Enterococcus faecalis Infection and Reactive Oxygen Species Down-Regulates the miR-17-92 Cluster in Gastric Adenocarcinoma Cell Culture

Abstract
:1. Introduction
2. Experimental
2.1. Cell Culture, E. faecalis, and Growth Conditions
2.2. Infection of Gastric Cells
2.3. Tert-Butyl Hydroperoxide Stimulation
2.4. RNA Isolation
2.5. Microarray
2.6. Quantitative Reverse Transcription PCR
2.7. Statistical Analysis
3. Results and Discussion
3.1. Living E. faecalis Affect Global miRNA Expression
3.2. E. faecalis Infection Alters the Expression of the miR-17-92 Cluster, miR-106-363 Cluster, and Other ROS Responsive miRNAs
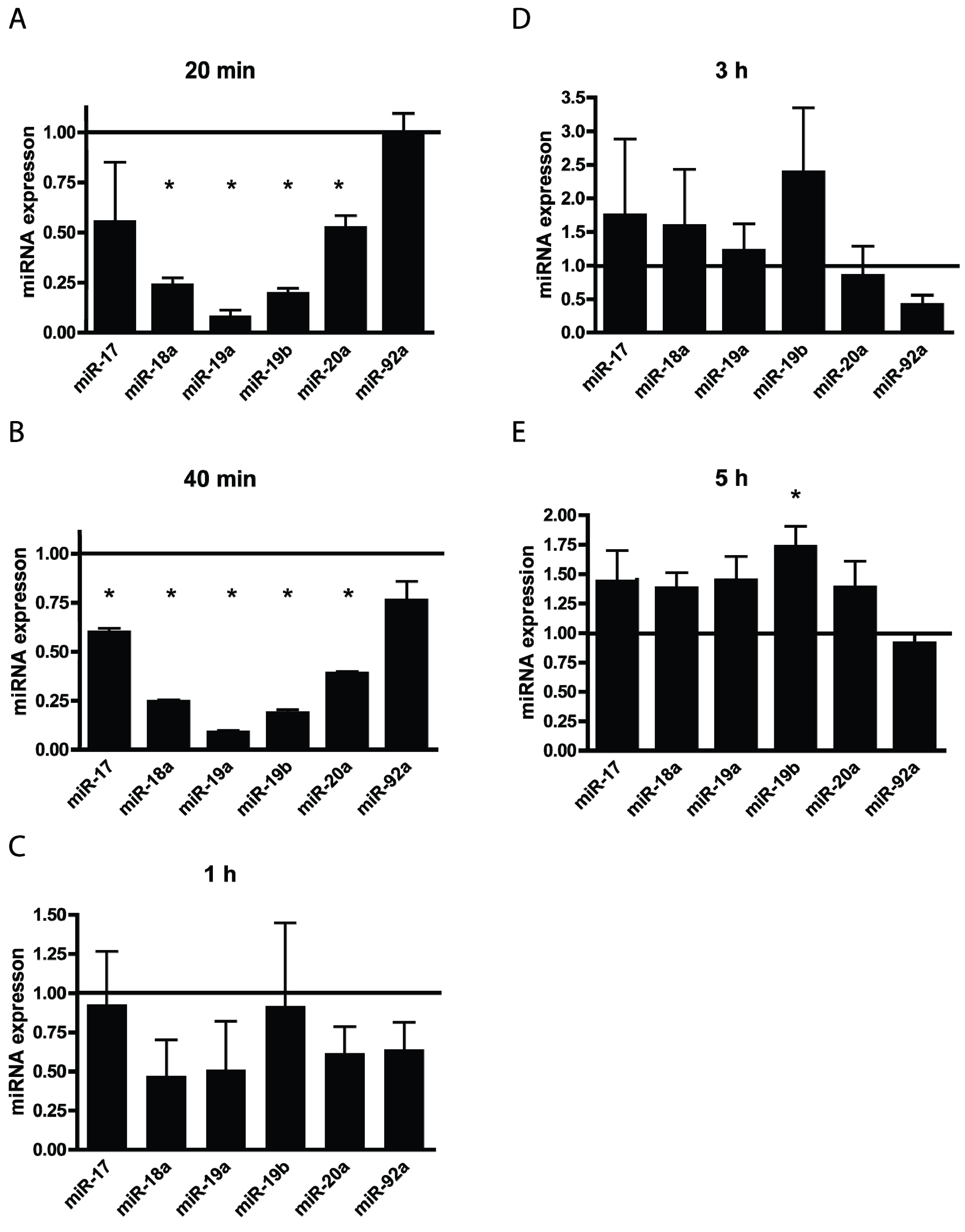
3.3. TBHP Reduce the Expression of miRNAs from the miR-17-92 Cluster in a Time Dependent Manner

{kind=link}
{kind=link}
| miRNA | Fold Change | Difference of Arbitrary Expression | p-Value | Corresponding miRNA Cluster |
|---|---|---|---|---|
| hsa-miR-17_st | −1.8 | 630.9 | 0.0001 | miR-17-92 cluster |
| hsa-miR-18a_st | −9.9 | 245.7 | >0.0001 | |
| hsa-miR-20a_st | −4.5 | 526.9 | >0.0001 | |
| hsa-miR-19b_st | −4.1 | 271.2 | 0.0002 | miR-17-92 and miR-106-363 cluster |
| hsa-miR-92a_st | 1.4 | −563.5 | 0.0023 | |
| hsa-miR-18b_st | −15.8 | 33.4 | 0.0001 | miR-106-363 cluster |
| hsa-miR-20b_st | −8.3 | 92.2 | >0.0001 | |
| hsa-miR-106a_st | −1.9 | 580.9 | >0.0001 | |
| hsa-miR-24-2-star_st | −3.6 | 53.4 | 0.0011 | ROS responsive miRs |
| hsa-miR-27a-star_st | −14.7 | 54.0 | 0.0001 |
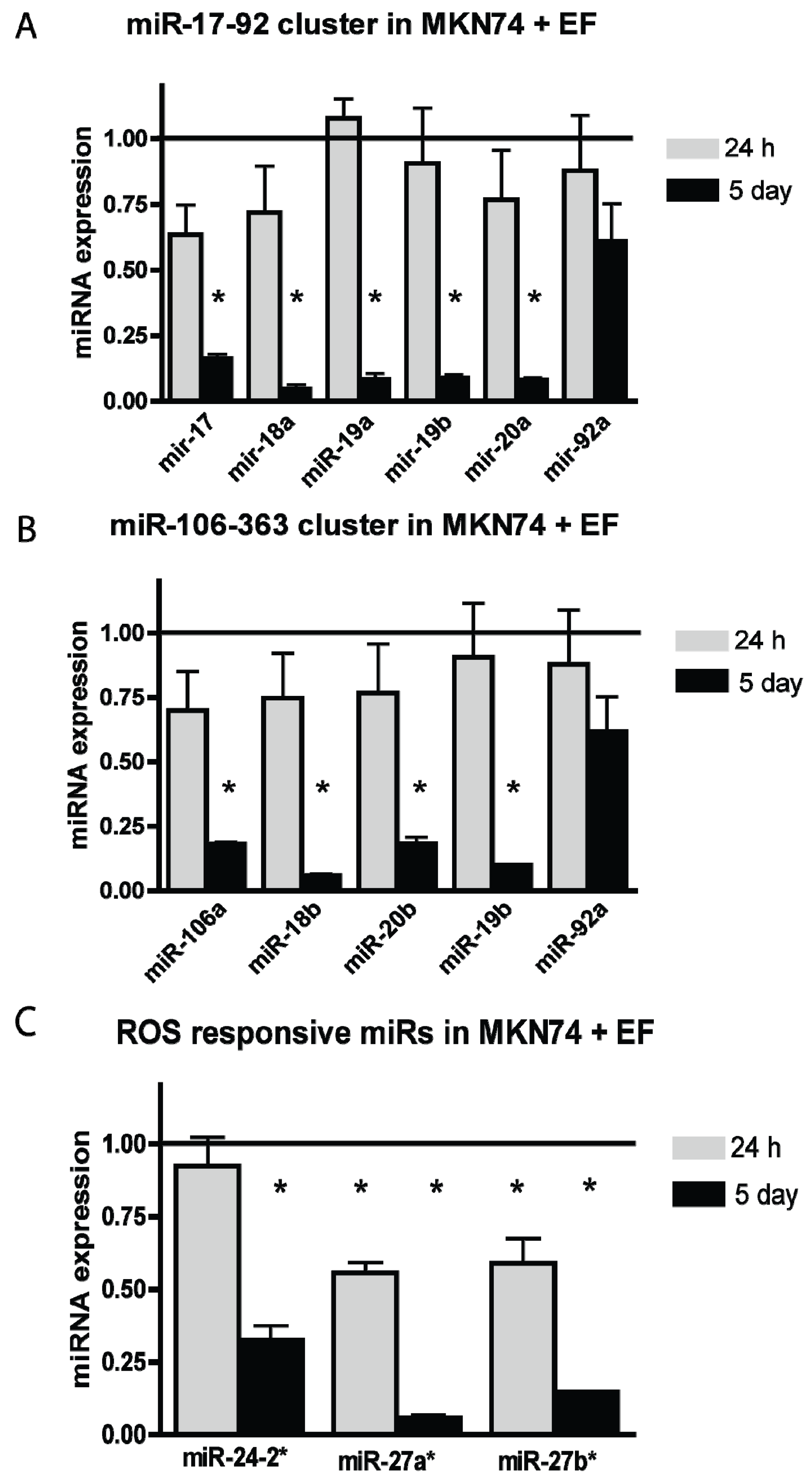
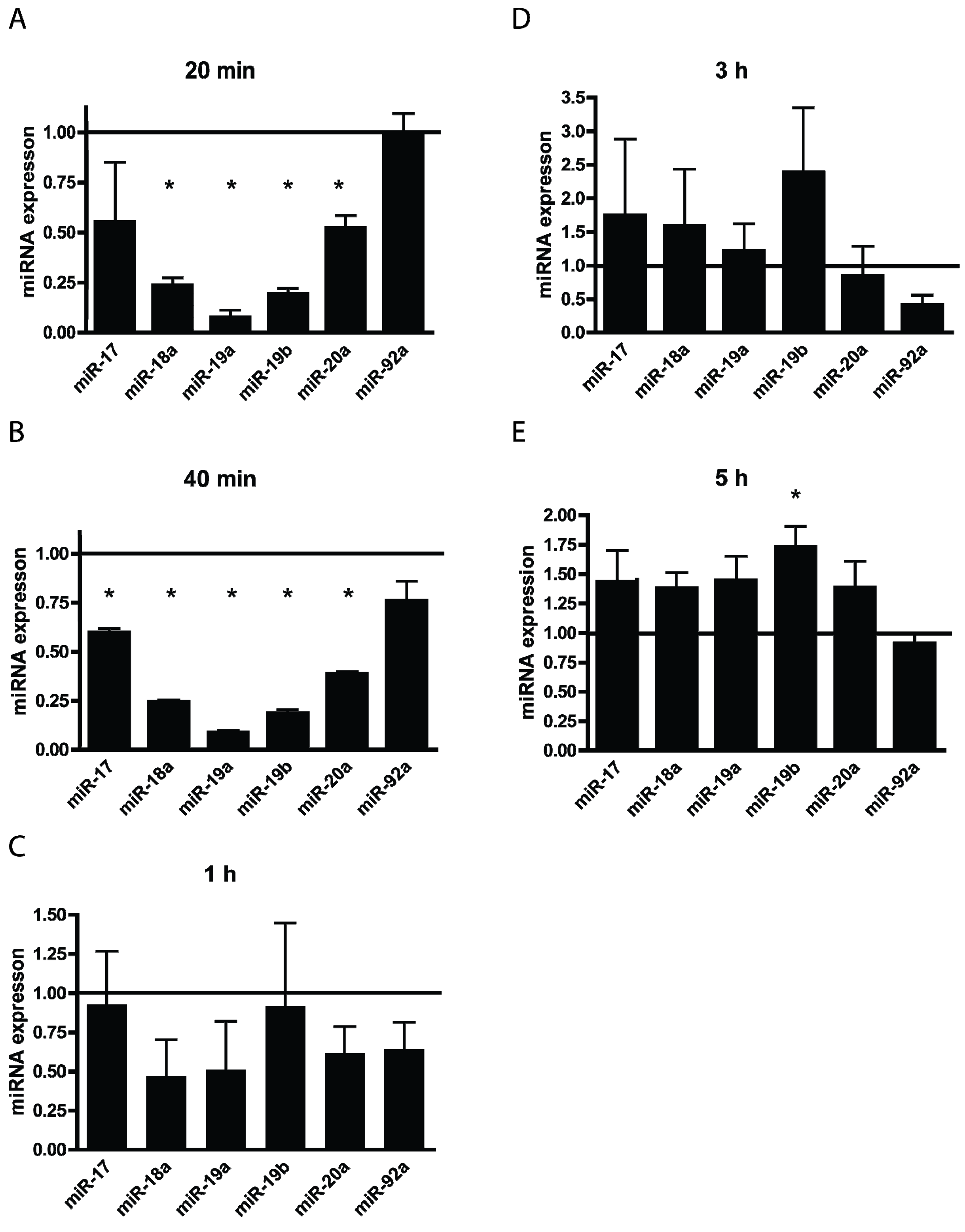
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ferlay, J.; Parkin, D.M.; Steliarova-Foucher, E. Estimates of cancer incidence and mortality in europe in 2008. Eur. J. Cancer 2010, 46, 765–781. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.P.; Harris, C.C. Inflammation and cancer: An ancient link with novel potentials. Int. J. Cancer 2007, 121, 2373–2380. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.Q.; Sridhar, S.; Chen, Y.; Hunt, R.H. Meta-analysis of the relationship between Helicobacter pylori seropositivity and gastric cancer. Gastroenterology 1998, 114, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Amieva, M.R.; El-Omar, E.M. Host-bacterial interactions in Helicobacter pylori infection. Gastroenterology 2008, 134, 306–323. [Google Scholar] [CrossRef] [PubMed]
- Dicksved, J.; Lindberg, M.; Rosenquist, M.; Enroth, H.; Jansson, J.K.; Engstrand, L. Molecular characterization of the stomach microbiota in patients with gastric cancer and in controls. J. Med. Microbiol. 2009, 58, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Fisher, K.; Phillips, C. The ecology, epidemiology and virulence of enterococcus. Microbiology 2009, 155, 1749–1757. [Google Scholar] [CrossRef] [PubMed]
- Friis-Hansen, L.; Rieneck, K.; Nilsson, H.O.; Wadstrom, T.; Rehfeld, J.F. Gastric inflammation, metaplasia, and tumor development in gastrin-deficient mice. Gastroenterology 2006, 131, 246–258. [Google Scholar]
- Wang, X.; Allen, T.D.; May, R.J.; Lightfoot, S.; Houchen, C.W.; Huycke, M.M. Enterococcus faecalis induces aneuploidy and tetraploidy in colonic epithelial cells through a bystander effect. Cancer Res. 2008, 68, 9909–9917. [Google Scholar] [CrossRef] [PubMed]
- Huycke, M.M.; Joyce, W.; Wack, M.F. Augmented production of extracellular superoxide by blood isolates of Enterococcus faecalis. J. Infect. Dis. 1996, 173, 743–746. [Google Scholar] [CrossRef] [PubMed]
- Strickertsson, J.A.; Desler, C.; Martin-Bertelsen, T.; Machado, A.M.; Wadstrom, T.; Winther, O.; Rasmussen, L.J.; Friis-Hansen, L. Enterococcus faecalis infection causes inflammation, intracellular oxphos-independent ros production, and DNA damage in human gastric cancer cells. PLoS One 2013, 8, e63147. [Google Scholar] [CrossRef] [PubMed]
- Waris, G.; Ahsan, H. Reactive oxygen species: Role in the development of cancer and various chronic conditions. J. Carcinog. 2006, 5. [Google Scholar] [CrossRef]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic Res 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Ziech, D.; Franco, R.; Pappa, A.; Panayiotidis, M.I. Reactive oxygen species (ROS)—Induced genetic and epigenetic alterations in human carcinogenesis. Mutat Res. 2011, 711, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Kantner, H.P.; Warsch, W.; Delogu, A.; Bauer, E.; Esterbauer, H.; Casanova, E.; Sexl, V.; Stoiber, D. ETV6/RUNX1 induces reactive oxygen species and drives the accumulation of DNA damage in B cells. Neoplasia 2013, 15, 1292–1300. [Google Scholar] [PubMed]
- Yang, Y.; Karakhanova, S.; Werner, J.; Bazhin, A.V. Reactive oxygen species in cancer biology and anticancer therapy. Curr. Med. Chem. 2013, 20, 3677–3692. [Google Scholar] [CrossRef] [PubMed]
- Jutooru, I.; Guthrie, A.S.; Chadalapaka, G.; Pathi, S.; Kim, K.; Burghardt, R.; Jin, U.H.; Safe, S. Mechanism of action of phenethylisothiocyanate and other reactive oxygen species-inducing anticancer agents. Mol. Cell. Biol. 2014, 34, 2382–2395. [Google Scholar] [CrossRef] [PubMed]
- Chadalapaka, G.; Jutooru, I.; Safe, S. Celastrol decreases specificity proteins (SP) and fibroblast growth factor receptor-3 (FGFR3) in bladder cancer cells. Carcinogenesis 2012, 33, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Di Leva, G.; Croce, C.M. Roles of small RNAs in tumor formation. Trends Mol. Med. 2010, 16, 257–267. [Google Scholar]
- Volinia, S.; Calin, G.A.; Liu, C.G.; Ambs, S.; Cimmino, A.; Petrocca, F.; Visone, R.; Iorio, M.; Roldo, C.; Ferracin, M.; et al. A microrna expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. USA 2006, 103, 2257–2261. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Volinia, S.; Okumura, H.; Shimizu, M.; Taccioli, C.; Rossi, S.; Alder, H.; Liu, C.G.; Oue, N.; Yasui, W.; et al. Relation between microrna expression and progression and prognosis of gastric cancer: A microrna expression analysis. Lancet Oncol. 2010, 11, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Miao, Y.; Xiao, B.; Huan, R.; Jiang, Z.; Meng, D.; Wang, Y. Differential expression of microRNA species in human gastric cancer versus non-tumorous tissues. J. Gastroenterol. Hepatol. 2009, 24, 652–657. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.K.; Lee, C.W.; Cho, C.H.; Fan, D.; Wu, K.; Yu, J.; Sung, J.J. microRNA dysregulation in gastric cancer: A new player enters the game. Oncogene 2010, 29, 5761–5771. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. microRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Kim, V.N.; Han, J.; Siomi, M.C. Biogenesis of small RNAs in animals. Nat. Rev. Mol. Cell Biol. 2009, 10, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Wan, G.; Liu, Y.; Han, C.; Zhang, X.; Lu, X. Noncoding RNAs in DNA repair and genome integrity. Antioxid. Redox Signal. 2014, 20, 655–677. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.C. Micrornas in cancer—From research to therapy. Biochim. Biophys. Acta 2010, 1805, 209–217. [Google Scholar]
- Cho, W.C. microRNAs: Potential biomarkers for cancer diagnosis, prognosis and targets for therapy. Int. J. Biochem. Cell Biol. 2010, 42, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.; Wu, J. Feud or friend? The role of the miR-17-92 cluster in tumorigenesis. Curr. Genomics 2010, 11, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Mogilyansky, E.; Rigoutsos, I. The miR-17/92 cluster: A comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ. 2013, 20, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, S.; Hsu, S.D.; Huang, W.Y.; Huang, H.Y.; Chen, W.; Weng, S.L.; Huang, H.D. A systematic review of microRNA expression profiling studies in human gastric cancer. Cancer Med. 2014, 3, 878–888. [Google Scholar] [CrossRef] [PubMed]
- Genechip® Command Console® Software 3.0 Version, Affymetrix, Inc.: Santa Clara, CA, USA, 2009.
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Thulasingam, S.; Massilamany, C.; Gangaplara, A.; Dai, H.; Yarbaeva, S.; Subramaniam, S.; Riethoven, J.J.; Eudy, J.; Lou, M.; Reddy, J. miR-27b*, an oxidative stress-responsive microRNA modulates nuclear factor-kb pathway in raw 264.7 cells. Mol. Cell. Biochem. 2011, 352, 181–188. [Google Scholar] [CrossRef]
- Ebi, H.; Sato, T.; Sugito, N.; Hosono, Y.; Yatabe, Y.; Matsuyama, Y.; Yamaguchi, T.; Osada, H.; Suzuki, M.; Takahashi, T. Counterbalance between RB inactivation and miR-17-92 overexpression in reactive oxygen species and DNA damage induction in lung cancers. Oncogene 2009, 28, 3371–3379. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.L.; Xue, G.; Mei, Q.; Wang, Y.Z.; Ding, F.X.; Liu, M.F.; Lu, M.H.; Tang, Y.; Yu, H.Y.; Sun, S.H. Repression of the miR-17-92 cluster by p53 has an important function in hypoxia-induced apoptosis. EMBO J. 2009, 28, 2719–2732. [Google Scholar] [CrossRef] [PubMed]
- Grillari, J.; Hackl, M.; Grillari-Voglauer, R. miR-17-92 cluster: Ups and downs in cancer and aging. Biogerontology 2010, 11, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Strickertsson, J.A.; Desler, C.; Rasmussen, L.J. Impact of bacterial infections on aging and cancer: Impairment of DNA repair and mitochondrial function of host cells. Exp. Gerontol. 2014. [CrossRef]
- Handa, O.; Naito, Y.; Yoshikawa, T. Helicobacter pylori: A ros-inducing bacterial species in the stomach. Inflamm Res. 2010, 59, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Pathi, S.S.; Jutooru, I.; Chadalapaka, G.; Sreevalsan, S.; Anand, S.; Thatcher, G.R.; Safe, S. Gt-094, a NO-NSAID, inhibits colon cancer cell growth by activation of a reactive oxygen species-microRNA-27a: ZBTB10-specificity protein pathway. Mol. Cancer Res. 2011, 9, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Takanashi, M.; Borjigin, N.; Ohno, S.I.; Fujita, K.; Hoshino, S.; Osaka, Y.; Tsuchida, A.; Kuroda, M. microRNA-18a modulates STAT3 activity through negative regulation of PIAS3 during gastric adenocarcinogenesis. Br. J. Cancer 2013, 108, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Li, T.; Zhang, B.; Li, H.; Wu, Q.; Yang, L.; Nie, Y.; Wu, K.; Shi, Y.; Fan, D. microRNA-19a/b regulates multidrug resistance in human gastric cancer cells by targeting pten. Biochem. Biophys. Res. Commun. 2013, 434, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, Z.; Yu, M.; Li, L.; Du, G.; Xiao, W.; Yang, H. Involvement of miR-20a in promoting gastric cancer progression by targeting early growth response 2 (EGR2). Int. J. Mol. Sci. 2013, 14, 16226–16239. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Wang, C.; Wang, M.; Li, Z.; Casimiro, M.C.; Liu, M.; Wu, K.; Whittle, J.; Ju, X.; Hyslop, T.; et al. A cyclin D1/microrna 17/20 regulatory feedback loop in control of breast cancer cell proliferation. J. Cell Biol. 2008, 182, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Zhou, H.; Xiao, H.; Liu, Z.; Tian, H.; Zhou, T. microRNA-92a functions as an oncogene in colorectal cancer by targeting pten. Dig. Dis. Sci. 2014, 59, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Machado, A.M.; Figueiredo, C.; Touati, E.; Maximo, V.; Sousa, S.; Michel, V.; Carneiro, F.; Nielsen, F.C.; Seruca, R.; Rasmussen, L.J. Helicobacter pylori infection induces genetic instability of nuclear and mitochondrial DNA in gastric cells. Clin. Cancer Res. 2009, 15, 2995–3002. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Strickertsson, J.A.B.; Rasmussen, L.J.; Friis-Hansen, L. Enterococcus faecalis Infection and Reactive Oxygen Species Down-Regulates the miR-17-92 Cluster in Gastric Adenocarcinoma Cell Culture. Genes 2014, 5, 726-738. https://doi.org/10.3390/genes5030726
Strickertsson JAB, Rasmussen LJ, Friis-Hansen L. Enterococcus faecalis Infection and Reactive Oxygen Species Down-Regulates the miR-17-92 Cluster in Gastric Adenocarcinoma Cell Culture. Genes. 2014; 5(3):726-738. https://doi.org/10.3390/genes5030726
Chicago/Turabian StyleStrickertsson, Jesper A. B., Lene Juel Rasmussen, and Lennart Friis-Hansen. 2014. "Enterococcus faecalis Infection and Reactive Oxygen Species Down-Regulates the miR-17-92 Cluster in Gastric Adenocarcinoma Cell Culture" Genes 5, no. 3: 726-738. https://doi.org/10.3390/genes5030726
APA StyleStrickertsson, J. A. B., Rasmussen, L. J., & Friis-Hansen, L. (2014). Enterococcus faecalis Infection and Reactive Oxygen Species Down-Regulates the miR-17-92 Cluster in Gastric Adenocarcinoma Cell Culture. Genes, 5(3), 726-738. https://doi.org/10.3390/genes5030726