A.T.; Lim-Melia, E.; Chandy, N.; Hill, R.S.; Partlow, J.N.; Al-Saffar, M.; et al. Deletions in GRID2 lead to a recessive syndrome of cerebellar ataxia and tonic upgaze in humans. Neurology 2013, 81, 1378–1386. [Google Scholar] [CrossRef] [PubMed]
- Van Schil, K.; Meire, F.; Karlstetter, M.; Bauwens, M.; Verdin, H.; Coppieters, F.; Scheiffert, E.; van Nechel, C.; Langmann, T.; Deconinck, N.; et al. Early-onset autosomal recessive cerebellar ataxia associated with retinal dystrophy: New human hotfoot phenotype caused by homozygous GRID2 deletion. Genet. Med. 2014. [Google Scholar] [CrossRef]
- Uebi, T.; Itoh, Y.; Hatano, O.; Kumagai, A.; Sanosaka, M.; Sasaki, T.; Sasagawa, S.; Doi, J.; Tatsumi, K.; Mitamura, K.; et al. Involvement of SIK3 in glucose and lipid homeostasis in mice. PLOS ONE 2012, 7, e37803. [Google Scholar] [CrossRef] [PubMed]
- Hemmings, H.C., Jr.; Yan, W.; Westphalen, R.I.; Ryan, T.A. The general anesthetic isoflurane depresses synaptic vesicle exocytosis. Mol. Pharmacol. 2005, 67, 1591–1599. [Google Scholar] [CrossRef] [PubMed]
- Rammes, G.; Danysz, W.; Parsons, C.G. Pharmacodynamics of memantine: An update. Curr. Neuropharmacol. 2008, 6, 55–78. [Google Scholar] [CrossRef] [PubMed]
- Seeman, P.; Caruso, C.; Lasaga, M. Memantine agonist action at dopamine D2High receptors. Synapse 2008, 62, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Park, H.J.; Choi, J.; Park, S.J.; Kang, H.; Kim, E.G. Comparison of ramosetron’s and ondansetron’s preventive anti-emetic effects in highly susceptible patients undergoing abdominal hysterectomy. Korean J. Anesthesiol. 2011, 61, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Khojasteh, A.; Sartiano, G.; Tapazoglou, E.; Lester, E.; Gandara, D.; Bernard, S.; Finn, A. Ondansetron for the prevention of emesis induced by high-dose cisplatin. A multi-center dose-response study. Cancer 1990, 66, 1101–1105. [Google Scholar] [CrossRef] [PubMed]
- Jayadev, S.; Bird, T.D. Hereditary ataxias: Overview. Genet. Med. 2013, 15, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Requena, T.; Espinosa-Sanchez, J.M.; Lopez-Escamez, J.A. Genetics of dizziness: Cerebellar and vestibular disorders. Curr. Opin. Neurol. 2014, 27, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Rosini, F.; Federighi, P.; Pretegiani, E.; Piu, P.; Leigh, R.J.; Serra, A.; Federico, A.; Rufa, A. Ocular-motor profile and effects of memantine in a familial form of adult cerebellar ataxia with slow saccades and square wave saccadic intrusions. PLOS ONE 2013, 8, e69522. [Google Scholar] [CrossRef] [PubMed]
- Shirai, Y.; Asano, K.; Takegoshi, Y.; Uchiyama, S.; Nonobe, Y.; Tabata, T. A simple machine vision-driven system for measuring optokinetic reflex in small animals. J. Physiol. Sci. 2013, 63, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Shutoh, F.; Ohki, M.; Kitazawa, H.; Itohara, S.; Nagao, S. Memory trace of motor learning shifts transsynaptically from cerebellar cortex to nuclei for consolidation. Neuroscience 2006, 139, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Zuo, J.; de Jager, P.L.; Takahashi, K.A.; Jiang, W.; Linden, D.J.; Heintz, N. Neurodegeneration in Lurcher mice caused by mutation in delta2 glutamate receptor gene. Nature 1997, 388, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Matsuda, S.; Drews, V.; Torashima, T.; Meisler, M.H.; Yuzaki, M. A hot spot for hotfoot mutations in the gene encoding the delta2 glutamate receptor. Eur. J. Neurosci. 2003, 17, 1581–1590. [Google Scholar] [CrossRef] [PubMed]
- Cull-Candy, S.G.; Wyllie, D.J. Glutamate-receptor channels in mammalian glial cells. Ann. N. Y. Acad. Sci. 1991, 633, 458–474. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.S.; Knierman, M.D.; Siuda, E.R.; Isaac, J.T.; Nisenbaum, E.S.; Bredt, D.S. Glutamate receptor delta2 associates with metabotropic glutamate receptor 1 (mGluR1), protein kinase Cgamma, and canonical transient receptor potential 3 and regulates mGluR1-mediated synaptic transmission in cerebellar Purkinje neurons. J. Neurosci. 2012, 32, 15296–15308. [Google Scholar] [CrossRef] [PubMed]
- Hirai, H.; Launey, T.; Mikawa, S.; Torashima, T.; Yanagihara, D.; Kasaura, T.; Miyamoto, A.; Yuzaki, M. New role of delta2-glutamate receptors in AMPA receptor trafficking and cerebellar function. Nat. Neurosci. 2003, 6, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, M.; Miyazaki, T.; Azechi, H.; Abe, M.; Natsume, R.; Hagiwara, T.; Aiba, A.; Mishina, M.; Sakimura, K.; Watanabe, M. Glutamate receptor delta2 is essential for input pathway-dependent regulation of synaptic AMPAR contents in cerebellar Purkinje cells. J. Neurosci. 2011, 31, 3362–3374. [Google Scholar] [CrossRef] [PubMed]
- Honore, T.; Davies, S.N.; Drejer, J.; Fletcher, E.J.; Jacobsen, P.; Lodge, D.; Nielsen, F.E. Quinoxalinediones: Potent competitive non-NMDA glutamate receptor antagonists. Science 1988, 241, 701–703. [Google Scholar] [CrossRef] [PubMed]
- Kakegawa, W.; Miyazaki, T.; Kohda, K.; Matsuda, K.; Emi, K.; Motohashi, J.; Watanabe, M.; Yuzaki, M. The N-terminal domain of GluD2 (GluRdelta2) recruits presynaptic terminals and regulates synaptogenesis in the cerebellum in vivo. J. Neurosci. 2009, 29, 5738–5748. [Google Scholar] [CrossRef] [PubMed]
- Rabacchi, S.; Bailly, Y.; Delhaye-Bouchaud, N.; Mariani, J. Involvement of the N-methyl d-aspartate (NMDA) receptor in synapse elimination during cerebellar development. Science 1992, 256, 1823–1825. [Google Scholar] [CrossRef] [PubMed]
- Garthwaite, J.; Brodbelt, A.R. Synaptic activation of N-methyl-d-aspartate and non-N-methyl-d-aspartate receptors in the mossy fibre pathway in adult and immature rat cerebellar slices. Neuroscience 1989, 29, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Yuzaki, M.; Forrest, D.; Verselis, L.M.; Sun, S.C.; Curran, T.; Connor, J.A. Functional NMDA receptors are transiently active and support the survival of Purkinje cells in culture. J. Neurosci. 1996, 16, 4651–4661. [Google Scholar] [PubMed]
- Balazs, R.; Jorgensen, O.S.; Hack, N. N-Methyl-d-aspartate promotes the survival of cerebellar granule cells in culture. Neuroscience 1988, 27, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Ortega, F.; Perez-Sen, R.; Morente, V.; Delicado, E.G.; Miras-Portugal, M.T. P2X7, NMDA and BDNF receptors converge on GSK3 phosphorylation and cooperate to promote survival in cerebellar granule neurons. Cell. Mol. Life Sci. 2010, 67, 1723–1733. [Google Scholar] [CrossRef] [PubMed]
- Kadotani, H.; Hirano, T.; Masugi, M.; Nakamura, K.; Nakao, K.; Katsuki, M.; Nakanishi, S. Motor discoordination results from combined gene disruption of the NMDA receptor NR2A and NR2C subunits, but not from single disruption of the NR2A or NR2C subunit. J. Neurosci. 1996, 16, 7859–7867. [Google Scholar] [PubMed]
- Marmolino, D.; Manto, M. Past, present and future therapeutics for cerebellar ataxias. Curr. Neuropharmacol. 2010, 8, 41–61. [Google Scholar] [CrossRef] [PubMed]
- Bormann, J. Memantine is a potent blocker of N-methyl-d-aspartate (NMDA) receptor channels. Eur. J. Pharmacol. 1989, 166, 591–592. [Google Scholar] [CrossRef] [PubMed]
- Strupp, M.; Brandt, T. Current treatment of vestibular, ocular motor disorders and nystagmus. Ther. Adv. Neurol. Disord. 2009, 2, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Katoh, A.; Ohtsuki, G.; Mishina, M.; Hirano, T. Oscillating Purkinje neuron activity causing involuntary eye movement in a mutant mouse deficient in the glutamate receptor delta2 subunit. J. Neurosci. 2004, 24, 2440–2448. [Google Scholar] [CrossRef] [PubMed]
- Faulstich, M.; van Alphen, A.M.; Luo, C.; du Lac, S.; De Zeeuw, C.I. Oculomotor plasticity during vestibular compensation does not depend on cerebellar LTD. J. Neurophysiol. 2006, 96, 1187–1195. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.W.; Uehlinger, J.; Dayani, N.; Talansky, B.E.; Gordon, M.; Rudomen, G.S.; Neumann, P.E. Analysis of the hotfoot (ho) locus by creation of an insertional mutation in a transgenic mouse. Dev. Biol. 1990, 137, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Kashiwabuchi, N.; Ikeda, K.; Araki, K.; Hirano, T.; Shibuki, K.; Takayama, C.; Inoue, Y.; Kutsuwada, T.; Yagi, T.; Kang, Y.; et al. Impairment of motor coordination, Purkinje cell synapse formation, and cerebellar long-term depression in GluR delta 2 mutant mice. Cell 1995, 81, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.B. Brain abnormalities in the lurcher (Lc) mutant mouse. Experientia 1975, 31, 220–221. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, J.; Matsuda, K.; Kakegawa, W.; Yamada, N.; Motohashi, J.; Mizushima, N.; Yuzaki, M. Reevaluation of neurodegeneration in lurcher mice: Constitutive ion fluxes cause cell death with, not by, autophagy. J. Neurosci. 2010, 30, 2177–2187. [Google Scholar] [CrossRef] [PubMed]
- Cendelin, J.; Tuma, J.; Korelusova, I.; Vozeh, F. The effect of genetic background on behavioral manifestation of Grid2(Lc) mutation. Behav. Brain Res. 2014, 271, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, E.J.; Rothman, J.S.; Dugue, G.P.; Diana, M.; Rousseau, C.; Silver, R.A.; Dieudonne, S. NMDA receptors with incomplete Mg(2)(+) block enable low-frequency transmission through the cerebellar cortex. J. Neurosci. 2012, 32, 6878–6893. [Google Scholar] [CrossRef] [PubMed]
- Hepp, R.; Hay, Y.A.; Aguado, C.; Lujan, R.; Dauphinot, L.; Potier, M.C.; Nomura, S.; Poirel, O.; El Mestikawy, S.; Lambolez, B.; et al. Glutamate receptors of the delta family are widely expressed in the adult brain. Brain Struct. Funct. 2014. [Google Scholar] [CrossRef]
- Glitsch, M.D. Calcium influx through N-methyl-d-aspartate receptors triggers GABA release at interneuron-Purkinje cell synapse in rat cerebellum. Neuroscience 2008, 151, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.J. Biphasic modulation of GABA release from stellate cells by glutamatergic receptor subtypes. J. Neurophysiol. 2007, 98, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Duguid, I.C.; Smart, T.G. Retrograde activation of presynaptic NMDA receptors enhances GABA release at cerebellar interneuron-Purkinje cell synapses. Nat. Neurosci. 2004, 7, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Watanabe, D.; Kawaguchi, S.Y.; Pastan, I.; Nakanishi, S. Roles of inhibitory interneurons in the cerebellar cortex. Ann. N. Y. Acad. Sci. 2002, 978, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Whittingham, D.G. Fertilization of mouse eggs in vitro. Nature 1968, 220, 592–593. [Google Scholar] [CrossRef] [PubMed]
- Tabata, T.; Sawada, S.; Araki, K.; Bono, Y.; Furuya, S.; Kano, M. A reliable method for culture of dissociated mouse cerebellar cells enriched for Purkinje neurons. J. Neurosci. Methods 2000, 104, 45–53. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumagai, A.; Fujita, A.; Yokoyama, T.; Nonobe, Y.; Hasaba, Y.; Sasaki, T.; Itoh, Y.; Koura, M.; Suzuki, O.; Adachi, S.; et al. Altered Actions of Memantine and NMDA-Induced Currents in a New Grid2-Deleted Mouse Line. Genes 2014, 5, 1095-1114. https://doi.org/10.3390/genes5041095
Kumagai A, Fujita A, Yokoyama T, Nonobe Y, Hasaba Y, Sasaki T, Itoh Y, Koura M, Suzuki O, Adachi S, et al. Altered Actions of Memantine and NMDA-Induced Currents in a New Grid2-Deleted Mouse Line. Genes. 2014; 5(4):1095-1114. https://doi.org/10.3390/genes5041095
Chicago/Turabian StyleKumagai, Ayako, Akira Fujita, Tomoki Yokoyama, Yuki Nonobe, Yasuhiro Hasaba, Tsutomu Sasaki, Yumi Itoh, Minako Koura, Osamu Suzuki, Shigeki Adachi, and et al. 2014. "Altered Actions of Memantine and NMDA-Induced Currents in a New Grid2-Deleted Mouse Line" Genes 5, no. 4: 1095-1114. https://doi.org/10.3390/genes5041095
APA StyleKumagai, A., Fujita, A., Yokoyama, T., Nonobe, Y., Hasaba, Y., Sasaki, T., Itoh, Y., Koura, M., Suzuki, O., Adachi, S., Ryo, H., Kohara, A., Tripathi, L. P., Sanosaka, M., Fukushima, T., Takahashi, H., Kitagawa, K., Nagaoka, Y., Kawahara, H., ... Takemori, H. (2014). Altered Actions of Memantine and NMDA-Induced Currents in a New Grid2-Deleted Mouse Line. Genes, 5(4), 1095-1114. https://doi.org/10.3390/genes5041095