Abstract
During the last few years microRNAs (miRNAs) have emerged as key mediators of post-transcriptional and epigenetic regulation of gene expression. MiRNAs targets, identified through gene expression profiling and studies in animal models, depict a scenario where miRNAs are fine-tuning metabolic pathways and genetic networks in both plants and animals. MiRNAs have shown to be differentially expressed in brain areas and alterations of miRNAs homeostasis have been recently correlated to pathological conditions of the nervous system, such as cancer and neurodegeneration. Here, we review and discuss the most recent insights into the involvement of miRNAs in the neurodegenerative mechanisms and their correlation with significant neurodegenerative disorders.
Abbreviations

| Ago2 = Argonaute-2 | ALS = Amyotrophic Lateral Sclerosis |
| APP = Amyloid precursor protein | BACE1 = beta-site APP-cleavage enzyme 1 |
| BTBD3 = BTB (POZ) domain containing 3 | COL2A1 = Collagen, type II, alpha 1 |
| CoREST = REST Corepressor | COXIV = cytochrome c oxidase IV subunit |
| dAgo1 = Drosophila Argonaute-1 | DAT = Dopamine transporter |
| DJ1 = Parkin-7 | E2F1/DP = E2F transcription factor 1 |
| EAAT2 = excitatory amino acid transporter 2 | TDP-43 = TAR DNA-binding protein 43 |
| GALC = galactosylceramide | GLT-1 = glutamate transporter |
| hAgo2 = human Argonaute-2 | HDAC4 = Histone Deacetylase 4 |
| HTT = Huntingtin | LSD = Lysosomal Disease |
| NEFL = Neurofilament light polypeptide | NfKB = nuclear factor kappa BNPC = Niemann Pick cells |
| PACT = protein activator of PKR | TBP = Tata Binding Protein |
| phospho-4E-BP1 = 4E binding protein1 | Pitx3 = paired-like homeodomain transcription factor 3 |
| PTBP2 = Polypyrimidine tract binding protein 2 | RLC = regulatory light chain |
| SH-SY5Y = Human Neuroblastoma Cells Line | SIRT1 = sirtuin 1 |
| SNP = single-nucleotide polymorphism | SPT = Serine palmitoyltransferase |
| TAp73 = Tumor protein p73 | ERK1 = Extracellular signal-regulated kinase 1FUS/TLS = fused in sarcoma/traslocated in liposarcoma |
| PGC-1α = Peroxisome Proliferator—Activated Receptor Gamma Coactivator 1 | TGFBI = transforming growth factor, beta 1TLR-7 = Toll-like receptor 7 |
| TRBP = Tar RNA binding proteinTRIM2 = tripartite motif containing 2 |
1. Biogenesis and Role of microRNAs
MicroRNAs (miRNAs) are a class of ~22 nucleotides non-coding RNA molecules representing a superior mechanism by which to regulate gene expression.
MiRNAs biosynthesis is conserved during evolution and consists of two steps that take place in the nucleus and the cytoplasm, respectively [1,2].
In the nucleus, miRNAs are largely transcribed by RNA polymerase II as primary-(pri-)miRNAs. At this stage, pri-miRNAs present several hairpin structures, each consisting of a stem and a terminal loop, and are subject to a 5'-capping, 3'-polyadenylation, editing, and splicing processing [3,4]. The processed pri-miRNAs are next “cropped” into smallest hairpin-structures precursor of ~70 nucleotides (called pre-miRNAs) by a nuclear microprocessor complex composed of Drosha, an RNase III enzyme (RNASEN), and DGCR8 (DiGeorge Critical Region 8) protein. The last is also called Pasha (Partner of Drosha) in D. melanogaster and C. elegans [5]. These proteins form a complex with several cofactors (e.g., DEAD box RNA helicases p68 [DDX5]; p72 [DDX17]; heterogeneous nuclear ribonucleo-proteins [hnRNPs]) important for the specificity of Drosha activity [6]. Due to RNase activity, Drosha cleaves the 5' and 3' arms of the pri-miRNA hairpin [7], while DGCR8 is necessary for the interaction with the pri-miRNA for the site-specific cleavage [8]. Thus, Drosha cleaves 11 base pairs away from the single-/double-stranded RNAs at the level of the hairpin stem base [8]. The cleavage occurs co-transcriptionally [7,8,9,10] and generates a product with 2 nucleotides with 3' overhang that is specifically recognized by Exportin-5, which transports the pre-miRNAs into the cytoplasm via a Ran-GTP-dependent mechanism [4,11].
Alternatively, miRNAs may be generated by splicing and debranching of short hairpin introns [12,13] called “MiRtrons”, or by processing of small nucleolar RNAs (snoRNAs), transfer RNAs (tRNAs), and endogenous short hairpin RNAs (shRNAs) using a microprocessor complex independent route [14,15,16,17,18,19].
In the cytoplasm, the pre-miRNA enters into the RISC Loading Complex consisting of Dicer (RNase), the double-stranded RNA-binding domain proteins TRBP, PACT and the core component Ago2 [6,20,21,22]. Dicer, TRBP, and PACT process pre-miRNAs to ~22 nucleotides long miRNAs duplex [20,21,22,23]. The two miRNA strands are then separated and the guide strand is associated with an Argonaute protein within the RISC, where it is directly involved in the silencing of target messages.
Thermodynamically the miRNAs duplex is asymmetric [24,25]. As a consequence, miRNA strand whose 5'-end is less stably base-paired will usually be chosen as the strand guide. In contrast, the miRNA strand of which the 5'-end is more stably base-paired (the passenger strand) will be excluded from the RISC Loading Complex and generally degraded [3,4,26].
1.1. Canonical Function of microRNAs
MiRNAs drive RISC to complementary sites within the target mRNAs in order to mediate their repression at the post-transcriptional level trough RNA-RNA base pairing, or translational repression, and/or mRNA deadenylation and decay (Figure 1) [1,27,28,29,30].
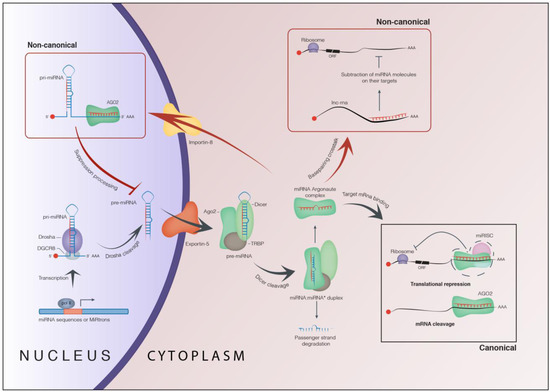
Figure 1.
Biogenesis and function of microRNAs. Image shows the most relevant nuclear and cytoplasm steps of the biogenesis of miRNAs together with the canonical and non-canonical activity of miRNAs (see main text for details).
MiRNAs bind to their cognate target mRNAs in the site-specific sequences, called miRNA Recognition Element (MRE), through a mechanism based on the pairing of the “seed” sequence involving ~6–8 nucleotides at the 5'-end of the miRNAs [31].
1.2. Non-Canonical Function of microRNAs
Recent studies have shown that miRNAs are also re-imported, perhaps, via exportin-1 or importin-8, from the cytoplasm to the nucleus through a combination with Argonaute proteins. Here, miRNAs could regulate gene expression at the transcriptional level (Figure 1) [32,33,34].
Additionally, evidence has highlighted a new regulatory circuit in which miRNAs can crosstalk each other through a new smart “biological alphabet” represented by the “MRE” sequences that “act as the letters whose different combinations may form an entire universe of words” (from Salmela et al. 2011 [35]). In detail, Pandolfi’s hypothesis has proposed that mRNAs, miRNAs, transcribed pseudogenes, and long noncoding RNAs (lncRNA, a class of non-protein coding transcripts, usually 200 to 1,000 of nucleotides in length) using MRE sequences “talk” to each other and suggested that this “competing endogenous RNA” (ceRNA) activity forms a large-scale regulatory network across the transcriptome [35], and acts as player in the human genome for regulating the distribution of miRNAs molecules toward specific targets. This mechanism is straightforward for physiological and pathological processes [35,36,37,38,39,40,41,42].
2. MicroRNAs and Neurodegeneration
Neurodegenerative diseases are a group of late onset progressive disorders of the nervous system, characterized by a complex pathogenesis that generally involves multiple basic cellular pathways alterations [43,44,45,46,47,48,49,50,51,52,53]. Thus, understanding the wide spectrum of cell mechanisms could be relevant for the development of more effective therapies for these disorders.
Emerging evidence addresses a key role of non-coding RNAs in neurogenesis and neurodegeneration [45,46,47,48]. This review discusses the current advancements on miRNAs and neurodegenerative processes. Here we summarized the most recent insights in the issues collected from some selected neurodegenerative diseases: Alzheimer’s disease (AD) [49], Parkinson’s disease (PD) [50], Amyotrophic Lateral Sclerosis (ALS) [51], and polyglutamine (polyQ) disorders such as Huntington’s disease (HD) [52] and Lysosomal Storage Disorders (LSD) [53].
Table 1 reports a landscape of miRNAs that are considered implicated at different levels in AD, PD, HD, ALS, and LSD pathogenesis. Overall, these findings highlight the critical impact of select miRNAs on regulating the expression of chief proteins in neurodegeneration (both pathogenesis and progression).
Table 1.
Reports a landscape of miRNAs involved in the pathogenesis of Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), Amyotrophic Lateral Sclerosis (ALS), and Lysosomal Storage Disorders (LSDs) not included in the main text.
| microRNA | Neurodegenerative Disease | Molecular Target | Effects | Reference |
|---|---|---|---|---|
| miR-15 | AD | ERK1 and Tau | ERK1 and Tau regulation | [54] |
| miR-16 | AD | APP | Overexpression reduced APP level | [55] |
| ERK1 and Tau | ERK1 and Tau regulation | [54] | ||
| miR-106a | AD | APP | APP repression | [56] |
| miR-106b | AD | APP | Aberrantly expressed in APPswe/PSE9 mice | [57] |
| miR-107 | AD | BACE1 | Downregulated. | [58] |
| Repression of Cofilin translation, a component of rod-like actin structures in the AD brain. | ||||
| miR-124 | AD | BACE1 | Suppressed induces over expression of BACE1 | [59] |
| miR-132 | AD | PTBP2 | Neuronal splicing regulator of Tau Exon 10 | [54] |
| miR-137 | AD | SPT | SPT and in turn Aβ levels up-regulate | [60] |
| miR-153 | AD | APP | Downregulated in modest AD pathology | [56] |
| miR-195 | AD | BACE1 | Overexpressed decreased BACE1 protein level | [61] |
| miR-497 | AD | ERK1 and Tau | ERK1 and Tau regulation | [54] |
| miR-520c | AD | APP | APP repression | [56] |
| Let-7b | AD | TLR-7 | Induce Toll-like receptor 7 activation | [62] |
| miR-7 | PD | α-synuclein mRNA | It can represses α-synuclein protein levels collaborating with miR-153 | [63] |
| miR-133b | PD | Pitx3 | Downregulated in PD brain tissue | [64] |
| miR-34b/c | PD | SH-SY5Y dopaminergic neuron | Downregulated | [65] |
| miR-let7 | PD | LRRK2 | Regulation of Drosophila e2f1 protein synthesis: repressed expression | [66] |
| miR-184 | PD | LRRK2 | Regulation of dp messenger RNAs synthesis: repressed expression | [66] |
| miR-433 | PD | SNP rs12720208 in the 3' UTR | Increased FGF20 expression and upregulation of alpha-synuclein | [67] |
| miR-9/miR-9* | HD | REST/COREST | Downregulated. Double negative feedback loop between the REST silencing complex and the miRNAs it regulates | [68] |
| [69] | ||||
| miR-29c | HD | REST | Downregulated | [70] |
| miR-34b | HD | p53 | Mysregulated causing by mHTT accumulation. Overexpressed in plasma of HD patients | [71] |
| miR-124 | HD | REST | Downregulated leads to an increases of their target level | [68] |
| miR-125b | HD | HTT | Downregulated | [72] |
| miR-222 | HD | REST | Downregulated. | [70] |
| miR-132 | HD | REST | Downregulated. Neurite sprouting | [68] |
| miR-135 | HD | REST | Downregulated | [68] |
| miR-137 | HD | REST | Aberrantly repressed directly mediated by REST | [73] |
| miR-146a | HD | TBP | Regulation of TBP by miR-146a may contribute to HD pathogenesis. Generally downregulated | [72] |
| miR-150 | HD | HTT | Downregulated | [72] |
| miR-153 | HD | REST | Downregulated | [73] |
| miR-200a | HD | Genes regulating synaptic function, neurodevelopment, and neuronal survival | Upregulated. Perturbed expression in HD patients. | [74] |
| miR-200c | [74] | |||
| miR-9 | ALS | NEFL | Downregulated | [75] |
| miR-23a | ALS | PGC-1 | Upregulated. It can reduce PGC-1α signalling, cytochome-b and COXIV protein levels | [76] |
| miR-29b | ALS | p53 | Upregulated | [76] |
| miR-124 | ALS | EAAT2/GLT1 | Indirect miR-124a-mediated regulation of GLT1 expression from neurons to astrocytes | [77] |
| miR-206 | ALS | HDAC4 | Upregulated in ALS end stage model to regenerate damaged neuromuscular synapses by HDAC4 reinnervation via | [78] |
| miR-455 | ALS | COL2A1 | Upregulated in skeletal muscles of ALS patients | [76] |
| Let-7b | ALS | TDP-43 | Downregulated | [79] |
| miR-663 | ALS | TDP-43 | Upregulated | [79] |
| miR-126 | LSD | GALC | Expressed in HSCs but not in differentiated cells | [80] |
| miR-130 | [80] | |||
| miR-196a | NPC | Lipid biosynthesis associated genes | Upregulated | [81] |
| miR-196b | NPC | Lipid biosynthesis associated genes | Upregulated | [81] |
| miR-296 | NPC | Lipid biosynthesis associated genes | Upregulated | [81] |
| miR-98 | NPC | Lipid biosynthesis associated genes | Downregulated.Lipid biosynthesis associated | [81] |
| miR-143 | NPC | Lipid biosynthesis associated genes | Downregulated. | [81] |
| Lipid biosynthesis associated |
2.1. MicroRNAs and Alzheimer’s Disease
The pathological hallmarks of AD are the deposition of intracellular neurofibrillary tangles containing Tau protein and the accumulation of extracellular plaques containing β-Amyloid (Aβ) peptides, beginning in the hippocampus, and spreading progressively throughout the brain [82,83,84]. The basic mechanisms generating Aβ are largely studied and now include microRNAs. This emerges by growing evidence suggesting that alterations in the miRNA network could contribute to risks for AD (Table 1). Here we discuss some recent significant examples.
The TargetScan program revealed that several members of the miR-16 family (miR-16, miR-15, miR-195, miR-497) could modulate endogenous ERK1 and Tau phosphorylation in neuronal cells in vitro. Of note, miR-15 directly targets ERK1. Given that ERK1 is a Tau kinase, alteration of miR-15 is considered as a potential cause of the abnormal Tau phosphorylation observed in AD (Figure 2) [54]. Indeed, the same research group demonstrated that miR-132 is also involved in the regulation of the splicing of the endogenous Tau exon 10 by targeting PTBP2 in the neurons [54].
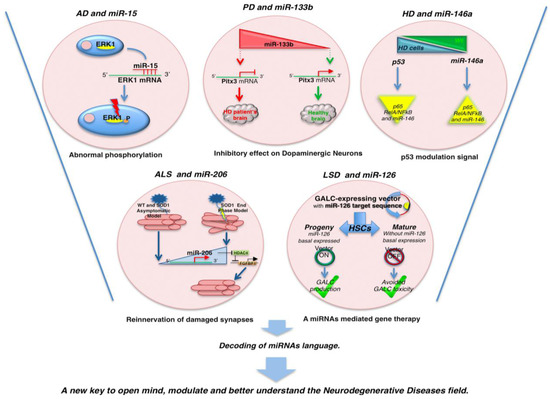
Figure 2.
MicroRNAs and neurodegenerative mechanisms. Image shows some selected example of miRNAs involved in the pathogenesis of AD, PD, HD, ALS, and LSDs. Details of each mechanism are described in the text.
miR-153 levels were significantly reduced in the brains of a subset of human AD specimens with mild AD pathology. The same group of patients also exhibited increased levels of β-Amyloid precursor protein (APP), compared to healthy controls. Using bioinformatics algorithms, Long et al. revealed that miR-153 had a target site within the 3'-untranslated region (3'-UTR) APP mRNA. These computational results were confirmed by in vitro and in vivo experiments. Interestingly, miR-153 also caused the reduction of the expression of the APLP2 (APP paralog), indicating that miR-153 could be a player for biological pathways involving both proteins [85,86].
Other studies indicated miR-34c as a potential marker for the cognitive disturbances associated with AD. miR-34c levels were elevated in the hippocampus of AD patients and corresponding mouse models (Figure 3). In fact, inhibition of miR-34c activity correlated with restoration of molecular mechanisms involved in the observed improvement of memory. Thus, it was suggested that miR-34c could be a negative control of memory consolidation [87]. Similar studies demonstrated the increase of the expression of miR-34a in the brain cortex of AD mouse models [88,89] and a specific interplay among miR-34a, TAp73, and synaptotagmin-1. TAp73 is a p53-family member and drives the expression of miR-34a, but not miR-34b and miR-34c, by acting on specific binding sites to the miR-34a promoter [89]. In brains of AD patients, the miR-34a, TAp73, and synaptotagmin-1 circuit is conserved although the aberrant expression of p73 (Figure 3) [89].
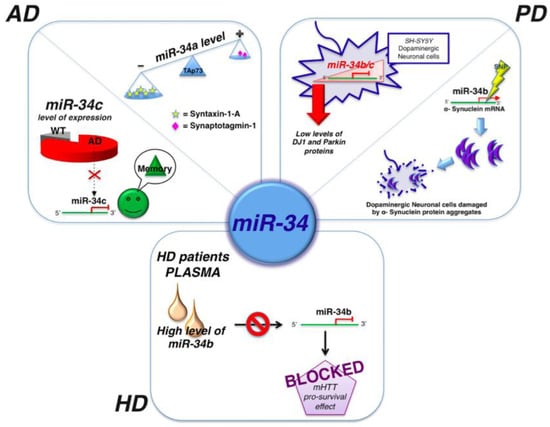
Figure 3.
miR-34 and neurodegeneration. Image shows the correlation of miR-34a, miR-34b, and miR-34c and AD, PD, and HD. Details of each mechanism are described in the text.
Several microRNAs target APP. In this context, miR-29a and miR-29b that target the APP and BACE1 mRNAs in their 3'-UTRs, are down regulated in a subset of AD patients. As a consequence, BACE1 protein expression is increased and the amyloidogenic peptide generated. This phenomenon was particularly evident in neuronal and glial cells indicating that the derepression of BACE1 expression that can occur in neurons or astrocytes [90]. Indeed, the BACE1 3'-UTR is also the target of miR-107, miR-124, and miR-195 (Table 1).
Interestingly, miR-107 controls the expression of other proteins relevant to AD pathology, such as cofilin [58], an actin-binding protein that accumulates in cytoplasmic inclusions known as Hirano bodies [91] (Hirano, 1994). This indicated that a single miRNA deregulation could activate several pathogenic cascades upstream to Aβ-accumulation pathway. Thus, miR-9 and miR-181c target 3'-UTRs mRNA of a group of TGFBI, TRIM2, SIRT1, and BTBD3 proteins, important for brain homeostasis and disease pathogenesis [92].
2.2. MicroRNAs and Parkinson’s Disease
Typical pathological features of PD, the second most common neurodegenerative disorder, are loss of dopaminergic neurons in the substantia nigra and presence of Lewy bodies, intracellular inclusions that cause impaired neuron survival in several brain areas [93].
MiRNAs may act as regulators of both known and novel biological processes leading to PD (Table 1) [94,95,96].
Several studies showed the role of several microRNAs as modulator of the expression of α-synuclein. Due to the amplification of its gene locus, this protein is accumulated within the neurons, thus representing a major indicator of autosomal dominant PD. This emerges also in transgenic animal models where the over-expression of human α-synuclein caused impaired function or decreased survival of dopaminergic neurons [97].
miR-7 represses α-synuclein protein by targeting the 3'-UTR of α-synuclein mRNA [63,98,99]. Interestingly, miR-7 cooperates with miR-153 to control the quantity of α-synuclein produced, both in the adult brain and during neuronal development [63,98,99]. Alteration of α-synuclein expression is also associated to the increase of the translation of the fibroblast growth factor-20. This event is a consequence of the risk allele for rs12720208 polymorphism that disrupts the binding site for miR-433 [100,101].
miR-133b is specifically expressed in healthy midbrain dopaminergic neurons whereas is deficient in midbrain tissue from patients with PD. miR-133b targets transcription factor Pitx3 mRNA and regulates the maturation and function of midbrain dopaminergic neurons within a negative feedback circuit where Pitx3 is included (Figure 2). In fact, overexpression of a Pitx3 transgene lacking 3'-UTR regulatory element was able to “rescue” the reduced midbrain dopaminergic neurons. Moreover, consistent with this, the reduction of the transcription of the Dopamine Transporter by miR-133b was partially suppressed by over-expression of Pitx3 lacking the 3'-UTR miR-133b MRE sequences. Therefore, a model was proposed in which miR-133b plays a role in a feedback loop pathway, where Pitx3 specifically induces transcription of miR-133b and Pitx3 activity is downregulated by miR-133b at post-transcriptional level [64,102]. More recently, de Mena et al., searching for DNA variants in miR-133 and PITX3 genes in PD patients (n = 777) and healthy controls (n = 650) from Spain, suggested that miR-133 and PITX3 gene variants did not contribute to the risk for PD, thus contrasting these data, at least in those specimens [67]. Additionally, miR-133b KO animal models were found to be devoid of dopaminergic neuron degeneration or impaired motor function, thereby indicating that a concert of miRNAs could be involved in a network that promote neuronal development and survival [103].
Down-regulation of miR-34b and miR-34c was associated to a decrease of Park-7 and Parkin, two proteins associated to familial and sporadic forms of PD (Figure 3) [65,66,104]. Additionally polymorphisms in the 3'-UTR of miR-34b and miR34c target the α-synuclein mRNA (Figure 3) [105].
Gain-of-function mutations in the leucine-rich repeat kinase 2 (LRRK2) mRNA cause age-dependent degeneration of dopaminergic neurons in both familial and sporadic PD. Interesting findings from LRRK2 studies in Drosophila demonstrated the interaction of LRRK2 with the RLC proteins dAgo1 or hAgo2. Within this network, in the brain of old fly, it was also observed that LRRK2 is involved in the negative regulation of dAgo1 levels and in the association of phospho-4E-BP1 with hAgo2 thus causing a deregulation of the synthesis of E2F1/DP and the impairment of miRNA pathway [66]. Of note was that the authors also demonstrated a concomitant role of let-7 or miR-184* in attenuating this aberrant circuit. Overall these findings delineate a trait of the pathogenic effect of LRRK2 [66].
2.3. MicroRNAs and Hungtinton’s Disease
Among the nine forms of Poly-glutamine (Poly-Q) disorders, HD is the most frequent. Here abnormal huntingtin (HTT) accumulation induces neuronal damage in brain patients. Increasing evidence revealed a key role of miRNAs in HD [70,74,106,107,108,109,110] (Table 1).
A widespread miRNAs dysregulation was mostly observed in the striatum and cortex [111], suggesting that miRNAs are involved in the impairment of transcriptome, which is a feature of HD [112]. In this regard, a pivotal role is exerted by RE1 Silencing Transcription Factor (REST), a direct and indirect regulator of a cohort of mRNAs and miRNAs altered in HD [68,112]. REST binds to several neuronal miRNA genes including miR-9/9*, miR-29b, miR-124, miR-132, miR-135, miR-137, and miR-153 [68,73]. Among the above miRNAs, miR-9 and miR-9* are direct target of REST and CoREST, respectively [69], whereas miR-222 is an indirect REST target [70].
Ghose et al. showed a down-regulation of miR-125b, miR-146a, and miR-150 in STHdhQ111/HdhQ111 cells (HD mouse cell model) compared to the wild type counterpart. They demonstrated that miR-150 and miR-125b target p53 that decreases the NFκB (RelA/NFκB) p65 subunit expression and causes the reduction of miR-146a, thereby indicating that p53 directly or indirectly regulates the expression of miR-146a (Figure 2) [72]. Indeed, p53 is also regulated by miR-34b [113], which is significantly increased in the plasma of HD patients. In vitro studies proposed that the dysregulation of miR-34b could be a consequence of mHTT accumulation, at least in pluripotent and neurons differentiated human cells (Figure 3) [71].
Finally, miR-200a and miR-200c were found significantly upregulated at the pre-symptomatic stage of an HD mouse model. Comparative analysis of networks suggested that these two miRNAs may control genes involved in neuronal dysfunction induced by mutant HTT, such as atypical synaptic transmission and altered neurogenesis [74].
2.4. MicroRNAs and Amyotrophic Lateral Sclerosis
ALS is a neurodegenerative disease characterized by the loss of motor neurons, denervation of target muscles, muscle atrophy, and paralysis. Therefore, elucidating ALS pathogenesis may require the exploration of the bidirectional signaling between motor neurons and skeletal muscle fibers at neuromuscular synapses [51].
MiRNAs machinery has been found compromised in ALS (Table 1). For instance the absence of processed miRNAs, due to Dicer deletion in spinal motor neurons, resulted in a mouse model with progressive paralysis, astrocytosis, and signs of axonopathy, classical features of ALS [75]. Authors identified a single miR-9-binding site on the neurofilament light polypeptide (NFLP) mRNA and observed that miR-9 was also downregulated in Spinal Muscular Atrophy models thereby suggesting direct evidence for miRNAs malfunction in motor neuron diseases [75]. In another study, it was shown that the nuclear factor TDP-43, a major component of the inclusions in ALS patients and Frontotemporal Lobar Degenerative Disorder, was found associated with Drosha complex, thereby involving miRNAs biogenesis. In TDP-43−/− mice let-7b was downregulated, and miR-663 upregulated [79]. This phenomenon was correlated with the expression of FUS/TLS suggesting that this complex acts as a cofactor involved in the biogenesis of a specific subset of miRNAs [114].
Of note, miRNAs dysregulation differ from the pre-symptomatic and the end-stage of ALS-SOD1 disease [115,116]. Analysis of the altered mitochondrial network genes in skeletal muscle revealed a contribution of miRNAs and that this kind of dysfunction plays a role in the progression of ALS. An accurate miRNAs expression study revealed a miR-23a, miR-29b, miR-206 and miR-455 upregulations in patients of ALS respect to control subjects [76]. miR-23a negatively regulates PGC-1α signaling, determining a significant reduction of PGC-1α, cytochome-b and COXIV protein levels in overexpressing miR-23a transgenic mice [76].
Recently, Morel et al. reported that the reduction of the expression of the glutamate transporter GLT1 in the end-stage SOD1 G93A mice, a mouse model of ALS, was a consequence of miR-124a activity. Interestingly they revealed an exosome-mediated transfer of microRNAs mechanism showing that miR-124a was transferred from neurons to astrocytes through neuronal exosomes [77].
miR-206 is required for efficient regeneration of neuromuscular synapses after acute nerve injury, which probably accounts for its salutary effects in ALS [117]. Evidence indicated that, although mice genetically lacking miR-206 were able to engage normal neuromuscular synapses during development, deficiency of miR-206 in the ALS mouse model accelerates the disease progression, at least in part through the histone deacetylase 4 (HDAC4) and fibroblast growth factor signaling pathways (Figure 2) [78]. Remarkably, the phenotypes of miR-206 and HDAC4 mutant mice indicated that miR-206 and HDAC4 have opposite effects on retrograde signals required for the reinnervation. Additionally, fibroblast growth factor-7 (FGF7), FGF10, and FGF22, known to be regulators of synapse formation [118,119], were unaltered in miR-206−/− mice compared to wild type animals, whereas the FGFBP1 protein (a factor that interacts with the FGF family members and enhance the FGF7 activity in rat L6 myoblasts by releasing the FGF up-taken by the extracellular matrix [120]) was downregulated in muscles of miR-206−/− mice and upregulated in muscles of HDAC4−/− mice after denervation. Collectively, these findings suggest that miR-206 and HDAC4 help and block the innervations of neuromuscular junction, respectively, via opposing effects on FGFBP1 [78].
2.5. MicroRNAs and Lysosomal Storage Disorders
LSDs are a group of inherited lysosomal metabolic disorders caused by deficiencies of lysosomal enzymes and of a series of lysosomal proteins, which are involved in the abnormal accumulation of undegraded substrates. These disorders are characterized by a broad spectrum of clinical variants and of progressive lethal neurodegeneration [53,121,122,123,124].
Recent studies point to miRNAs as tool for gene therapy application [117]. In this regard we have developed the hematopoietic stem cell (HSC)-based gene therapy for Krabbe disease, fatal LSD caused by mutations in the galactocerebrosidase (GALC) gene [125,126]. To be effective, this therapy needs the modulation of the transgene expression to avoid forced GALC toxicity [127]. The rationale of this approach was the inhibition of GALC toxicity in the HSCs used for GLD treatment in order to protect differentiated cells that could be able to carry efficiently their function. To this end, we took advantage of this by the observation that miR-126 and miR-130 were expressed in HSCs but not in differentiated progeny. By incorporating miR-126 target sequences into a GALC-expressing lentiviral vector we have suppressed GALC expression in HSCs maintaining a robust expression in mature hematopoietic cells [80].
In another approach, Osborn et al. have used the same strategy to achieve an opposite goal. To develop a Mucopolysaccharidosis type I (MPS I) treatment, a LSD characterized by a progressive accumulation of glycosaminoglycans due to the α-L-iduronidase (IDUA) gene mutation, they have included a miRNA sequence in a minicircle DNA vector with a tissue-specific promoter to obtain a slight improvement of its long-term expression respect to the same vector without the miRNA element. This therapy allowed the restoration of IDUA enzyme and the amelioration of pathology in MPS I mice although with the support of immune modulation by costimulatory blockade [128].
A late endosomal-lysosomal accumulation of multiple lipid molecules characterizes the Niemann-Pick type C (NPC) disease. MiRNAs pattern, both in health subjects than in NPC patients, have been analyzed in human fibroblasts by Ozsait et al. using a TaqMan Low-Density Array system containing 365 mature human miRNAs. They showed that NPC patient fibroblasts presented altered levels of miRNAs pattern respect to controls. Remarkably, they found that miR-196a, miR-196b, and miR-296 were upregulated whereas 38 miRNAs were significantly downregulated in NPC cells. Among these non-coding RNAs, miR-98 and miR-143, the lipid biosynthesis associated miRNA, were the most downregulated [81].
3. Concluding Remarks
The implications of miRNAs in neurodegenerative molecular mechanisms offer the opportunity to elucidate the pathogenesis of the nervous system diseases.
As described in this review, several miRNAs could be involved in the same molecular network. For instance, miR-150, miR-125b [72], and miR-34b [71], although with different mechanisms, altered the p53 homeostasis in HD. Additionally, miR-34b and miR-34c down-regulation was associated to PD [65,105], whereas miR-34c [87] and miR-34a [89] correlated with AD. Remarkably, the work by Liu and collaborators associated the activity of miR-34 on the control of age-associated events and long-term brain integrity in Drosophila, and suggested that miR-34 could be a molecular link between ageing and neurodegeneration [129].
Alternatively, abnormal expression of a single miRNA molecule could be responsible of a specific pathology. Thus, miR-206 seems to be implicated in ALS [78] while miR-133b could be involved in PD pathogenesis [64,102].
Of note, specific antisense antagomirs (miRNA inhibitors) could be used to affect the activity of specific miRNAs and to develop a therapeutic strategy for these disorders.
Therefore, miRNAs-based therapeutics has greatly advanced because of their capability to efficiently silence multiple messages concurrently within an entire disease pathway.
Acknowledgements
We thank all researchers cited in this review and we apologize to colleagues in the field for not being able to cite many important papers due to the limitation of the article.
We thank Nicoletta Francia (English Language Speaker) for proofreading of the manuscript.
This work is supported by a grant from Fondazione Cassa di Risparmio di Perugia, Italy (project n. 2010.011.0445) to S.M.
References
- Bartel, D.P. microRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Siomi, H.; Siomi, M.C. Posttranscriptionalregulation of microRNAbiogenesis in animals. Mol. Cell 2010, 38, 323–332. [Google Scholar] [CrossRef]
- Carthew, R.W.; Sontheimer, E.J. Origins and mechanism of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef]
- Kim, V.N.; Han, J.; Siomi, M.C. Biogenesis of small RNAs in animals. Nat. Rev. Mol. Cell Biol. 2009, 10, 126–139. [Google Scholar] [CrossRef]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Rådmark, O.; Kim, S.; Kim, V.N. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef]
- Gregory, R.I.; Yan, K.P.; Amuthan, G.; Chendrimada, T.; Doratotaj, B.; Cooch, N.; Shiekhattar, R. The Microprocessor complex mediates the genesis of microRNAs. Nature 2004, 432, 235–240. [Google Scholar]
- Han, J.; Lee, Y.; Yeom, K.H.; Kim, Y.K.; Jin, H.; Kim, V.N. The Drosha–DGCR8 complex in primary microRNA processing. Genes Dev. 2004, 18, 3016–3027. [Google Scholar] [CrossRef]
- Han, J.; Lee, Y.; Yeom, K.H.; Nam, J.W.; Heo, I.; Rhee, J.K.; Sohn, S.Y.; Cho, Y.; Zhang, B.T.; Kim, V.N. Molecular basis for the recognition of primary microRNAs by the Drosha–DGCR8 complex. Cell 2006, 125, 887–901. [Google Scholar] [CrossRef]
- Kim, Y.K.; Kim, V.N. Processing of intronic microRNAs. EMBO J. 2007, 26, 775–783. [Google Scholar]
- Morlando, M.; Ballarino, M.; Gromak, N.; Pagano, F.; Bozzoni, I.; Proudfoot, N.J. Primary microRNA transcripts are processed co-trascriptionally. Nat. Struct. Mol. Biol. 2008, 15, 902–909. [Google Scholar]
- Okada, C.; Yamashita, E.; Lee, S.J.; Shibata, S.; Katahira, J.; Nakagawa, A.; Yoneda, Y.; Tsukihara, T. A high-resolution structure of the pre-microRNA nuclear export machinery. Science 2009, 326, 1275–1279. [Google Scholar] [CrossRef]
- Okamura, K.; Hagen, J.W.; Duan, H.; Tyler, D.M.; Lai, E.C. The mirtron pathway generates microRNA-class regulatory RNAs in Drosophila. Cell 2007, 130, 89–100. [Google Scholar] [CrossRef]
- Ruby, J.G.; Jan, C.H.; Bartel, D.P. Intronic microRNA precursor that bypass Drosha processing. Nature 2007, 448, 83–86. [Google Scholar] [CrossRef]
- Babiarz, J.E.; Ruby, J.G.; Wang, Y.; Bartel, D.P.; Blelloch, R. Mouse ES cells ewpress endogenous shRNAs, siRNAs, and other Microprocesso-indipendent, Dicer-dependent small RNAs. Genes Dev. 2008, 22, 2773–2785. [Google Scholar] [CrossRef]
- Ender, C.; Krek, A.; Friedländer, M.R.; Beitzinger, M.; Weinmann, L.; Chen, W.; Pfeffer, S.; Rajewsky, N.; Meister, G. A human snoRNA with microRNA-like functions. Mol. Cell 2008, 32, 519–528. [Google Scholar] [CrossRef]
- Saraiya, A.A.; Wang, C.C. snoRNA, a novel precursor of microRNA in Giardia lamblia. PLoS Pathog. 2008, 4, e1000224. [Google Scholar] [CrossRef]
- Cole, C.; Sobala, A.; Lu, C.; Thatcher, S.R.; Bowman, A.; Brown, J.W.; Green, P.J.; Barton, G.J.; Hutvagner, G. Filtering of deep sequencing data reveals the existence of abundant Dicer-dependent small RNAs derived from tRNAs. RNA 2009, 15, 2147–2160. [Google Scholar] [CrossRef]
- Czech, B.; Zhou, R.; Erlich, Y.; Brennecke, J.; Binari, R.; Villalta, C.; Gordon, A.; Perrimon, N.; Hannon, G.J. Hierarchical rules for Argonaute loading in Drosophila. Mol. Cell 2009, 36, 445–456. [Google Scholar] [CrossRef]
- Miyoshi, K.; Miyoshi, T.; Hartig, J.V.; Siomi, H.; Siomi, M.C. Molecular mechanism that funnel RNA precursor into endogenous small-interfering RNA and microRNA biogenesis pathways in Drosophila. RNA 2010, 16, 506–515. [Google Scholar] [CrossRef]
- Haase, A.D.; Jaskiewicz, L.; Zhang, H.; Lainé, S.; Sack, R.; Gatignol, A.; Filipowicz, W. TRBP, a regulator of cellular PKR and HIV-1 virus expression, interacts with Dicer and functions in RNA silencing. EMBO Rep. 2005, 6, 961–967. [Google Scholar] [CrossRef]
- Lee, Y.; Hur, I.; Park, S.Y.; Kim, Y.K.; Suh, M.R.; Kim, V.N. The role of PACT in the RNA silencing pathway. EMBO J. 2006, 25, 522–532. [Google Scholar] [CrossRef]
- MacRae, I.J.; Ma, E.; Zhou, M.; Robinson, C.V.; Doudna, J.A. In vitro reconstitution of the human RISC-loading complex. Proc. Natl. Acad. Sci. USA 2008, 105, 512–517. [Google Scholar] [CrossRef]
- Chendrimada, T.P.; Gregory, R.I.; Kumaraswamy, E.; Norman, J.; Cooch, N.; Nishikura, K.; Shiekhattar, R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 2005, 436, 740–744. [Google Scholar] [CrossRef]
- Khvorova, A.; Reynolds, A.; Jayasena, S.D. Functional siRNAs and miRNAs exhibit strand bias. Cell 2003, 115, 209–216. [Google Scholar] [CrossRef]
- Schwarz, D.S.; Hutvágner, G.; Du, T.; Xu, Z.; Aronin, N.; Zamore, P.D. Asymmetry in the assembly of the RNAi enzyme complex. Cell 2003, 115, 199–208. [Google Scholar] [CrossRef]
- Siomi, H.; Siomi, M.C. On the road to reading the RNA-interference code. Nature 2009, 457, 396–404. [Google Scholar] [CrossRef]
- Hutvágner, G.; Zamore, P.D. A microRNA in a multiple-turnover RNAi enzyme complex. Science 2002, 297, 2056–2060. [Google Scholar] [CrossRef]
- Zeng, Y.; Wagner, E.J.; Cullen, B.R. Both natural designed micro RNAs can inhibit expression of cognate mRNAs when expressed in human cells. Mol. Cell 2002, 9, 1327–1333. [Google Scholar] [CrossRef]
- Doench, J.G.; Petersen, C.P.; Sharp, P.A. siRNAs can function as miRNAs. Genes Dev. 2003, 17, 438–442. [Google Scholar] [CrossRef]
- Djuranovic, S.; Nahvi, A.; Green, R. miRNA-mediated gene silencing by translational repression followed by mRNA deadenylation and decay. Science 2012, 336, 237–240. [Google Scholar] [CrossRef]
- Shukla, G.C.; Singh, J.; Barik, S. microRNAs: Processing, maturation, target recognition and regulatory functions. Mol. Cell Pharmacol. 2011, 3, 83–92. [Google Scholar]
- Kim, D.H.; Saetrom, P.; Snøve, O., Jr.; Rossi, J.J. MicroRNA-directed transcriptional gene silencing in mammalian cells. Proc. Natl. Acad. Sci. USA 2008, 105, 16230–16235. [Google Scholar]
- Chen, X.; Liang, H.; Zhang, C.Y.; Zen, K. miRNA regulates noncoding RNA: A noncanonical function model. Trends Biochem. Sci. 2012, 37, 457–459. [Google Scholar] [CrossRef]
- Xia, J.; Joyce, C.E.; Bowcock, A.M.; Zhang, W. Noncanonical microRNAs and endogenous siRNAs in normal and psoriatic human skin. Hum. Mol. Genet. 2013, 22, 737–748. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–368. [Google Scholar]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al.; FANTOM Consortium; RIKEN Genome Exploration Research Group and Genome Science Group (Genome Network Project Core Group) The transcriptional landscape of the mammalian genome. Science 2005, 309, 1559–1563. [Google Scholar] [CrossRef]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar]
- Chi, S.W.; Zang, J.B.; Mele, A.; Darnell, R.B. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature 2009, 460, 479–486. [Google Scholar]
- Licatalosi, D.D.; Mele, A.; Fak, J.J.; Ule, J.; Kayikci, M.; Chi, S.W.; Clark, T.A.; Schweitzer, A.C.; Blume, J.E.; Wang, X.; et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 2008, 456, 464–469. [Google Scholar] [CrossRef]
- Karreth, F.A.; Tay, Y.; Perna, D.; Ala, U.; Tan, S.M.; Rust, A.G.; DeNicola, G.; Webster, K.A.; Weiss, D.; Perez-Mancera, P.A.; et al. In vivo identification of tumor-suppressive PTEN ceRNAs in an oncogenic BRAF-induced mouse model of melanoma. Cell 2011, 147, 382–395. [Google Scholar] [CrossRef]
- Tay, Y.; Kats, L.; Salmena, L.; Weiss, D.; Tan, S.M.; Ala, U.; Karreth, F.; Poliseno, L.; Provero, P.; di Cunto, F.; et al. Coding-independent regulation of the tumor suppressor PTEN by competing endogenous mRNAs. Cell 2011, 147, 344–357. [Google Scholar] [CrossRef]
- Orlacchio, A.; Bernardi, G.; Orlacchio, A.; Martino, S. Stem cells: An overview of the current status of therapies for central and peripheral nervous system diseases. Curr. Med. Chem. 2010, 17, 595–608. [Google Scholar] [CrossRef]
- Orlacchio, A.; Bernardi, G.; Orlacchio, A.; Martino, S. Stem cells and neurological diseases. Discov. Med. 2010, 9, 546–553. [Google Scholar]
- Abe, M.; Bonini, N.M. microRNAs and neurodegeneration: Role and impact. Trends Cell. Biol. 2013, 23, 30–36. [Google Scholar] [CrossRef]
- Costa, V.; Esposito, R.; Aprile, M.; Ciccodicola, A. Non-coding RNA and pseudogenes in neurodegenerative diseases: “The (un)Usual Suspects”. Front. Genet. 2012, 3, 231. [Google Scholar]
- Kosik, K.S. The neuronal microRNA system. Nat. Rev. Neurosci. 2006, 7, 911–920. [Google Scholar] [CrossRef]
- Martino, S.; di Girolamo, I.; Orlacchio, A.; Datti, A.; Orlacchio, A. microRNA implications across neurodevelopment and neuropathology. J. Biomed. Biotechnol. 2009, 2009, 654346. [Google Scholar]
- Arevalo-Rodriguez, I.; Pedraza, O.L.; Rodríguez, A.; Sánchez, E.; Gich, I.; Solà, I.; Bonfill, X.; Alonso-Coello, P. Alzheimer’s disease dementia guidelines for diagnostic testing: A systematic review. Am. J. Alzheimers Dis. Other Demen. 2013, 28, 111–119. [Google Scholar]
- Surmeier, D.J.; Sulzer, D. The pathology roadmap inParkinson disease. Prion 2013, 7, 85–91. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Jones, A.; Troakes, C.; King, A.; Al-Sarraj, S.; van den Berg, L.H. The genetics and neuropathology ofamyotrophic lateral sclerosis. Acta Neuropathol. 2012, 124, 339–352. [Google Scholar] [CrossRef]
- Tierney, T.S.; Vasudeva, V.S.; Weir, S.; Hayes, M.T. Neuromodulation for neurodegenerative conditions. Front. Biosci. (Elite Ed.) 2013, 5, 490–499. [Google Scholar]
- Platt, F.M.; Boland, B.; van der Spoel, A.C. The cell biology of disease: Lysosomal storage disorders: The cellular impact of lysosomal dysfunction. J. Cell. Biol. 2012, 199, 723–734. [Google Scholar] [CrossRef]
- Hébert, S.S.; Sergeant, N.; Buée, L. microRNAs and the regulation oftau metabolism. Int. J. Alzheimers Dis. 2012, 2012, 406561. [Google Scholar]
- Liu, W.; Liu, C.; Zhu, J.; Shu, P.; Yin, B.; Gong, Y.; Qiang, B.; Yuan, J.; Peng, X. MicroRNA-16 targets amyloid precursor protein to potentially modulate Alzheimer’s-associated pathogenesis in SAMP8 mice. Neurobiol. Aging 2012, 33, 522–534. [Google Scholar] [CrossRef]
- Long, J.M.; Lahiri, D.K. Current drug targets for modulating Alzheimer’s amyloid precursor protein: Role of specific micro-RNA species. Curr. Med. Chem. 2011, 18, 3314–3321. [Google Scholar] [CrossRef]
- Wang, H.; Liu, J.; Zong, Y.; Xu, Y.; Deng, W.; Zhu, H.; Liu, Y.; Ma, C.; Huang, L.; Zhang, L.; Qin, C. miR-106b is aberrantly expressed in a double transgenic mouse model for Alzheimer’s desease targets TGF-B typeII receptor. Brain Res. 2010, 1357, 166–174. [Google Scholar]
- Yao, J.; Hennessey, T.; Flynt, A.; Lai, E.; Beal, M.F.; Lin, M.T. microRNA-related cofilin abnormality in Alzheimer’s disease. PLoS One 2010, 5, e15546. [Google Scholar]
- Fang, M.; Wang, J.; Zhang, X.; Geng, Y.; Hu, Z.; Rudd, J.A.; Ling, S.; Chen, W.; Han, S. The miR-124 regulates the expression of BACE1/β-secretase correlated with cell death in Alzheimer’s disease. Toxicol. Lett. 2011, 209, 94–105. [Google Scholar]
- Geekiyanage, H.; Chan, C. microRNA-137/181c regulates serine palmitoyltransferase and in turn amyloid β, novel targets in sporadic Alzheimer’s disease. J. Neurosci. 2011, 31, 14820–14830. [Google Scholar] [CrossRef]
- Zhu, H.C.; Wang, L.M.; Wang, M.; Song, B.; Tan, S.; Teng, J.F.; Duan, D.X. microRNA-195 downregulates Alzheimer’sdisease amyloid-beta production by targeting BACE1. Brain Res. Bull. 2012, 88, 596–601. [Google Scholar] [CrossRef]
- Lehmann, S.M. An unconventional role for miRNA: Let-7 activates Toll-like receptor 7 and causes neurodegeneration. Nat. Neurosci. 2012, 15, 827–835. [Google Scholar]
- Junn, E.; Lee, K.W.; Jeong, B.S.; Chan, T.W.; Im, J.Y.; Mouradian, M.M. Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc Natl. Acad. Sci. USA 2009, 106, 13052–13057. [Google Scholar]
- Kim, J.; Inoue, K.; Ishii, J.; Vanti, W.B.; Voronov, S.V.; Murchison, E.; Hannon, G.; Abeliovich, A. A MicroRNA feedback circuit in midbrain dopamine neurons. Science 2007, 317, 1220–1224. [Google Scholar]
- Miñones-Moyano, E.; Porta, S.; Escaramís, G.; Rabionet, R.; Iraola, S.; Kagerbauer, B.; Espinosa-Parrilla, Y.; Ferrer, I.; Estivill, X.; Martí, E. MicroRNA profiling of Parkinson’s disease brains identifies early downregulation of miR-34b/c which modulate mitochondrial function. Hum. Mol. Genet. 2011, 20, 3067–3078. [Google Scholar]
- Gehrke, S.; Imai, Y.; Sokol, N.; Lu, B. Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature 2010, 466, 637–641. [Google Scholar] [CrossRef]
- De Mena, L.; Coto, E.; Cardo, L.F.; Díaz, M.; Blázquez, M.; Ribacoba, R.; Salvador, C.; Pastor, P.; Samaranch, L.; Moris, G.; et al. Analysis of the Micro-RNA-133 and PITX3 genes in Parkinson’s disease. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2010, 153B, 1234–1239. [Google Scholar]
- Johnson, R.; Zuccato, C.; Belyaev, N.D.; Guest, D.J.; Cattaneo, E.; Buckley, N.J. A microRNA-based gene dysregulation pathway in Huntington’s disease. Neurobiol. Dis. 2008, 29, 438–445. [Google Scholar]
- Packer, A.N.; Xing, Y.; Harper, S.Q.; Jones, L.; Davidson, B.L. The bifunctional microRNA miR-9/miR-9* regulates REST and CoREST and is downregulated in Huntington’s disease. J. Neurosci. 2008, 28, 14341–14346. [Google Scholar]
- Lee, S.T.; Chu, K.; Im, W.S.; Yoon, H.J.; Im, J.Y.; Park, J.E.; Park, K.H.; Jung, K.H.; Lee, S.K.; Kim, M.; et al. Altered microRNA regulation in Huntington’s disease models. Exp. Neurol. 2011, 227, 172–179. [Google Scholar]
- Gaughwin, P.M.; Ciesla, M.; Lahiri, N.; Tabrizi, S.J.; Brundin, P.; Björkqvist, M. Hsa-miR-34b is a plasma-stable microRNA that is elevated in pre-manifest Huntington’s disease. Hum. Mol. Genet. 2011, 20, 2225–2237. [Google Scholar] [CrossRef]
- Ghose, J.; Sinha, M.; Das, E.; Jana, N.R.; Bhattacharyya, N.P. Regulation of miR-146a by RelA/NFkB and p53 in STHdh(Q111)/Hdh(Q111) cells, a cell model of Huntington’s disease. PLoS One 2011, 6, e23837. [Google Scholar]
- Soldati, C.; Bithell, A.; Johnston, C.; Wong, K.Y.; Stanton, L.W.; Buckley, N.J. Dysregulation of REST-regulated coding and non-coding RNAs in a cellular model of Huntington’s disease. J. Neurochem. 2012, 124, 418–430. [Google Scholar]
- Jin, J.; Cheng, Y.; Zhang, Y.; Wood, W.; Peng, Q.; Hutchison, E.; Mattson, M.P.; Becker, K.G.; Duan, W. Interrogation of brain miRNA and mRNA expression profiles reveals a molecular regulatory network that is perturbed by mutant huntingtin. J. Neurochem. 2012, 123, 477–490. [Google Scholar] [CrossRef]
- Haramati, S.; Chapnik, E.; Sztainberg, Y.; Eilam, R.; Zwang, R.; Gershoni, N.; McGlinn, E.; Heiser, P.W.; Wills, A.M.; Wirguin, I.; et al. miRNA malfunction causes spinal motor neuron disease. Proc. Natl. Acad. Sci. USA 2010, 107, 13111–13116. [Google Scholar]
- Russell, A.P.; Wada, S.; Vergani, L.; Hock, M.B.; Lamon, S.; Léger, B.; Ushida, T.; Cartoni, R.; Wadley, G.D.; Hespel, P.; et al. Disruption of skeletal muscle mitochondrial network genes and miRNAs in amyotrophic lateral sclerosis. Neurobiol. Dis. 2012, 49C, 107–117. [Google Scholar]
- Morel, L.; Regan, M.; Higashimori, H.; Ng, S.K.; Esau, C.; Vidensky, S.; Rothstein, J.; Yang, Y. Neuronal exosomal miRNA-dependent translational regulation of astroglial glutamate transporter GLT1. J. Biol. Chem. 2013, 288, 7105–7116. [Google Scholar] [CrossRef]
- Williams, A.H.; Valdez, G.; Moresi, V.; Qi, X.; McAnally, J.; Elliott, J.L.; Bassel-Duby, R.; Sanes, J.R.; Olson, E.N. microRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science 2009, 326, 1549–1554. [Google Scholar]
- Buratti, E.; de Conti, L.; Stuani, C.; Romano, M.; Baralle, M.; Baralle, F. Nuclear factor TDP-43 can affect selected microRNA levels. FEBS J. 2010, 277, 2268–2281. [Google Scholar] [CrossRef]
- Gentner, B.; Visigalli, I.; Hiramatsu, H.; Lechman, E.; Ungari, S.; Giustacchini, A.; Schira, G.; Amendola, M.; Quattrini, A.; Martino, S.; et al. Identification of hematopoietic stem cell-specific miRNAs enables gene therapy of globoid cell leukodystrophy. Sci. Transl. Med. 2010, 2, 58ra84. [Google Scholar] [CrossRef]
- Ozsait, B.; Komurcu-Bayrak, E.; Levula, M.; Erginel-Unaltuna, N.; Kähönen, M.; Rai, M.; Lehtimäki, T.; Laaksonen, R. Niemann-Pick type C fibroblasts have a distinct microRNA profile related to lipid metabolism and certain cellular components. Biochem. Biophys. Res. Commun. 2010, 403, 316–321. [Google Scholar] [CrossRef]
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s disease. Lancet 2011, 377, 1019–1031. [Google Scholar]
- Selkoe, D.; Mandelkow, E.; Holtzman, D. Deciphering Alzheimer disease. Cold Spring Harb. Perspect Med. 2012, 2, a011460. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef]
- Long, J.M.; Ray, B.; Lahiri, D.K. microRNA-153 physiolofically inhibits expression of amyloid-B precursor protein in cultured human fetal brain cells and is dysregulated in a subset of Alzheimer disease patients. J. Biol. Chem. 2012, 287, 31298–31310. [Google Scholar]
- Liang, C.; Zhu, H.; Xu, Y.; Huang, L.; Ma, C.; Deng, W.; Liu, Y.; Qin, C. microRNA-153 negatively regulates the expression of amyloid precursor protein and amyloid precursor-like protein 2. Brain Res. 2012, 1455, 103–113. [Google Scholar] [CrossRef]
- Zovoilis, A.; Agbemenyah, H.Y.; Agis-Balboa, R.C.; Stilling, R.M.; Edbauer, D.; Rao, P.; Farinelli, L.; Delalle, I.; Schmitt, A.; Falkai, P.; et al. microRNA-34c is a novel target to treat dementias. EMBO J. 2011, 30, 4299–4308. [Google Scholar] [CrossRef]
- Wang, X.; Liu, P.; Zhu, H.; Xu, Y.; Ma, C.; Dai, X.; Huang, L.; Liu, Y.; Zhang, L.; Qin, C. miR-34a, a microRNA up-regulated in a double transgenic mouse model of Alzheimer’s disease, inhibits bcl2 translation. Brain Res. Bull. 2009, 80, 268–273. [Google Scholar] [CrossRef]
- Agostini, M.; Tucci, P.; Killick, R.; Candi, E.; Sayan, B.S.; Rivetti di Val Cervo, P.; Nicotera, P.; McKeon, F.; Knight, R.A.; Mak, T.W.; et al. Neuronal differentiation by TAp73 is mediated by microRNA-34a regulation of synaptic protein targets. Proc. Natl. Acad. Sci. USA 2011, 108, 21093–21098. [Google Scholar]
- Hébert, S.S.; Horré, K.; Nicolaï, L.; Papadopoulou, A.S.; Mandemakers, W.; Silahtaroglu, A.N.; Kauppinen, S.; Delacourte, A.; de Strooper, B. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase expression. Proc. Natl. Acad. Sci. USA 2008, 105, 6415–6420. [Google Scholar] [CrossRef]
- Hirano, A. Hirano bodies and related neuronal inclusions. Neuropathol. Appl. Neurobiol. 1994, 20, 3–11. [Google Scholar] [CrossRef]
- Schonrock, N.; Humphreys, D.T.; Preiss, T.; Götz, J. Target gene repression mediated by miRNAs miR-181c and miR-9 both of which are down-regulated by amyloid-β. J. Mol. Neurosci. 2012, 46, 324–335. [Google Scholar]
- Hoehn, M.; Yahr, M.D. Parkinsonism: Onset, progression, and mortality. Neurology 2001, 57, 11–26. [Google Scholar]
- Reinhardt, A.; Feuillette, S.; Cassar, M.; Callens, C.; Thomassin, H.; Birman, S.; Lecourtois, M.; Antoniewski, C.; Tricoire, H. Lack of miRNA misregulation at early pathological stages in drosophila neurodegenerative disease models. Front. Genet. 2012, 3, 226. [Google Scholar]
- Shtilbans, A.; Henchcliffe, C. Biomarkers in Parkinson’s disease: An update. Curr. Opin. Neurol. 2012, 25, 460–465. [Google Scholar] [CrossRef]
- Kapushesky, M.; Adamusiak, T.; Burdett, T.; Culhane, A.; Farne, A.; Filippov, A.; Holloway, E.; Klebanov, A.; Kryvych, N.; Kurbatova, N.; et al. Gene Expression Atlas update—A value-added database of microarray and sequencing-based functional genomics experiments. Nucleic Acids Res. 2012, 40, D1077–D1081. [Google Scholar] [CrossRef]
- Thomas, B.; Beal, M.F. Parkinson’s disease. Hum. Mol. Genet. 2007, 16, R183–R194. [Google Scholar] [CrossRef]
- Khodr, C.E.; Pedapati, J.; Han, Y.; Bohn, M.C. Inclusion of a portion of the native SNCA 3'UTR reduces toxicity of human S129A SNCA on striatal-projecting dopamine neurons in rat substantia nigra. Dev. Neurobiol. 2012, 72, 906–917. [Google Scholar]
- Doxakis, E. Post-transcriptional regulation of alpha-synuclein expression by mir-7 and mir-153. J. Biol. Chem. 2010, 285, 12726–12734. [Google Scholar] [CrossRef]
- Santosh, P.S.; Arora, N.; Sarma, P.; Pal-Bhadra, M.; Bhadra, U. Interaction map and selection of microRNA targets in Parkinson’s disease-related genes. J. Biomed. Biotechnol. 2009, 2009, 363145. [Google Scholar]
- Wang, G.; van der Walt, J.M.; Mayhew, G.; Li, Y.J.; Züchner, S.; Scott, W.K.; Martin, E.R.; Vance, J.M. Variation in the miRNA-433 binding site of FGF20 confers risk for Parkinson disease by overexpression of alpha-synuclein. Am. J. Hum. Genet. 2008, 82, 283–289. [Google Scholar] [CrossRef]
- Hébert, S.S.; de Strooper, B. Molecular biology miRNAs in neurodegeneration. Science 2007, 317, 1179–1180. [Google Scholar] [CrossRef]
- Heyer, M.P.; Pani, A.K.; Smeyne, R.J.; Kenny, P.J.; Fleng, G. Normal midbrain dopaminergic neuron development and function in miR133b mutant mice. J. Neurosci. 2012, 32, 10887–10894. [Google Scholar]
- Margis, R.; Margis, R.; Rieder, C.R. Identification of blood microRNAs associated to Parkinson’s disease. J. Biotechnol. 2011, 152, 96–101. [Google Scholar]
- Mouradian, M.M. MicroRNAs in Parkinson’s disease. Neurobiol. Dis. 2012, 46, 279–284. [Google Scholar] [CrossRef]
- Gascon, E.; Gao, F.B. Cause or effect: Misregulation of microRNA pathways in Neurodegeneration. Front. Neurosci. 2012, 6, 48. [Google Scholar]
- Ha, T.Y. microRNAs in human diseases: From autoimmune diseases to skin. Psychiatr. Neurodegener. Dis. Immune Netw. 2011, 11, 227–244. [Google Scholar]
- Fiszer, A.; Olejniczak, M.; Switonski, P.M.; Wroblewska, J.P.; Wisniewska-Kruk, J.; Mykowska, A.; Krzyzosiak, W.J. An evaluation of oligonucleotide-based therapeutic strategies for polyQ diseases. BMC Mol. Biol. 2012, 13, 6. [Google Scholar]
- Witkos, T.M.; Koscianska, E.; Krzyzosiak, W.J. Practical Aspects of microRNA Target Prediction. Curr. Mol. Med. 2011, 11, 93–109. [Google Scholar]
- Hu, J.; Liu, J.; Corey, D.R. Allele-selective inhibition of huntingtin expression by switching to an miRNA-like RNAi mechanism. Chem. Biol. 2010, 17, 1183–1188. [Google Scholar]
- Hodges, A.; Strand, A.D.; Aragaki, A.K.; Kuhn, A.; Sengstag, T.; Hughes, G.; Elliston, L.A.; Hartog, C.; Goldstein, D.R.; Thu, D.; et al. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum. Mol. Genet. 2006, 15, 965–977. [Google Scholar]
- Buckley, N.J.; Johnson, R.; Zuccato, C.; Bithell, A.; Cattaneo, E. The role of REST in transcriptional and epigenetic dysregulation in Huntington’s disease. Neurobiol. Dis. 2010, 39, 28–39. [Google Scholar]
- Corney, D.C.; Flesken-Nikitin, A.; Godwin, A.K.; Wang, W.; Nikitin, A.Y. microRNA-34b and MicroRNA-34c are targets of P53 and cooperate in control of cell proliferation and adhesion-independent growth. Cancer Res. 2007, 67, 8433–8438. [Google Scholar] [CrossRef]
- Morlando, M.; Dini Modigliani, S.; Torrelli, G.; Rosa, A.; di Carlo, V.; Caffarelli, E.; Bozzoni, I. FUS stimulates microRNA biogenesis by facilitating co-transcriptional Drosha recruitment. EMBO J. 2012, 31, 4502–4510. [Google Scholar] [CrossRef]
- Butovsky, O.; Siddiqui, S.; Gabriely, G.; Lanser, A.J.; Dake, B.; Murugaiyan, G.; Doykan, C.E.; Wu, P.M.; Gali, R.R.; Iyer, L.K.; et al. Modulating inflammatory monocytes with a unique microRNA gene signature ameliorates murine ALS. J. Clin. Invest. 2012, 122, 3063–3087. [Google Scholar]
- De Felice, B.; Guida, M.; Guida, M.; Coppola, C.; de Mieri, G.; Cotrufo, R. A miRNA signature in leukocytes from sporadic amyotrophic lateral sclerosis. Gene 2009, 508, 35–40. [Google Scholar]
- Lin, N.; Friedlander, R.M. Regeneration of neuromuscular synapses: Action of microRNA-206. Neurosurgery 2010, 66, N19–N20. [Google Scholar] [CrossRef]
- Fox, M.A.; Sanes, J.R.; Borza, D.B.; Eswarakumar, V.P.; Fässler, R.; Hudson, B.G.; John, S.W.; Ninomiya, Y.; Pedchenko, V.; Pfaff, S.L.; et al. Distinct target-derived signals organize formation, maturation, and maintenance of motor nerve terminals. Cell 2007, 129, 179–193. [Google Scholar] [CrossRef]
- Umemori, H.; Sanes, J.R. Signal regulatory proteins (SIRPS) are secreted presynaptic organizing molecules. J. Biol. Chem. 2008, 283, 34053–34061. [Google Scholar] [CrossRef]
- Beer, H.D.; Bittner, M.; Niklaus, G.; Munding, C.; Max, N.; Goppelt, A.; Werner, S. The fibroblast growth factor binding protein is a novel interaction partner of FGF-7, FGF-10 and FGF-22 and regulates FGF activity: Implications for epithelial repair. Oncogene 2005, 24, 5269–5277. [Google Scholar] [CrossRef]
- Santambrogio, S.; Ricca, A.; Maderna, C.; Ieraci, A.; Aureli, M.; Sonnino, S.; Kulik, W.; Aimar, P.; Bonfanti, L.; Martino, S.; et al. The galactocerebrosidase enzyme contributes to maintain a functional neurogenic niche during early post-natal CNS development. Hum. Mol. Genet. 2012, 21, 4732–4750. [Google Scholar] [CrossRef]
- Neri, M.; Ricca, A.; di Girolamo, I.; Alcala’-Franco, B.; Cavazzin, C.; Orlacchio, A.; Martino, S.; Naldini, L.; Gritti, A. Neural stem cell gene therapy ameliorates pathology and function in a mouse model of globoid cell leukodystrophy. Stem Cells 2011, 29, 1559–1571. [Google Scholar] [CrossRef]
- Lattanzi, A.; Neri, M.; Maderna, C.; di Girolamo, I.; Martino, S.; Orlacchio, A.; Amendola, M.; Naldini, L.; Gritti, A. Widespread enzymatic correction of CNS tissues by a single intracerebral injection of therapeutic lentiviral vector in leukodystrophy mouse models. Hum. Mol. Genet. 2010, 19, 2208–2227. [Google Scholar] [CrossRef]
- Martino, S.; Consiglio, A.; Cavalieri, C.; Tiribuzi, R.; Costanzi, E.; Severini, G.M.; Emiliani, C.; Bordignon, C.; Orlacchio, A. Expression and purification of a human, soluble Arylsulfatase A for Metachromatic Leukodystrophy enzyme replacement therapy. J. Biotechnol. 2005, 117, 243–251. [Google Scholar]
- Orchard, P.J.; Wagner, J.E. Leukodystrophy and gene therapy with a dimmer switch. N. Engl. J. Med. 2011, 364, 572–573. [Google Scholar] [CrossRef]
- Martino, S.; Tiribuzi, R.; Tortori, A.; Conti, D.; Visigalli, I.; Lattanzi, A.; Biffi, A.; Gritti, A.; Orlacchio, A. Specific determination of beta-galactocerebrosidase activity via competitive inhibition of beta-galactosidase. Clin. Chem. 2009, 55, 541–548. [Google Scholar] [CrossRef]
- Visigalli, I.; Ungari, S.; Martino, S.; Park, H.; Cesani, M.; Gentner, B.; Sergi Sergi, L.; Orlacchio, A.; Naldini, L.; Biffi, A. The galactocerebrosidase enzyme contributes to the maintenance of a functional hematopoietic stem cell niche. Blood 2010, 116, 1857–1866. [Google Scholar] [CrossRef]
- Osborn, M.J.; McElmurry, R.T.; Lees, C.J.; DeFeo, A.P.; Chen, Z.Y.; Kay, M.A.; Naldini, L.; Freeman, G.; Tolar, J.; Blazar, B.R. Minicircle DNA-based gene therapy coupled with immune modulation permits long-term expression of α-L-iduronidase in mice with mucopolysaccharidosis type I. Mol. Ther. 2011, 19, 450–460. [Google Scholar]
- Liu, N.; Landreh, M.; Cao, K.; Abe, M.; Hendriks, G.J.; Kennerdell, J.R.; Zhu, Y.; Wang, L.S.; Bonini, N. The microRNA miR-34 modulates ageing and neurodegeneration in Drosophila. Nature 2012, 482, 519–523. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).