Ecological and Temporal Constraints in the Evolution of Bacterial Genomes


{kind=link}
{kind=link}
{kind=link}
Abstract
: Studies on the experimental evolution of microorganisms, on their in vivo evolution (mainly in the case of bacteria producing chronic infections), as well as the availability of multiple full genomic sequences, are placing bacteria in the playground of evolutionary studies. In the present article we review the differential contribution to the evolution of bacterial genomes that processes such as gene modification, gene acquisition and gene loss may have when bacteria colonize different habitats that present characteristic ecological features. In particular, we review how the different processes contribute to evolution in microbial communities, in free-living bacteria or in bacteria living in isolation. In addition, we discuss the temporal constraints in the evolution of bacterial genomes, considering bacterial evolution from the perspective of processes of short-sighted evolution and punctual acquisition of evolutionary novelties followed by long stasis periods.1. Introduction
The classical theory of evolution is mainly based on the study of multicellular organisms. However, most of the biosphere is composed of unicellular microorganisms, which were not well known at the time when the theory of evolution came about. On the other hand, whereas experimental evolution studies are not easily achievable for multicellular organisms [1], due to the extensive time lapse required, these studies can be performed on microorganisms, which can present very large population sizes and extremely short generation times. Therefore, studies on bacterial evolution are of increasing interest to understand the basic mechanisms of evolution.
Bacterial experimental evolution studies as those performed by the groups of Lenski [2-7], Rainey [8-13], Levin [14,15] or Kolter [16-19] among several others, have served to experimentally test several general evolutionary processes, which would not be easily tested using multicellular organisms as models. These processes include sympatric diversification, punctuated evolution, kin selection, prey-predator interactions, bet-hedging or the effect of cheating on group selection among others.
Along with the studies on experimental evolution, the analysis on the in vivo evolution of microorganisms provides relevant information for understanding general aspects of the theory of evolution. Particularly relevant in this respect are the studies on the evolution of bacterial pathogens that produce long-lasting chronic infections. An example of this situation is the evolution experienced by Pseudomonas aeruginosa when this organism produces chronic infections. P. aeruginosa is an opportunistic pathogen that can colonize the lung of cystic fibrosis patients during decades and evolves during this colonization [20]. Studies on population biology dealing with the acquisition of antibiotic resistance by bacterial pathogens have also provided valuable information for understanding evolution [21].
The increased availability of the full genome sequences of prototypic strains of several bacterial species allows the detailed analysis of the differential effect that processes such as mutation and horizontal gene transfer (HGT) may have on the evolution of bacterial genomes. More recently, efforts have focused on sequencing several isolates belonging to the same bacterial species, in order to get a closer view to the process of bacterial diversification. These analyses, together with ecological studies that link the habitat of each species/isolate to its corresponding genomic ecotype might allow a more in depth understanding of the mechanisms driving bacterial evolution [22].
One important issue to be mentioned here is the fact that, in addition to the universal principle of evolution based in the selection of gradual modified descendants (mutants) claimed by Darwin [23] and the proponents of the Modern Synthesis [24], HGT, which allows fast, stepwise adaptation by quantum leaps [25], is a major evolutionary force in bacteria [26], and an example of punctuated evolution [27]. Of course, the acquisition of genes from other organisms can occur in all living beings, and indeed transposons were discovered in corn [28], but the relevance that HGT, as driver for acquisition of important adaptive traits [29-34], has on microbial evolution seems to be much higher than for other organisms [35-37].
Bacterial genome evolution is thus modulated by two main mechanisms: mutation (and recombination), which is common to the evolution of all living beings, and genome remodeling that results from gene acquisition and gene loss, and is more relevant for bacteria. It is important to note here that gene acquisition is only possible when microorganisms form part of communities, which contain members that may act as donors and recipients of the transferred elements. Mutation however is the unique mechanisms of variation for those organisms growing in isolation. Finally, gene loss is frequent for bacteria as endosymbionts that colonize a single ecosystem, where the physicochemical conditions are very constant through time.
In this article, we will review how these different processes contribute to the evolution of bacterial genomes (considering as bacterial genome both the chromosomal element and the mobilome or ensemble of mobile elements [32]), in relation to the different ecological conditions under which bacterial evolution occurs.
2. Tracking Phylogenetic Relationships in Bacteria
Molecular methods for tracking the phylogenetic relationship, and hence the evolution tree of organisms, are mainly based on the analysis of sequences of ortholog genes, being those encoding ribosomal RNAs the most popular to distinguish between species, to the point that this method has come to be considered the blueprint for reconstructing phylogenies [38]. However, whereas for higher organisms the trees generated using different orthologs are generally congruent, this is not necessarily so in bacterial species, where gene trees for different orthologs frequently show incongruencies [38,39] among them and with the aforementioned rDNA tree. HGT has been postulated to explain these incongruent trees and today; the acquisition of genes, plasmids and other genetic elements by horizontal gene transfer is accepted as an important mechanism for driving the evolution of bacterial genomes [35-37]. Evolution might be driven as well by gene duplication, divergence of paralogs and consequently genome expansion. Indeed, it has been suggested that this is an important mechanism for the evolution of myxobacteria [40], a group of social eubacterial predators characterized by the large size of their genomes [41]. Nevertheless, recent studies have shown that that this type of evolution is not very relevant in other bacterial species. As a consequence, it has been suggested that gene duplication and ulterior diversification of paralog genes play a much less important role in bacterial gene diversification than the acquisition of xenologs by HGT [42]. Altogether, this means that, although some vertical phylogenetic signal can still be obtained for microorganisms [43], genetic exchange has led to networks models when tracking evolutionary patterns [44].
The methods currently in use for distinguishing clones or populations in a given bacterial species can give insights into the relevance of HGT and mutation on these organisms. The golden standards for determining clonal relationships are pulse field gel electrophoresis (PFGE) and multilocus sequencing typing (MLST), each of which measures a different feature of bacterial genomes [45]. PFGE analyses the overall structure of bacterial genomes, and can therefore measure intragenomic recombination and gene trafficking (gene acquisition by HGT and gene loss), whereas MLST measures mutation in genes that are common to all members of a given species, otherwise known as the core genome [46]. One important aspect of these technologies with regards to bacterial evolution is the consistency of the attained results.
Bacteria have the outstanding capacity of modifying their genome either by mutation or by HGT. This capacity, together with the short generation times and considerably large populations of bacteria may enable considerable diversification. Indeed, bacteria colonize all known ecosystems in the biosphere, including extreme habitats. Consequently, bacterial species exhibit a great variety in the length of their genomes and in the type of genes they harbor. However, and despite this large ecological and genomic variability, the sequences of genes belonging to the core genome are usually greatly conserved for each bacterial species, even at the third position of the codons, which indicates that purifying selection is likely playing a relevant role in the evolution of bacterial populations. As will be discussed further on, short-sighted evolution [45,47] might be more able to justify the long lasting stability of bacterial core genomes than mutation clearance that is frequent in populations with sexual reproduction.
3. Sympatric Diversification Driven by Mutation and HGT
The presence of HGT-acquired elements in bacterial chromosomes makes the definition of bacterial species to be a fuzzy concept [22]. Because of this, different species concepts have been proposed for the bacterial world, among which is the proposal that ecotypes, defined as those bacteria presenting the same ecological behavior and a similar core genome, constitute valid taxonomic groups [48,49].
Whereas the core genome, that presents few changes among closely related bacteria, might define what is common, and thus the taxonomic root, ecotypes will define specific adaptations to particular ecosystems, a mode of sympatric diversification. Sympatric evolution of bacteria has been studied using experimental evolution models [50]. Furthermore, studies on the population dynamics of bacterial species based on the analysis of full-genome sequences allow to establish the role of sympatric diversification in the evolution of natural bacteria populations [51].
In vitro experiments have shown that the free-living bacterium Pseudomonas fluorescens can evolve rapidly when confronted with new environmental conditions to generate a repertoire of mutants that are capable of colonizing different habitats in a structured environment [9,11]. Since, during the experiment, bacteria grew in isolation, without any other counterpart that might be a donor of DNA, mutation is the only mechanism for achieving this diversification. It is important to note that the same types of mutants are selected when the experiment is repeated, which demonstrates that the adaptation process is not completely stochastic in the sense that adapting to the same ecosystem will involve the selection of the same variants.
These conclusions, derived from studies based on experimental evolution models, have been confirmed by the analysis of the in vivo evolution of P. aeruginosa, when this bacterial species colonizes the lungs of cystic fibrosis patients and presents a fast adaptation process [52-54]. People suffering from cystic fibrosis are frequently infected by P. aeruginosa and the same clone remains in the lung for decades. Since P. aeruginosa is an environmental microorganism, chronic infection requires the adaptation to a new ecosystem, in this case, the human lung. As in the case of the above mentioned in vitro experiment, P. aeruginosa diversifies into distinct populations as a consequence of mutations. Notably, the same set of mutations is found in isolates from different patients [55,56], and a similar adaptation is observed in patients suffering chronic obstructive pulmonary disease [57], indicating again that the process is not completely stochastic and that similar mutations may be expected to occur each time bacteria are confronted with same new environment. Overall this indicates that the processes of mutation and selection on their own may lead to the rapid diversification of bacterial populations.
Does this have a relevant implication in the long-term evolution of bacterial genomes? There is no simple answer to this question. In the case of free-living bacteria as those mentioned above, adaptation to a given environment would mean de-adaptation from another. In this situation, the stability of the genomes is guaranteed by periodic selection. On the other hand, diversification occasionally involves the emergence of cheaters, that are more fit than the evolved variant, but that can lead to the disruption of the whole community [9]. Because of this, in several occasions, fast mutation driven-diversification does not lead to long-term evolution, but is an example of short-sighted evolution (see below).
A different situation might take place when genes are transferred by HGT. In this case, bacteria can acquire a full set of proteins in a single step that enable them to colonize a new ecosystem. Nevertheless, and along with this potential adaptive advantage, the acquisition of new DNA confers a fitness cost to the new host because of the need to maintain, replicate, transcribe and translate the novel genetic elements [58,59]. Furthermore, the introduction of new proteins will require their adaptation to the host's metabolic and regulatory networks.
Under these circumstances, HGT-acquired genes will be rapidly lost unless they render relevant fitness benefits. Two examples that provide information on the trade-offs between fitness gain and fitness costs derived from the acquisition of novel genes by HGT are the acquisition of DNA conferring resistance to a toxic compound and the acquisition of DNA conferring the capability to colonize new environments. The first example has been explored with regard to antibiotic resistance. In an antibiotic-rich environment, bacteria are required to be drug resistant [60]. This means that when susceptible and resistant microorganisms are exposed to this type of strong selection, only the resistant ones will survive and the susceptible (parental) microorganisms will disappear. Nevertheless, since acquisition of DNA implies a fitness cost, it can therefore be predicted that once selection is over, the element conferring resistance will be lost, in a new example of periodic selection [21,61]. However, several plasmids have easy-to-get, hard-to-lose elements, either because they can contain toxin-antitoxin elements or because they harbor relevant elements that can be co-selected [21]. On the other hand, it has been demonstrated that bacteria can acquire mutations that compensate for the costs associated with resistance [62], which means that in habitats without the toxic selector agent, both susceptible and resistant bacteria can co-exist [63-65].
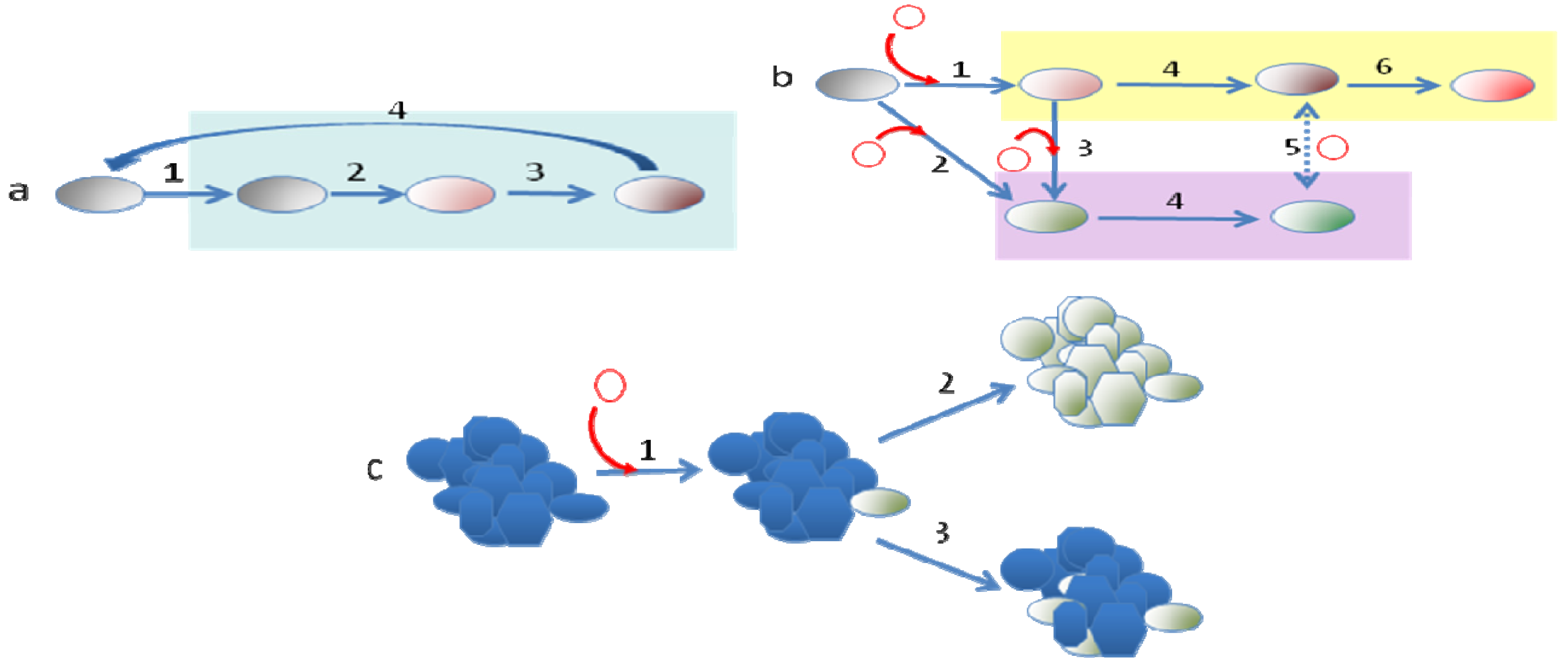
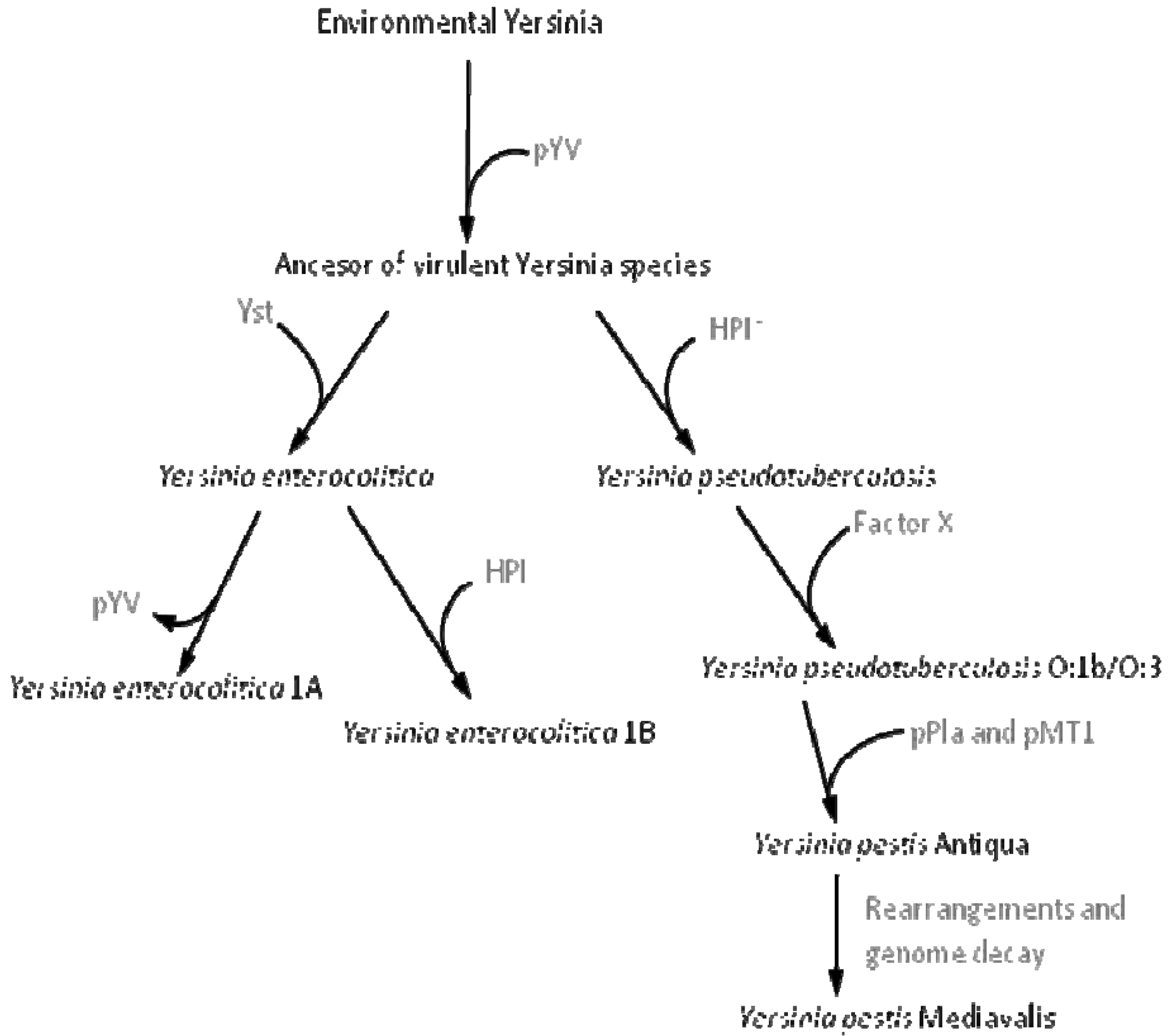
The situation observed when bacteria acquire DNA, which enables them to colonize a new ecosystem, might have a higher relevance for their long-term evolution. Indeed it has been shown that the acquisition and/or loss of DNA regions (genomic islands) not forming part of the core genome contributes to the diversification and adaptation of bacteria to colonize novel niches [29,31,33,66]. Since orfan genes and those coding hypothetical proteins are frequently specific for each bacterial species, it has been suggested that they might play a determinant role in the adaptation of the microorganisms to different habitats [67]. A good example of how the incorporation of novel DNA by HGT into a bacterial genome can trigger speciation is the acquisition of pathogenicity islands [29,34,68] by, Yersinia pestis [69-71], the cause of the plague. The genus Yersinia encompasses 15 species, three of which (Y. pestis, Y. pseudotuberculosis and Y. enterocolitica) are pathogenic to humans. All three pathogenic members of the Yersinia genus target the lymph tissues during infection because they carry the pYV virulence plasmid, which is needed for infecting these tissues and for overcoming the host defense mechanisms. This first event of HGT is the one that allowed the pathogenic Yersinia species to access a new habitat that lacked frequent bacterial competitors and was the initial quantum leap required for the speciation of these pathogens, making them able to access to a new niche (infected host). This evolution in quantum leaps has been followed by the incorporation of further elements, the loss of dispensable genes and mutations leading to the fine-tuning of the acquired determinants with the pre-existing bacterial regulatory and metabolic networks, all of which have led to the speciation of the Yersinia genus (see Figure 1 for details of this evolutionary process).
4. Evolution in Microbial Communities
Microbial communities, such as mammal gut microbioma or plants' rhizosphere, constitute dense and biodiverse ecosystems where different bacteria can support similar selective pressures. It is therefore not strange that in these communities, acquired genes conferring a relevant adaptive advantage can spread across the community members. This might be particularly important when the community must respond to the presence of a toxic compound such as an antibiotic. In these circumstances, each member of the community is required to be resistant, and the spread of antibiotic resistance genes will be favored by second-order selection processes [73,74]. This means that the introduction in a microbial community, in which members share the capability of exchanging DNA (genetic exchange communities [75]), of a given determinant that is useful for each of the members of the community, will lead to the presence of a strong selective force to the dissemination of the determinant among all members of the community. This might be the reason why genomes from coexisting microbes tend to be more similar than would be expected by chance [76]. For instance, some studies have recently shown that a high degree of interphyllum genetic exchange exists in Thermotogales or Aquificales bacterial groups that allow the widespread adaptation to shared environments between genealogically distinct bacteria [77,78]
Nevertheless, the acquisition of a new determinant by HGT does not necessarily imply its dissemination among all members of the population. This can be the case for the acquisition of genes encoding agarases or other carbohydrates present in algae and absent in terrestrial organisms by the human gut bacterium Bacteroides plebius in Japanese people [79]. This acquisition seems to have been mediated by the ingestion of marine bacteria associated to algal diet in the human population. Despite their acquisition by B. plebius, these elements have not disseminated among other microorganisms living in the same habitat. This may happen because, once a member of the community acquires the biodegradative characteristics required to use a novel resource, the stable food trade-offs already established among the population allow the use of this novel resource without the need of incorporating DNA by all members of the community. This possibility fits well with the idea that group selection, besides kin selection, might be an important evolutionary event in establishing cooperation among different organisms inhabiting complex, yet stable, communities [80].
The dissemination of HGT-acquired genes can be restricted by their epigenetic compatibility with the recipient genome. The products encoded by these novel genes need to be incorporated into regulatory and metabolic networks in recipient bacteria [73,81,82]. This process in not always easy (presence of similar products in the recipient network competing with the new product, problems in the expression of new genes, connectivity degree with other elements in the network, etc.), and this situation might impede the fixation of HGT-acquired determinants [81], in such a way that most new incoming sequences are rapidly eliminated [83]. However in some cases, acquired genes are of instant use [84], allowing recipient bacteria to instantaneously exploit the evolutionary novelty provided by the new elements acquired by HGT. The trade-offs between fitness costs (including epigenetic compatibility) and fitness gain, understood as the capacity to exploit a new habitat, constitute a major bottleneck for the fixation of HGT-acquired genes.
5. Free-Living Bacteria, When Size Matters
Free-living bacteria are those capable of colonizing a variety of ecosystems. For these bacterial species, two different types of evolution can be foreseen. In the first, all members of each given species are able to colonize the different habitats, which means that this bacterial species will have large core genomes and limited accessory ones.
This is likely to be the situation for P. aeruginosa, a bacterial species capable of colonizing a variety of terrestrial and aquatic habitats, and producing infections in different hosts ranging from plants to humans. It is noteworthy that the same virulence determinants required to infect plants serve to infect humans [85]. This suggests that all members of these bacterial species have the capacity of colonize environmental and clinical ecosystems [86]. The detailed genetic analysis of several isolates from different ecosystems has shown that indeed there is not a clear cut-off between environmental and clinical isolates of this bacterial species [45,87].
Furthermore, the genome of P. aeruginosa is large and enriched in sensory and regulatory elements [88], indicating the need of sensing different ecosystems and respond accordingly by all members of the species. This population structure does not mean however that P. aeruginosa is genetically static. Although the core genome is large and presents an overall conserved synteny, a few loci are still subject to diversifying selection. These loci present a non-random association of genotypes, such as defining clonal regions within the genome. It is important to notice that different genotypes are associated to specific repertoires of accessory genes, showing that the association of specific clones to given accessory elements is an ancient event in the evolution of P. aeruginosa [45,87], and does not seem to be particularly relevant towards enhancing the capacity of specific clones to colonize a given ecosystem.
In sharp contrast to this situation, genome analysis of several E. coli isolates shows a different type of evolution. In this bacterial species, the core genome is much smaller than that of P. aeruginosa and the accessory genome is much larger [89]. Furthermore, whereas some E. coli strains are commensals, some others are pathogens, and the differences between one and the other mainly rely on the elements acquired by HGT. This genetic structure of E. coli populations allows defining this bacterial species as a multi-specialist in the sense that the whole species can colonize different ecosystems, but each lineage has acquired a specific genetic repertoire, allowing its specialization for colonizing a given environment.
A more drastic example of this situation concerns the plankton components Prochlorococcus marinus and Pelagibacter ubique [90-92], which are supposed to be among the most abundant components of the biosphere. These organisms present extremely compact genomes, a characteristic that is common for intracellular endosymbionts but supposed to be rare in free-living organisms, because the genomic reduction observed in free living bacteria cannot easily be explained by genetic drift [93], as is the case for intracellular endosymbionts (see below). The study of different isolates of Prochlorococcus, suggests that they diversify to occupy a variety of oceanic environments with different light, temperature and salinity conditions. However, despite the authors statement that the observed genomic shrinkage and the existence of different lineages of Prochlorococcus are not remnants of genetic drift, but potential outcomes of a niche-oriented stepwise diversification, the reasons for gene-loss are still unclear. One possibility might be that the physicochemical conditions of each of these niches may be as stable as those of endosymbionts and under these conditions, a bacterial autotroph, not requiring as much metabolic versatility as an heterotroph, would have evolved towards genome reduction. In a nutrient-poor (mainly in N and P elements) environment such as sea surface water, the replication, transcription and translation of unnecessary genes is costly and therefore natural selection will favor the deletion of these genes.
The constant increase in the number of genome sequences enables the estimation of the size of core genome, which is defined as the gene repertoire that is common to all members of a given bacterial species and the so called pan-genome [37,94], defined as the full genetic repertoire of this species. It is important to mention that the core genome and the accessory genome frequently present different G+C composition, which indicates a different phylogenetic origin [46].
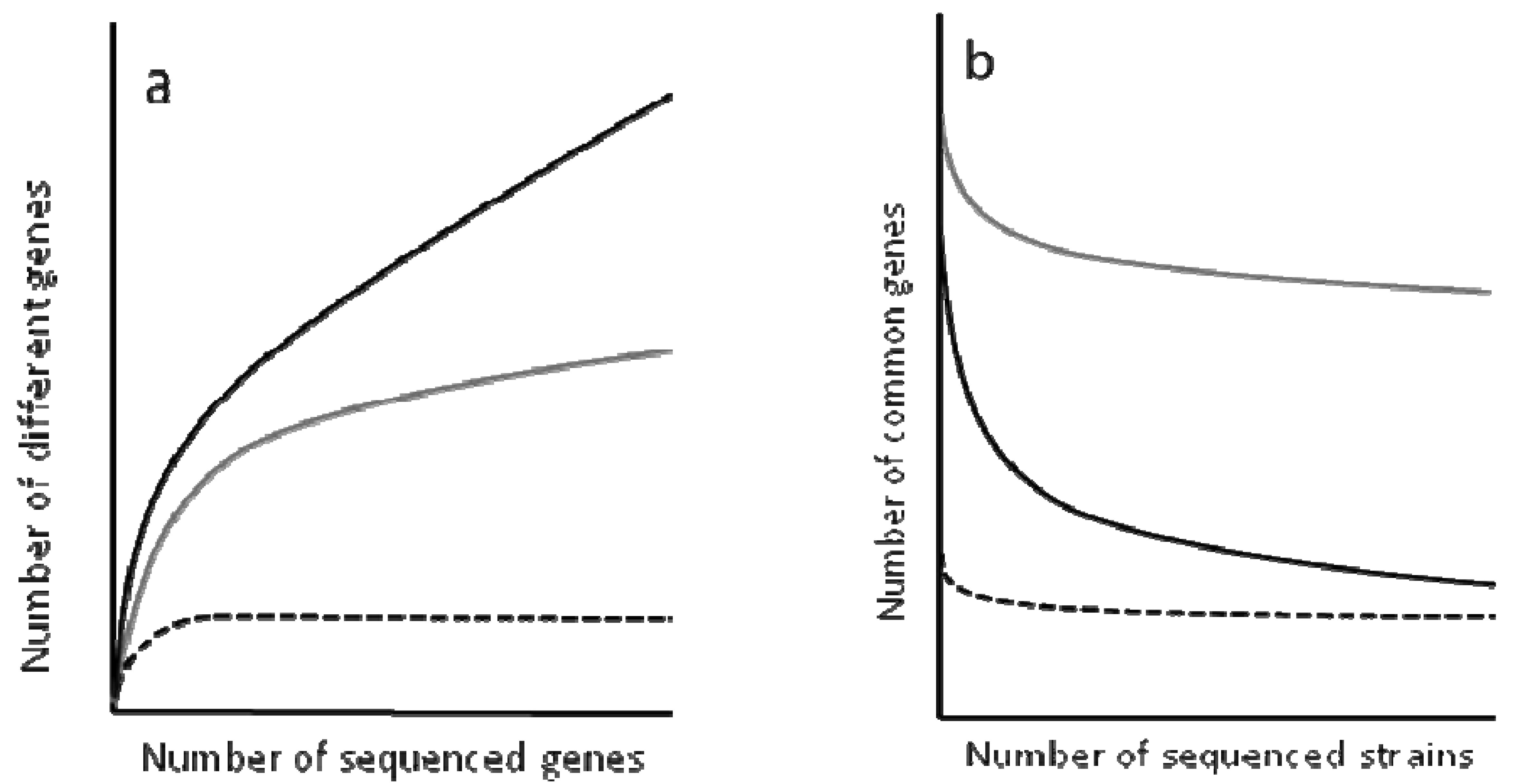
To determine the complexity of the pan-genomes, the results are analyzed using rarefaction curves (Figure 2). These curves allow the distinction between closed and open genomes [94-98]. The former are those presenting a non-asymptotic curve, which means that upon sequencing a given number of genomes, the number of novel genes does not increase. One example of closed genome is the endosymbiont Buchnera aphidicola (see the next section for a discussion of evolution in endosymbiotic bacteria). This bacterial species occupies an isolated and restricted niche that hampers its possibilities of acquiring new genes and the sequence of four strains has shown that any novel isolate to be sequenced would not contain any different gene to those that have already been found in analyed genomes.
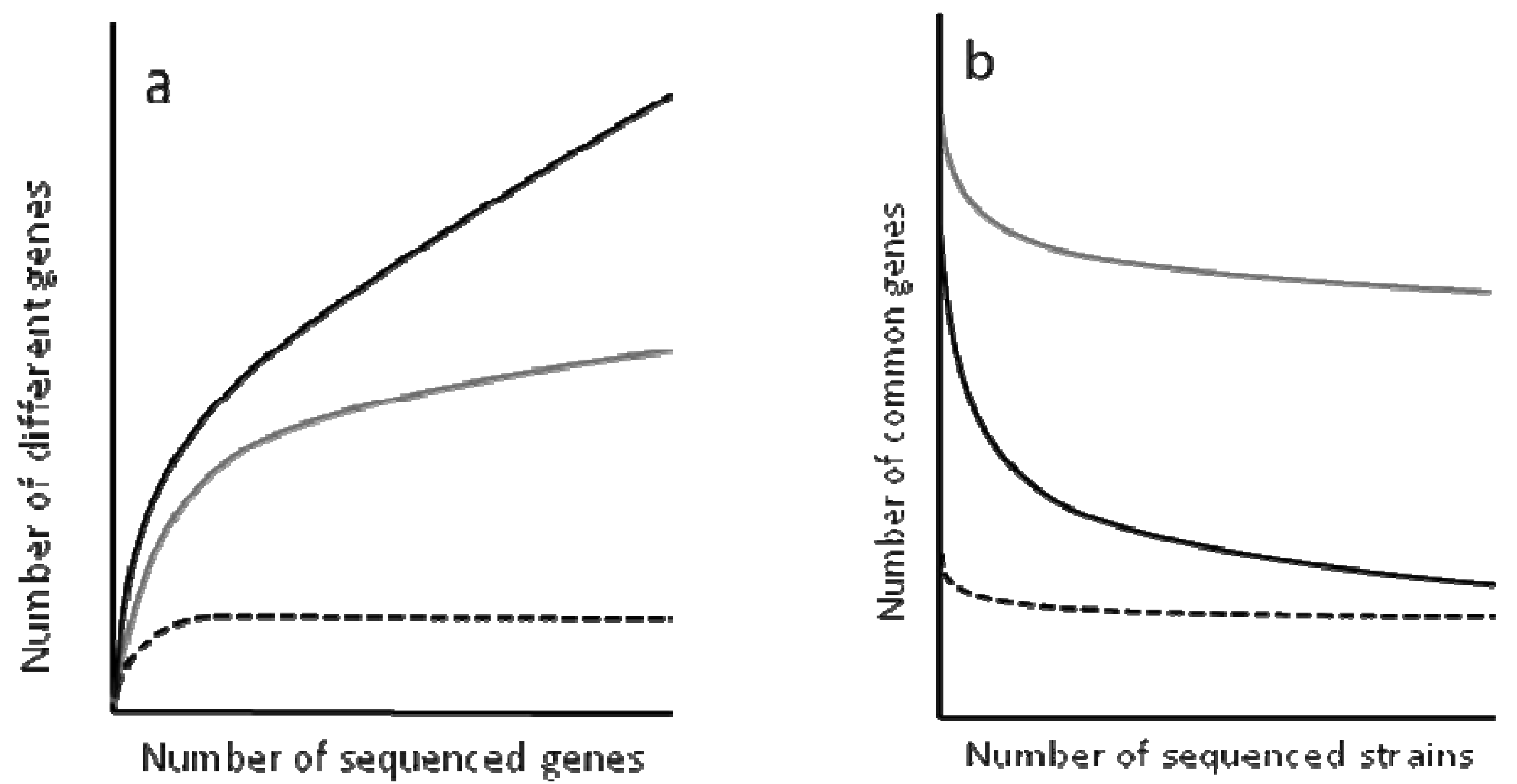
In contrast to the situation observed for B. aphidicola, the pan-genomes of several bacterial species present asymptotic rarefaction curves, indicating that the number of genes that can be shared by the population is theoretically infinite. However, the shape of the curve is not the same for all genomes and can provide an estimation of the size of the pan-genome. For instance, each new sequenced strain of Streptococcus pneumoniae adds around 30 genes to its pan-genome, and the number decreases with each new sequenced strain, whereas each new strain of E. coli adds over 300 genes to the its pangenome. Based on these analyses, it has been predicted that the pan-genome of S. pneumonia encompasses around 3300 to 5000 genes, 1647 of which make up its core genome [100]. This likely reflects that S. pneumoniae colonize a restricted habitat, but is not in isolation as Buchnera.
6. Bacteria Living in Isolation
Not all bacteria live in communities. Bacteria inhabiting either deep or poor soils, pathogens causing infections in otherwise sterile habitats (such as blood) or insect endosymbiotic bacteria live in relative isolation from other bacteria. In these circumstances, the possibilities of gene exchange are low and bacterial genome evolution is determined mainly by mutation or by genome rearrangements [101], including genome reductive evolution.
One important issue concerning mutation-driven evolution is to know whether adaptability itself can evolve [102]. In this regard, it has been shown that toxic agents such as antibiotics can transiently increase mutation rates and hence help to select resistant strains [103-105]. Increasing mutability under stress is still a controversial issue. Some models indicate that bacteria under stress (stationary phase, antibiotics) can undergo gene amplification [106-109], which is resumed upon stress removal. Other models however postulate that increased mutability under stress is a tightly regulated process [110] that involves the production of error-prone DNA polymerases and the induction of the SOS system among other processes [111,112]. Independently of which model best explains these results, it seems that mutation itself is not a fixed value and can be regulated depending on bacterial growing conditions.
The description of bacterial strains (mutators) presenting much higher mutation rates than those of the overall population [113,114] indicates that mutability is a selectable trait that can drive the evolvability [102,115] of bacterial populations. Indeed, the percentage of hypermutator strains is higher in the case of bacteria causing chronic infections, as described in [57,116,117]. This enrichment is supposed to be as a consequence of a second-order selection process by which those isolates with higher probability of acquiring antibiotic resistance (mutators) in the treated patient are selected [118]. The finding that co-evolution with viruses [119] increases in bacterial population the fraction of isolates presenting high mutation rates, further supports the idea that elevated stress levels select for mutators through second-order selection processes.
Besides mutations, in a scenario lacking DNA donors, genome rearrangements emerge as a source of novelties, leading to operon shuffling and the emergence of new operons [120] or association of different mobile elements in order to generate novel integrative elements such as multi-resistance determinants [121].
Mutations are relevant in processes of transient adaptation as those experienced by human pathogens producing chronic infections and in ensuring stable adaptation leading to long lasting evolution processes and eventually speciation as those observed in the case of endosymbionts. In the first case, the adapted strain leaves the infected host and must compete with their non-evolved counterparts. Under these circumstances, it is expected that the adapted mutant strain will be outcompeted and mutations will not be fixed (short-sighted evolution [47]). This situation has been theoretically discussed and a source-sink model for explaining the dynamics of virulence in opportunistic pathogens have been proposed, in which the evolution of such pathogens can be considered from the standpoint of continuous switching between permanent environmental (source) and transient infective (sink) habitats [122]. It is important to mention here that this mode of evolution is only possible when the opportunistic pathogen already harbors the determinants required for producing the infection in the compromised patient. Otherwise, a first step allowing access to the new niche will be required for the formerly non-pathogenic bacteria (see discussion on Y. pestis above). An exception for this situation may occur when the mutations allow epidemicity or are acquired by an epidemic clone that is already well adapted to clinical settings, in which case the clone can be maintained in the clinical setting, without competing with their counterparts that live in the species'natural habitats, and mutations can be fixed. However, this situation, although described for some P. aeruginosa clones [123], is infrequent.
In the second evolutionary process, mutation enables access to the resources of a novel environment or is secondary to the acquisition by HGT of the elements required for entering in this ecosystem. This means that, once the organism have acquired the required capacity to colonize a given ecosystem, the optimization of its metabolism is triggered towards reducing fitness costs that results from the presence in its genome of un-needed genes, and therefore leads to genome reduction [124]. This situation is observed in intracellular bacteria, especially in endosymbionts. The access to the intracellular milieu is stressful and because of this, few organisms can colonize this habitat. However, once this habitat is colonized it becomes a rather stable environment in which no other competitors are present. Insect endosymbiotic bacteria are an example. They exhibit highly derived genomes characterized by an important genomic reduction accompanied by a biased base composition toward A/T rich genomes as a consequence of their particular life mode and host dependence [125,126]. An extreme case of genomic reduction is observed in the genome of Carsonella rudii, that harbors just 182 open reading frames and exhibits the total loss of genes for numerous categories [127].
Genome shrinkage is mainly observed in obligate endosymbionts, whose genomes exhibit a high genomic reduction, an important structural stasis and high sequence evolution rate [126,128], whereas facultative endosymbionts exhibit more dynamic genomes and more gene conservation. In addition, facultative symbionts have more repetitive sequences and mobile elements than obligate symbionts, which enable the former to have higher gene plasticity.
Genome reduction follows a temporal pattern in which gene loss is fast during the first stages of the evolutionary process, diminishing afterwards and leading to stasis with few changes in the genomes once optimal adaptation is reached. One important aspect is that, besides reduction, some other changes that allow a better adaptation can be observed. For instance, the amplification of genes encoding the pathways for tryptophan and leucine biosynthesis in Buchnera [129] constitute an example of adaptation to improve provisioning of host nutrients that involve genome expansion, and not genome reduction. Although the final structure of the genome would be very similar if the process is repeated because the same unnecessary genes would be eliminated, the evolutionary trajectories might present some degree of stochasticity, with genes being eliminated in a different order. In this regard, small population size determines a reduction in the role of natural selection in endosymbiotic bacteria and hence genetic drift modulates genome evolution in the endosymbiotic scenario driving to the fixation of mildly deleterious mutations (for example amino acid changes that reduce the stability of proteins), and the elimination of nonessential genes [130]. Nevertheless, recent theoretical and experimental studies indicate that gene loss is not completely random and that metabolic constraints play a relevant role in the evolution of genome reduction of bacteria endosymbionts, indicating that both random gene drift and natural selection are partners in the process of endosymbionts evolution [81].
7. Homologous Recombination and Microevolution in Bacteria
In previous sections we have reviewed the main forces shaping the evolution of bacterial genomes as a function of the different ecosystems that bacteria might face. In addition to the different forces discussed, homologous recombination also plays an important role in bacterial evolution, allowing the exchange between closely related bacteria of small genomic regions. Two types of homologous recombination can be important for the evolution of bacterial genomes. One is when recombination occurs in a given genome without acquiring DNA from another cell (intragenomic recombination). Here, the process might produce genome reorganization and gene duplication. The other type of homologous recombination happens when a recipient cell acquires, by HGT, DNA presenting a large degree of similarity with its own genome from closely related bacteria.
Intragenomic homologous recombination can be important in the evolution of paralog genes derived from gene duplication and indeed it has been demonstrated that genome expansion of mixobacteria is driven by this mechanism (see above). However, despite several studies [131,132] suggesting a role for gene duplication events in the evolution of bacterial genomes, few duplicated genes are found in phylogenetically controlled studies for most bacterial species, and recent studies indicate that gene acquisition by HGT, more than duplication, drives functional diversification of protein families in bacteria [42,133], and hence colonization of novel ecosystems.
Another possible effect of intragenomic homologous recombination could be the generation of deletions by recombining repetitive sequences. However, studies on genome size reduction by experimental evolution suggest that extensive bacterial genome reduction can occur on a short evolutionary time scale and that homologous recombination only plays a limited role in this process [134].
Although homologous recombination between different individuals harboring different alleles of the same gene is supposed to be a mechanism of exchange of gene variants more than a mechanism for the acquisition of evolutionary novelties, it can also introduce genetic variation without the need of gene duplication One example of this situation is the development of resistance to beta-lactam antibiotics by Streptococus pneumoniae and Neisseria ghonorroeae due to the acquisition of DNA from closely related bacteria and its recombination, which allows the formation of antibiotic resistant mosaic Penicillin Binding Proteins [135,136]. Homologous recombination between members of the same species can also be an important mechanism modulating the bacterial evolution [137]. For instance, it has been proposed that the evolution of virulent E. coli clones is driven by the acquisition of pathogenicity islands, which, as in the case of Y. pestis, permits access into a new niche (infected patient), followed by homologous recombination in some parts of the genome [29,137-139] likely to evade the host immune response by generating antigenic variability, a feature described in other bacterial pathogens like Mycobacterium tuberculosis [140].
Since homologous recombination only occurs between very closely related DNA sequences, it has been suggested that the barriers to homologous recombination can be suitable markers for the classification of bacterial species [141-143]. This implies that restrictions to homologous recombination can contribute to the speciation process in bacteria [22,48]. In this regard, the finding that different clonal complexes of Salmonella enterica present restrictions for recombining has been interpreted as evidence that these lineages might represent incipient species [144].
Besides contributing to diversification, homologous recombination contributes, as in individuals with sexual reproduction, to convergent evolution and to the buffering of deleterious mutations. One example of this statement can be found in the recombination between Salmonella paratyphi A and Salmonella typhi, which has allowed the exchange of gene variants that are important for their adaptation to a common ecological niche, which is the human host [145].
One final aspect by which homologous recombination influences the evolution of bacteria is through its role in the combination of the different modules that form a mobile genetic element, which is transferred as a whole. One good example of this situation is the evolution of integrons [146]. These elements consist of arrays of gene cassettes, each one flanked by imperfect inverted repeats (att regions). Recruitment of new gene cassettes is due to the site-specific recombination [147] of the att regions, a process mediated by an integrase, also present in the integron. In recent years, several integrons, presenting different arrays of antibiotic resistance genes that are co-transferred have been described, showing that homologous recombination of short sequences flanking non-homologous genes can be a source of adaptability [148]; in this case towards the selective pressure imposed by antibiotics belonging to different structural families [149].
One important issue to discuss here is the fact that homologous recombination can be relaxed under certain conditions, thus allowing the interchange of genetic material between bacterial strains belonging to different species. This is mainly important in the case of mutator strains presenting defects in the mismatch repair (MMR) system, which present both high mutation rates (see above) and less stringent homologous recombination [150-152]. This indicates that the mutator phenotype will favor fast adaption by both increasing mutation rates and recombination. The finding that MMR genes present higher sequence mosaicism as compared to housekeeping genes in different E. coli lineages correlates with the hyper-recombination phenotype of MMR-deficient mutators, and supports a mechanism of evolution that involves modulation of mutation and recombination rates by recurrent losses and reacquisitions of MMR gene functions [153].
Considering its role in bacterial speciation, homologous recombination is a force most likely acting in the different ecological scenarios discussed in this review
8. Temporal Constraints in the Evolution of Bacterial Genomes: Punctuated Equilibrium and Short-Sighted Evolution
All along the review, we have stated that bacteria present an impressive capacity of adaptation to any given change. This adaptation is given by their high population sizes and the strength of the processes that allow diversification, which include mutation, homologous recombination and HGT. The enhanced mutability and recombination shown by bacteria under stress, as well as the existence of strains presenting high mutation rates (mutators), increases the capacity of variation of bacterial genomes and consequently the adaptability of bacterial populations. This high adaptability potential implies that when bacteria face a new selective force, their evolution should be fast. Indeed, information on the evolution of bacterial pathogens after the discovery of antibiotics indicates that this is true. A few decades after introducing antibiotics to therapy, bacteria have acquired a variety of resistance mechanisms and resistant organisms, which were susceptible before the use of antibiotics, became widespread [64,65,154-156]. However, despite this fast adaptation, bacterial core genomes are rather stable (see above), and it can be stated that the evolution of bacterial genome is an example of punctuated evolution in which periods of fast evolution are followed by stasis [27]. The periods of fast evolution are associated with the colonization of a new habitat [157] or with the presence of a new selective force. Stasis occurs after this process of fast adaptation, unless the ecosystem/selective force changes again. Following on from the example of the antibiotics, a paradigmatic example of this situation is the history of the TEM 1 β-lactamase that confers resistance to first generation β-lactams. This plasmid-encoded enzyme was rapidly disseminated in plasmids among Enterobacteriaceae (fast evolution) and remained without any change in the population (stasis) until inhibitors of its activity were launched into the market (novel selective force), i.e., the moment at which a strong allelic diversification occurred [158].
Another aspect to discuss here is whether the different processes of evolution follow a temporal pattern. This seems to be the case in the evolution of bacterial pathogens that begin with the acquisition by HGT of the elements that allow entrance into the new host, and the consequent spread in this novel niche of this evolved clone (clonal expansion), followed by the acquisition of other elements that might work to produce different types of infections and the fine-tuning of the bacterial physiological networks, driven by mutation and homologous recombination, to allow its adjustment to the conditions of the new host and permit a good integration of the newly acquired determinants (Figures 1 and 3). When the trade-offs of cost-benefits for accessing the new host impede the return of the evolved bacteria to its original habitat, a strong genome reduction by purifying selection can be foreseen [93]. The most drastic reduction in genome size occurs in obligate mutualistic endosymbionts, which form a common metabolic network. Although, it has been described that the process of gene loss has no clearly defined limit [159], most changes occur in the first stages of adaptation to a new habitat.
A final aspect of the temporal constraints of bacterial evolution, regards adaptive changes that are not fixed and, thus, do not contribute to the long-term evolution of bacterial populations. This situation has received the name of short-sighted evolution and reflects the fact that bacteria can evolve to better adapt to a given niche, but, if this adaptation represents de-adaptation to the original ecosystem and the new habitat is not stable enough to allow long-term evolution, the changes are not fixed and thus constitute futile adaptation cycles in the evolution of otherwise rather stable bacterial genomes [122].
Unlike what happens in other organisms presenting smaller population sizes, even if the adaptation of a given bacterial strain to a specific habitat compromises its fitness in the previous ecosystem, this situation will not compromise the fitness of the overall population, which is by far much larger. Because of this, short-sighted evolution is a fruitful mechanism of bacterial adaptation that allows the adjustment of a given strain to a specific niche just during time-lapse in which the strain colonizing this habitat, despite the fact that the evolutionary novelty is not fixed [47].
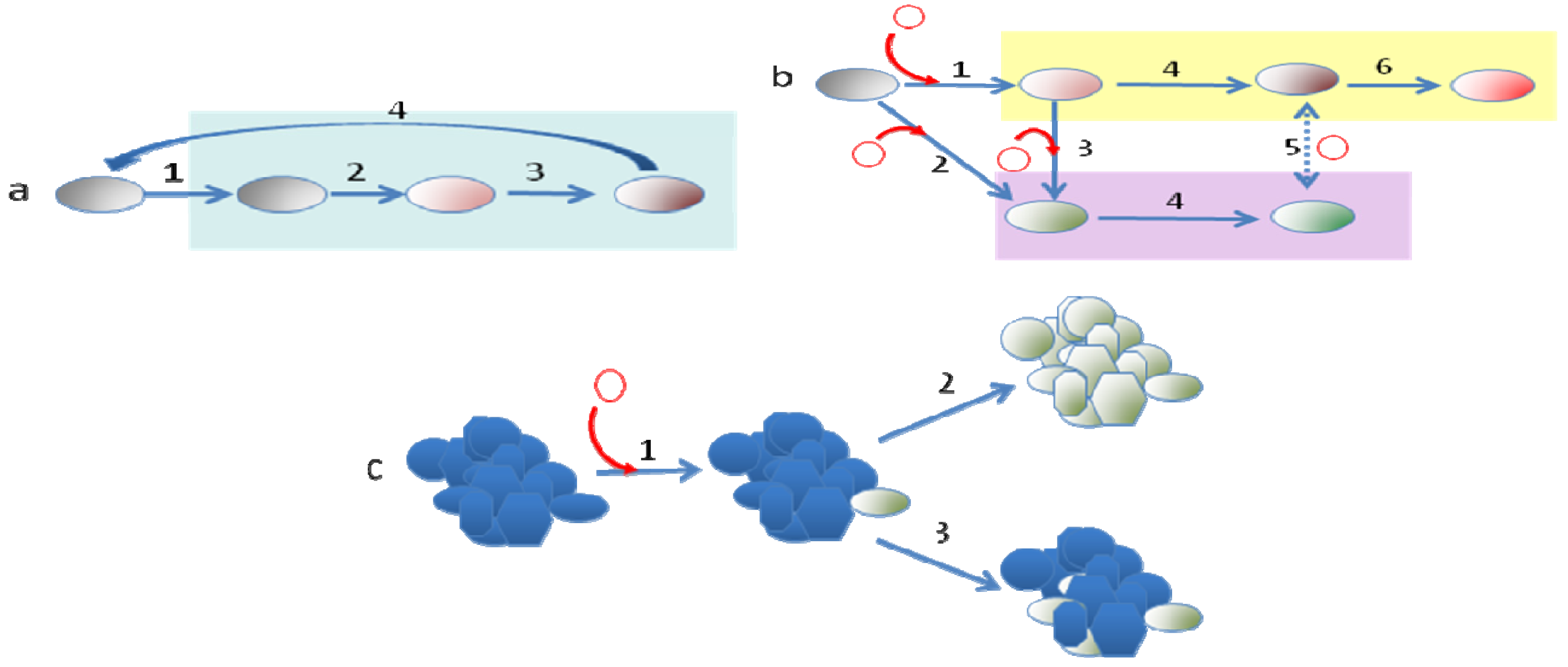
9. Concluding Remarks
An emergent view of evolution is that a plurality of mechanisms and processes drive the diversification of living beings. The study of bacterial evolution has contributed to reinforce this idea, showing the coexistence of multiple mechanisms (gene modification, gene gain and loss, genome rearrangement, utilization of genetic available resources, homologous recombination, etc.) driving microevolutionary processes and long term diversification of bacteria (Figure 3). The relative importance of these mechanisms for bacterial evolution is determined by the ecological scenarios in which bacteria live and follows specific temporal patterns such as those driven by punctuated equilibrium and short-sighted evolution.
Acknowledgments
Authors thank J. Peter W. Young for his kind invitation for writing this paper. In addition, authors thank two anonymous referees for their valuable suggestions. Thanks are given to Elena Bulmer for English editing assistance. Author's laboratories are supported by grants BIO2008-00090 from the Spanish Ministry of Science and Innovation, CGL 2010-15231 from Spanish DGI and KBBE-227258 (BIOHYPO), HEALTH-F3-2010-241476 (PAR) and EVOTAR from European Union.
References
- Navas, A.; Cobas, G.; Talavera, M.; Ayala, J.A.; Lopez, J.A.; Martinez, J.L. Experimental validation of Haldane's hypothesis on the role of infection as an evolutionary force for Metazoans. Proc. Natl. Acad. Sci. USA 2007, 104, 13728–13731. [Google Scholar]
- Elena, S.F.; Cooper, V.S.; Lenski, R.E. Punctuated evolution caused by selection of rare beneficial mutations. Science 1996, 272, 1802–1804. [Google Scholar]
- Woods, R.J.; Barrick, J.E.; Cooper, T.F.; Shrestha, U.; Kauth, M.R.; Lenski, R.E. Second-order selection for evolvability in a large Escherichia coli population. Science 2011, 331, 1433–1436. [Google Scholar]
- Lenski, R.E.; Rose, M.R.; Simpson, S.C.; Tadler, S.C. Long-term experimental evolution in Escherichia coli. I. Adaptation and divergence during 2,000 generations. Am. Nat. 1991, 138, 1315–1341. [Google Scholar]
- Blount, Z.D.; Borland, C.Z.; Lenski, R.E. Historical contingency and the evolution of a key innovation in an experimental population of Escherichia coli. Proc. Natl. Acad. Sci. USA 2008, 105, 7899–7906. [Google Scholar]
- Papadopoulos, D.; Schneider, D.; Meier-Eiss, J.; Arber, W.; Lenski, R.E.; Blot, M. Genomic evolution during a 10,000-generation experiment with bacteria. Proc. Natl. Acad. Sci. USA 1999, 96, 3807–3812. [Google Scholar]
- Lenski, R.E.; Travisano, M. Dynamics of adaptation and diversification: A 10,000-generation experiment with bacterial populations. Proc. Natl. Acad. Sci. USA 1994, 91, 6808–6814. [Google Scholar]
- Hansen, S.K.; Rainey, P.B.; Haagensen, J.A.; Molin, S. Evolution of species interactions in a biofilm community. Nature 2007, 445, 533–536. [Google Scholar]
- Rainey, P.B.; Rainey, K. Evolution of cooperation and conflict in experimental bacterial populations. Nature 2003, 425, 72–74. [Google Scholar]
- Spiers, A.J.; Buckling, A.; Rainey, P.B. The causes of Pseudomonas diversity. Microbiology 2000, 146, 2345–2350. [Google Scholar]
- Rainey, P.B.; Travisano, M. Adaptive radiation in a heterogeneous environment. Nature 1998, 394, 69–72. [Google Scholar]
- Rainey, P.B.; Kerr, B. Cheats as first propagules: A new hypothesis for the evolution of individuality during the transition from single cells to multicellularity. Bioessays 2010, 32, 872–880. [Google Scholar]
- Beaumont, H.J.; Gallie, J.; Kost, C.; Ferguson, G.C.; Rainey, P.B. Experimental evolution of bet hedging. Nature 2009, 462, 90–93. [Google Scholar]
- Steinberg, K.M.; Levin, B.R. Grazing protozoa and the evolution of the Escherichia coli O157:H7 Shiga toxin-encoding prophage. Proc. Biol. Sci. 2007, 274, 1921–1929. [Google Scholar]
- Jensen, M.A.; Faruque, S.M.; Mekalanos, J.J.; Levin, B.R. Modeling the role of bacteriophage in the control of cholera outbreaks. Proc. Natl. Acad. Sci. USA 2006, 103, 4652–4657. [Google Scholar]
- Vulic, M.; Kolter, R. Evolutionary cheating in Escherichia coli stationary phase cultures. Genetics 2001, 158, 519–526. [Google Scholar]
- Finkel, S.E.; Kolter, R. Evolution of microbial diversity during prolonged starvation. Proc. Natl. Acad. Sci. USA 1999, 96, 4023–4027. [Google Scholar]
- Zambrano, M.M.; Kolter, R. GASPing for life in stationary phase. Cell 1996, 86, 181–184. [Google Scholar]
- Zambrano, M.M.; Siegele, D.A.; Almiron, M.; Tormo, A.; Kolter, R. Microbial competition: Escherichia coli mutants that take over stationary phase cultures. Science 1993, 259, 1757–1760. [Google Scholar]
- Jelsbak, L.; Johansen, H.K.; Frost, A.L.; Thogersen, R.; Thomsen, L.E.; Ciofu, O.; Yang, L.; Haagensen, J.A.; Hoiby, N.; Molin, S. Molecular epidemiology and dynamics of Pseudomonas aeruginosa populations in lungs of cystic fibrosis patients. Infect. Immun. 2007, 75, 2214–2224. [Google Scholar]
- Martinez, J.L.; Fajardo, A.; Garmendia, L.; Hernandez, A.; Linares, J.F.; Martinez-Solano, L.; Sanchez, M.B. A global view of antibiotic resistance. FEMS Microbiol. Rev. 2009, 33, 44–65. [Google Scholar]
- Fraser, C.; Alm, E.J.; Polz, M.F.; Spratt, B.G.; Hanage, W.P. The bacterial species challenge: Making sense of genetic and ecological diversity. Science 2009, 323, 741–746. [Google Scholar]
- Darwin, C. On the Origin of Species by Means of Natural Selection or the Preservation of Favored Races in the Struggle of Life; John Murray: London, UK, 1859. [Google Scholar]
- Dobzhansky, T. Genetics and the Origin of Species; Columbia University Press: New York, NY, USA, 1937. [Google Scholar]
- Groisman, E.A.; Ochman, H. Pathogenicity islands: Bacterial evolution in quantum leaps. Cell 1996, 87, 791–794. [Google Scholar]
- Levin, B.R.; Bergstrom, C.T. Bacteria are different: Observations, interpretations, speculations, and opinions about the mechanisms of adaptive evolution in prokaryotes. Proc. Natl. Acad. Sci. USA 2000, 97, 6981–6985. [Google Scholar]
- Gould, S.J.; Eldredge, N. Punctuated equilibrium comes of age. Nature 1993, 366, 223–227. [Google Scholar]
- Mc, C.B. The origin and behavior of mutable loci in maize. Proc. Natl. Acad. Sci. USA 1950, 36, 344–355. [Google Scholar]
- Dobrindt, U.; Hochhut, B.; Hentschel, U.; Hacker, J. Genomic islands in pathogenic and environmental microorganisms. Nat. Rev. Microbiol. 2004, 2, 414–424. [Google Scholar]
- Hacker, J.; Carniel, E. Ecological fitness, genomic islands and bacterial pathogenicity. A Darwinian view of the evolution of microbes. EMBO Rep. 2001, 2, 376–381. [Google Scholar]
- Dobrindt, U.; Hacker, J. Whole genome plasticity in pathogenic bacteria. Curr. Opin. Microbiol. 2001, 4, 550–557. [Google Scholar]
- Frost, L.S.; Leplae, R.; Summers, A.O.; Toussaint, A. Mobile genetic elements: The agents of open source evolution. Nat. Rev. Microbiol. 2005, 3, 722–732. [Google Scholar]
- Ochman, H.; Lawrence, J.G.; Groisman, E.A. Lateral gene transfer and the nature of bacterial innovation. Nature 2000, 405, 299–304. [Google Scholar]
- Morschhauser, J.; Kohler, G.; Ziebuhr, W.; Blum-Oehler, G.; Dobrindt, U.; Hacker, J. Evolution of microbial pathogens. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2000, 355, 695–704. [Google Scholar]
- Gogarten, J.P.; Townsend, J.P. Horizontal gene transfer, genome innovation and evolution. Nat. Rev. Microbiol. 2005, 3, 679–687. [Google Scholar]
- Ragan, M.A.; Beiko, R.G. Lateral genetic transfer: Open issues. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009, 364, 2241–2251. [Google Scholar]
- Boto, L. Horizontal gene transfer in evolution: Facts and challenges. Proc. Biol. Sci. 2010, 277, 819–827. [Google Scholar]
- Woese, C.R.; Kandler, O.; Wheelis, M.L. Towards a natural system of organisms: Proposal for the domains Archaea, Bacteria, and Eucarya. Proc. Natl. Acad. Sci. USA 1990, 87, 4576–4579. [Google Scholar]
- Whitehouse, D.B.; Tomkins, J.; Lovegrove, J.U.; Hopkinson, D.A.; McMillan, W.O. A phylogenetic approach to the identification of phosphoglucomutase genes. Mol. Biol. Evol. 1998, 15, 456–462. [Google Scholar]
- Goldman, B.S.; Nierman, W.C.; Kaiser, D.; Slater, S.C.; Durkin, A.S.; Eisen, J.A.; Ronning, C.M.; Barbazuk, W.B.; Blanchard, M.; Field, C.; et al. Evolution of sensory complexity recorded in a myxobacterial genome. Proc. Natl. Acad. Sci. USA 2006, 103, 15200–15205. [Google Scholar]
- Schneiker, S.; Perlova, O.; Kaiser, O.; Gerth, K.; Alici, A.; Altmeyer, M.O.; Bartels, D.; Bekel, T.; Beyer, S.; Bode, E.; et al. Complete genome sequence of the myxobacterium Sorangium cellulosum. Nat. Biotechnol. 2007, 25, 1281–1289. [Google Scholar]
- Treangen, T.J.; Rocha, E.P. Horizontal transfer, not duplication, drives the expansion of protein families in prokaryotes. PLoS Genet. 2011, 7. [Google Scholar] [CrossRef]
- Beiko, R.G.; Harlow, T.J.; Ragan, M.A. Highways of gene sharing in prokaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 14332–14337. [Google Scholar]
- Doolittle, W.F. Phylogenetic classification and the universal tree. Science 1999, 284, 2124–2129. [Google Scholar]
- Morales, G.; Wiehlmann, L.; Gudowius, P.; van Delden, C.; Tummler, B.; Martinez, J.L.; Rojo, F. Structure of Pseudomonas aeruginosa populations analyzed by single nucleotide polymorphism and pulsed-field gel electrophoresis genotyping. J. Bacteriol. 2004, 186, 4228–4237. [Google Scholar]
- Young, J.P.; Crossman, L.C.; Johnston, A.W.; Thomson, N.R.; Ghazoui, Z.F.; Hull, K.H.; Wexler, M.; Curson, A.R.; Todd, J.D.; Poole, P.S.; et al. The genome of Rhizobium leguminosarum has recognizable core and accessory components. Genome Biol. 2006, 7. [Google Scholar] [CrossRef]
- Levin, B.R.; Bull, J.J. Short-sighted evolution and the virulence of pathogenic microorganisms. Trends Microbiol. 1994, 2, 76–81. [Google Scholar]
- Cohan, F.M. What are bacterial species? Annu. Rev. Microbiol. 2002, 56, 457–487. [Google Scholar]
- Prosser, J.I.; Bohannan, B.J.; Curtis, T.P.; Ellis, R.J.; Firestone, M.K.; Freckleton, R.P.; Green, J.L.; Green, L.E.; Killham, K.; Lennon, J.J.; et al. The role of ecological theory in microbial ecology. Nat. Rev. Microbiol. 2007, 5, 384–392. [Google Scholar]
- Friesen, M.L.; Saxer, G.; Travisano, M.; Doebeli, M. Experimental evidence for sympatric ecological diversification due to frequency-dependent competition in Escherichia coli. Evolution 2004, 58, 245–260. [Google Scholar]
- Bailly, X.; Giuntini, E.; Sexton, M.C.; Lower, R.P.; Harrison, P.W.; Kumar, N.; Young, J.P. Population genomics of Sinorhizobium medicae based on low-coverage sequencing of sympatric isolates. ISME J. 2011, 5, 1722–1734. [Google Scholar]
- Wilder, C.N.; Allada, G.; Schuster, M. Instantaneous within-patient diversity of Pseudomonas aeruginosa quorum-sensing populations from cystic fibrosis lung infections. Infect. Immun. 2009, 77, 5631–5639. [Google Scholar]
- Bragonzi, A.; Paroni, M.; Nonis, A.; Cramer, N.; Montanari, S.; Rejman, J.; di Serio, C.; Doring, G.; Tummler, B. Pseudomonas aeruginosa microevolution during cystic fibrosis lung infection establishes clones with adapted virulence. Am. J. Respir. Crit. Care Med. 2009, 180, 138–145. [Google Scholar]
- Renders, N.; Verbrugh, H.; van Belkum, A. Dynamics of bacterial colonisation in the respiratory tract of patients with cystic fibrosis. Infect. Genet. Evol. 2001, 1, 29–39. [Google Scholar]
- Mena, A.; Smith, E.E.; Burns, J.L.; Speert, D.P.; Moskowitz, S.M.; Perez, J.L.; Oliver, A. Genetic adaptation of Pseudomonas aeruginosa to the airways of cystic fibrosis patients is catalyzed by hypermutation. J. Bacteriol. 2008, 190, 7910–7917. [Google Scholar]
- Smith, E.E.; Buckley, D.G.; Wu, Z.; Saenphimmachak, C.; Hoffman, L.R.; D'Argenio, D.A.; Miller, S.I.; Ramsey, B.W.; Speert, D.P.; Moskowitz, S.M.; et al. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc. Natl. Acad. Sci. USA 2006, 103, 8487–8492. [Google Scholar]
- Martinez-Solano, L.; Macia, M.D.; Fajardo, A.; Oliver, A.; Martinez, J.L. Chronic Pseudomonas aeruginosa infection in chronic obstructive pulmonary disease. Clin. Infect. Dis. 2008, 47, 1526–1533. [Google Scholar]
- Bouma, J.E.; Lenski, R.E. Evolution of a bacteria/plasmid association. Nature 1988, 335, 351–352. [Google Scholar]
- Levin, B.R.; Stewart, F.M. The population biology of bacterial plasmids: A priori conditions for the existence of mobilizable nonconjugative factors. Genetics 1980, 94, 425–443. [Google Scholar]
- Martinez, J.L.; Baquero, F. Interactions among strategies associated with bacterial infection: Pathogenicity, epidemicity, and antibiotic resistance. Clin. Microbiol. Rev. 2002, 15, 647–679. [Google Scholar]
- Fajardo, A.; Linares, J.F.; Martinez, J.L. Towards an ecological approach to antibiotics and antibiotic resistance genes. Clin. Microbiol. Infect. 2009, 15, 14–16. [Google Scholar]
- Bjorkman, J.; Nagaev, I.; Berg, O.G.; Hughes, D.; Andersson, D.I. Effects of environment on compensatory mutations to ameliorate costs of antibiotic resistance. Science 2000, 287, 1479–1482. [Google Scholar]
- Martinez, J.L. The role of natural environments in the evolution of resistance traits in pathogenic bacteria. Proc. Biol. Sci. 2009, 276, 2521–2530. [Google Scholar]
- Martinez, J.L. Environmental pollution by antibiotics and by antibiotic resistance determinants. Environ. Pollut. 2009, 157, 2893–2902. [Google Scholar]
- Martinez, J.L. Antibiotics and antibiotic resistance genes in natural environments. Science 2008, 321, 365–367. [Google Scholar]
- Brzuszkiewicz, E.; Gottschalk, G.; Ron, E.; Hacker, J.; Dobrindt, U. Adaptation of pathogenic E. coli to various niches: Genome flexibility is the key. Genome Dyn. 2009, 6, 110–125. [Google Scholar]
- van Passel, M.W.; Marri, P.R.; Ochman, H. The emergence and fate of horizontally acquired genes in Escherichia coli. PLoS Comput. Biol. 2008, 4. [Google Scholar] [CrossRef]
- Hacker, J.; Blum-Oehler, G.; Muhldorfer, I.; Tschape, H. Pathogenicity islands of virulent bacteria: Structure, function and impact on microbial evolution. Mol. Microbiol. 1997, 23, 1089–1097. [Google Scholar]
- Zhou, D.; Yang, R. Molecular Darwinian evolution of virulence in Yersinia pestis. Infect. Immun. 2009, 77, 2242–2250. [Google Scholar]
- Achtman, M.; Morelli, G.; Zhu, P.; Wirth, T.; Diehl, I.; Kusecek, B.; Vogler, A.J.; Wagner, D.M.; Allender, C.J.; Easterday, W.R.; et al. Microevolution and history of the plague bacillus, Yersinia pestis. Proc. Natl. Acad. Sci. USA 2004, 101, 17837–17842. [Google Scholar]
- Morelli, G.; Song, Y.; Mazzoni, C.J.; Eppinger, M.; Roumagnac, P.; Wagner, D.M.; Feldkamp, M.; Kusecek, B.; Vogler, A.J.; Li, Y.; et al. Yersinia pestis genome sequencing identifies patterns of global phylogenetic diversity. Nat. Genet. 2010, 42, 1140–1143. [Google Scholar]
- Wren, B.W. The yersiniae--a model genus to study the rapid evolution of bacterial pathogens. Nat. Rev. Microbiol. 2003, 1, 55–64. [Google Scholar]
- Baquero, F. From pieces to patterns: Evolutionary engineering in bacterial pathogens. Nat. Rev. Microbiol. 2004, 2, 510–518. [Google Scholar]
- Baquero, F.; Alvarez-Ortega, C.; Martinez, J.L. Ecology and evolution of antibiotic resistance. Environ. Microbiol. Rep. 2009, 1, 469–476. [Google Scholar]
- Skippington, E.; Ragan, M.A. Lateral genetic transfer and the construction of genetic exchange communities. FEMS Microbiol. Rev. 2011. in press. [Google Scholar]
- Chaffron, S.; Rehrauer, H.; Pernthaler, J.; von Mering, C. A global network of coexisting microbes from environmental and whole-genome sequence data. Genome Res. 2010, 20, 947–959. [Google Scholar]
- Zhaxybayeva, O.; Swithers, K.S.; Lapierre, P.; Fournier, G.P.; Bickhart, D.M.; DeBoy, R.T.; Nelson, K.E.; Nesbo, C.L.; Doolittle, W.F.; Gogarten, J.P.; et al. On the chimeric nature, thermophilic origin, and phylogenetic placement of the Thermotogales. Proc. Natl. Acad. Sci. USA 2009, 106, 5865–5870. [Google Scholar]
- Boussau, B.; Gueguen, L.; Gouy, M. Accounting for horizontal gene transfers explains conflicting hypotheses regarding the position of aquificales in the phylogeny of Bacteria. BMC Evol. Biol. 2008, 8. [Google Scholar] [CrossRef]
- Hehemann, J.H.; Correc, G.; Barbeyron, T.; Helbert, W.; Czjzek, M.; Michel, G. Transfer of carbohydrate-active enzymes from marine bacteria to Japanese gut microbiota. Nature 2010, 464, 908–912. [Google Scholar]
- Marshall, J.A. Group selection and kin selection: Formally equivalent approaches. Trends Ecol. Evol. 2011, 26, 325–332. [Google Scholar]
- Cohen, O.; Gophna, U.; Pupko, T. The complexity hypothesis revisited: Connectivity rather than function constitutes a barrier to horizontal gene transfer. Mol. Biol. Evol. 2011, 28, 1481–1489. [Google Scholar]
- Morosini, M.I.; Ayala, J.A.; Baquero, F.; Martinez, J.L.; Blazquez, J. Biological cost of AmpC production for Salmonella enterica serotype Typhimurium. Antimicrob. Agents Chemother. 2000, 44, 3137–3143. [Google Scholar]
- Kuo, C.H.; Ochman, H. The fate of new bacterial genes. FEMS Microbiol. Rev. 2009, 33, 38–43. [Google Scholar]
- Schliep, K.; Lopez, P.; Lapointe, F.J.; Bapteste, E. Harvesting evolutionary signals in a forest of prokaryotic gene trees. Mol. Biol. Evol. 2011, 28, 1393–1405. [Google Scholar]
- Rahme, L.G.; Stevens, E.J.; Wolfort, S.F.; Shao, J.; Tompkins, R.G.; Ausubel, F.M. Common virulence factors for bacterial pathogenicity in plants and animals. Science 1995, 268, 1899–1902. [Google Scholar]
- Alonso, A.; Rojo, F.; Martinez, J.L. Environmental and clinical isolates of Pseudomonas aeruginosa show pathogenic and biodegradative properties irrespective of their origin. Environ. Microbiol. 1999, 1, 421–430. [Google Scholar]
- Wiehlmann, L.; Wagner, G.; Cramer, N.; Siebert, B.; Gudowius, P.; Morales, G.; Kohler, T.; van Delden, C.; Weinel, C.; Slickers, P.; et al. Population structure of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2007, 104, 8101–8106. [Google Scholar]
- Cases, I.; de Lorenzo, V.; Ouzounis, C.A. Transcription regulation and environmental adaptation in bacteria. Trends Microbiol. 2003, 11, 248–253. [Google Scholar]
- Lukjancenko, O.; Wassenaar, T.M.; Ussery, D.W. Comparison of 61 sequenced Escherichia coli genomes. Microb. Ecol. 2010, 60, 708–720. [Google Scholar]
- Dufresne, A.; Garczarek, L.; Partensky, F. Accelerated evolution associated with genome reduction in a free-living prokaryote. Genome Biol. 2005, 6, R14. [Google Scholar]
- Marais, G.A.; Calteau, A.; Tenaillon, O. Mutation rate and genome reduction in endosymbiotic and free-living bacteria. Genetica 2008, 134, 205–210. [Google Scholar]
- Giovannoni, S.J.; Tripp, H.J.; Givan, S.; Podar, M.; Vergin, K.L.; Baptista, D.; Bibbs, L.; Eads, J.; Richardson, T.H.; Noordewier, M.; Rappe, M.S.; Short, J.M.; Carrington, J.C.; Mathur, E.J. Genome streamlining in a cosmopolitan oceanic bacterium. Science 2005, 309, 1242–1245. [Google Scholar]
- Kuo, C.H.; Moran, N.A.; Ochman, H. The consequences of genetic drift for bacterial genome complexity. Genome Res. 2009, 19, 1450–1454. [Google Scholar]
- Tettelin, H.; Riley, D.; Cattuto, C.; Medini, D. Comparative genomics: The bacterial pan-genome. Curr. Opin. Microbiol. 2008, 11, 472–477. [Google Scholar]
- Medini, D.; Donati, C.; Tettelin, H.; Masignani, V.; Rappuoli, R. The microbial pan-genome. Curr. Opin. Genet. Dev. 2005, 15, 589–594. [Google Scholar]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S.; et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar]
- Hiller, N.L.; Janto, B.; Hogg, J.S.; Boissy, R.; Yu, S.; Powell, E.; Keefe, R.; Ehrlich, N.E.; Shen, K.; Hayes, J.; et al. Comparative genomic analyses of seventeen Streptococcus pneumoniae strains: Insights into the pneumococcal supragenome. J. Bacteriol. 2007, 189, 8186–8195. [Google Scholar]
- Hogg, J.S.; Hu, F.Z.; Janto, B.; Boissy, R.; Hayes, J.; Keefe, R.; Post, J.C.; Ehrlich, G.D. Characterization and modeling of the Haemophilus influenzae core and supragenomes based on the complete genomic sequences of Rd and 12 clinical nontypeable strains. Genome Biol. 2007, 8. [Google Scholar] [CrossRef]
- Mira, A.; Martin-Cuadrado, A.B.; D'Auria, G.; Rodriguez-Valera, F. The bacterial pan-genome:a new paradigm in microbiology. Int. Microbiol. 2010, 13, 45–57. [Google Scholar]
- Donati, C.; Hiller, N.L.; Tettelin, H.; Muzzi, A.; Croucher, N.J.; Angiuoli, S.V.; Oggioni, M.; Dunning Hotopp, J.C.; Hu, F.Z.; Riley, D.R.; et al. Structure and dynamics of the pan-genome of Streptococcus pneumoniae and closely related species. Genome Biol. 2010, 11. [Google Scholar] [CrossRef]
- Mira, A.; Klasson, L.; Andersson, S.G. Microbial genome evolution: Sources of variability. Curr. Opin. Microbiol. 2002, 5, 506–512. [Google Scholar]
- Radman, M.; Matic, I.; Taddei, F. Evolution of evolvability. Ann. N. Y. Acad. Sci. 1999, 870, 146–155. [Google Scholar]
- Martinez, J.L.; Baquero, F. Mutation frequencies and antibiotic resistance. Antimicrob. Agents Chemother. 2000, 44, 1771–1777. [Google Scholar]
- Alonso, A.; Campanario, E.; Martinez, J.L. Emergence of multidrug-resistant mutants is increased under antibiotic selective pressure in Pseudomonas aeruginosa. Microbiology 1999, 145, 2857–2862. [Google Scholar]
- Blazquez, J.; Oliver, A.; Gomez-Gomez, J.M. Mutation and evolution of antibiotic resistance: Antibiotics as promoters of antibiotic resistance? Curr. Drug Targets 2002, 3, 345–349. [Google Scholar]
- Andersson, D.I.; Slechta, E.S.; Roth, J.R. Evidence that gene amplification underlies adaptive mutability of the bacterial lac operon. Science 1998, 282, 1133–1135. [Google Scholar]
- Kugelberg, E.; Kofoid, E.; Reams, A.B.; Andersson, D.I.; Roth, J.R. Multiple pathways of selected gene amplification during adaptive mutation. Proc. Natl. Acad. Sci. USA 2006, 103, 17319–17324. [Google Scholar]
- Pettersson, M.E.; Andersson, D.I.; Roth, J.R.; Berg, O.G. The amplification model for adaptive mutation: Simulations and analysis. Genetics 2005, 169, 1105–1115. [Google Scholar]
- Smith, K.A.; Chernova, O.B.; Groves, R.P.; Stark, M.B.; Martinez, J.L.; Davidson, J.N.; Trent, J.M.; Patterson, T.E.; Agarwal, A.; Duncan, P.; et al. Multiple mechanisms of N-phosphonacetyl-L-aspartate resistance in human cell lines: Carbamyl-P synthetase/aspartate transcarbamylase/dihydro-orotase gene amplification is frequent only when chromosome 2 is rearranged. Proc. Natl. Acad. Sci. USA 1997, 94, 1816–1821. [Google Scholar]
- Gomez-Gomez, J.M.; Blazquez, J.; Baquero, F.; Martinez, J.L. H-NS and RpoS regulate emergence of Lac Ara+ mutants of Escherichia coli MCS2. J. Bacteriol. 1997, 179, 4620–4622. [Google Scholar]
- McKenzie, G.J.; Harris, R.S.; Lee, P.L.; Rosenberg, S.M. The SOS response regulates adaptive mutation. Proc. Natl. Acad. Sci. USA 2000, 97, 6646–6651. [Google Scholar]
- McKenzie, G.J.; Rosenberg, S.M. Adaptive mutations, mutator DNA polymerases and genetic change strategies of pathogens. Curr. Opin. Microbiol. 2001, 4, 586–594. [Google Scholar]
- LeClerc, J.E.; Li, B.; Payne, W.L.; Cebula, T.A. High mutation frequencies among Escherichia coli and Salmonella pathogens. Science 1996, 274, 1208–1211. [Google Scholar]
- Baquero, M.R.; Nilsson, A.I.; Turrientes Mdel, C.; Sandvang, D.; Galan, J.C.; Martinez, J.L.; Frimodt-Moller, N.; Baquero, F.; Andersson, D.I. Polymorphic mutation frequencies in Escherichia coli: Emergence of weak mutators in clinical isolates. J. Bacteriol. 2004, 186, 5538–5542. [Google Scholar]
- Wagner, A. Robustness and evolvability: A paradox resolved. Proc. Biol. Sci. 2008, 275, 91–100. [Google Scholar]
- Oliver, A.; Canton, R.; Campo, P.; Baquero, F.; Blazquez, J. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science 2000, 288, 1251–1254. [Google Scholar]
- Turrientes, M.C.; Baquero, M.R.; Sanchez, M.B.; Valdezate, S.; Escudero, E.; Berg, G.; Canton, R.; Baquero, F.; Galan, J.C.; Martinez, J.L. Polymorphic mutation frequencies of clinical and environmental Stenotrophomonas maltophilia populations. Appl. Environ. Microbiol. 2010, 76, 1746–1758. [Google Scholar]
- Macia, M.D.; Blanquer, D.; Togores, B.; Sauleda, J.; Perez, J.L.; Oliver, A. Hypermutation is a key factor in development of multiple-antimicrobial resistance in Pseudomonas aeruginosa strains causing chronic lung infections. Antimicrob. Agents Chemother. 2005, 49, 3382–3386. [Google Scholar]
- Pal, C.; Macia, M.D.; Oliver, A.; Schachar, I.; Buckling, A. Coevolution with viruses drives the evolution of bacterial mutation rates. Nature 2007, 450, 1079–1081. [Google Scholar]
- Koonin, E.V.; Wolf, Y.I. Genomics of bacteria and archaea: The emerging dynamic view of the prokaryotic world. Nucleic Acids Res. 2008, 36, 6688–6719. [Google Scholar]
- Rice, L.B. Association of different mobile elements to generate novel integrative elements. Cell Mol. Life Sci. 2002, 59, 2023–2032. [Google Scholar]
- Sokurenko, E.V.; Gomulkiewicz, R.; Dykhuizen, D.E. Source-sink dynamics of virulence evolution. Nat. Rev. Microbiol. 2006, 4, 548–555. [Google Scholar]
- Yang, L.; Jelsbak, L.; Molin, S. Microbial ecology and adaptation in cystic fibrosis airways. Environ. Microbiol. 2011, 13, 1682–1689. [Google Scholar]
- Ochman, H. Genomes on the shrink. Proc. Natl. Acad. Sci. USA 2005, 102, 11959–11960. [Google Scholar]
- Moran, N.A. Microbial minimalism: Genome reduction in bacterial pathogens. Cell 2002, 108, 583–586. [Google Scholar]
- Moran, N.A.; McCutcheon, J.P.; Nakabachi, A. Genomics and evolution of heritable bacterial symbionts. Annu. Rev. Genet. 2008, 42, 165–190. [Google Scholar]
- Nakabachi, A.; Yamashita, A.; Toh, H.; Ishikawa, H.; Dunbar, H.E.; Moran, N.A.; Hattori, M. The 160-kilobase genome of the bacterial endosymbiont Carsonella. Science 2006, 314, 267. [Google Scholar]
- Tamas, I.; Klasson, L.; Canback, B.; Naslund, A.K.; Eriksson, A.S.; Wernegreen, J.J.; Sandstrom, J.P.; Moran, N.A.; Andersson, S.G. 50 million years of genomic stasis in endosymbiotic bacteria. Science 2002, 296, 2376–2379. [Google Scholar]
- Lai, C.Y.; Baumann, L.; Baumann, P. Amplification of trpEG: Adaptation of Buchnera aphidicola to an endosymbiotic association with aphids. Proc. Natl. Acad. Sci. USA 1994, 91, 3819–3823. [Google Scholar]
- Bracho, A.M.; Martinez-Torres, D.; Moya, A.; Latorre, A. Discovery and molecular characterization of a plasmid localized in Buchnera sp. bacterial endosymbiont of the aphid Rhopalosiphum padi. J. Mol. Evol. 1995, 41, 67–73. [Google Scholar]
- Jordan, I.K.; Makarova, K.S.; Spouge, J.L.; Wolf, Y.I.; Koonin, E.V. Lineage-specific gene expansions in bacterial and archaeal genomes. Genome Res. 2001, 11, 555–565. [Google Scholar]
- Snel, B.; Bork, P.; Huynen, M.A. Genomes in flux: the evolution of archaeal and proteobacterial gene content. Genome Res. 2002, 12, 17–25. [Google Scholar]
- Rocha, E.P. Evolutionary patterns in prokaryotic genomes. Curr. Opin. Microbiol. 2008, 11, 454–460. [Google Scholar]
- Nilsson, A.I.; Koskiniemi, S.; Eriksson, S.; Kugelberg, E.; Hinton, J.C.; Andersson, D.I. Bacterial genome size reduction by experimental evolution. Proc. Natl. Acad. Sci. USA 2005, 102, 12112–12116. [Google Scholar]
- Dowson, C.G.; Coffey, T.J.; Kell, C.; Whiley, R.A. Evolution of penicillin resistance in Streptococcus pneumoniae; the role of Streptococcus mitis in the formation of a low affinity PBP2B in S. pneumoniae. Mol. Microbiol. 1993, 9, 635–643. [Google Scholar]
- Spratt, B.G.; Bowler, L.D.; Zhang, Q.Y.; Zhou, J.; Smith, J.M. Role of interspecies transfer of chromosomal genes in the evolution of penicillin resistance in pathogenic and commensal Neisseria species. J. Mol. Evol. 1992, 34, 115–125. [Google Scholar]
- Schubert, S.; Darlu, P.; Clermont, O.; Wieser, A.; Magistro, G.; Hoffmann, C.; Weinert, K.; Tenaillon, O.; Matic, I.; Denamur, E. Role of intraspecies recombination in the spread of pathogenicity islands within the Escherichia coli species. PLoS Pathog. 2009, 5. [Google Scholar] [CrossRef]
- Wirth, T.; Falush, D.; Lan, R.; Colles, F.; Mensa, P.; Wieler, L.H.; Karch, H.; Reeves, P.R.; Maiden, M.C.; Ochman, H.; et al. Sex and virulence in Escherichia coli: An evolutionary perspective. Mol. Microbiol. 2006, 60, 1136–1151. [Google Scholar]
- Papke, R.T.; Koenig, J.E.; Rodriguez-Valera, F.; Doolittle, W.F. Frequent recombination in a saltern population of Halorubrum. Science 2004, 306, 1928–1929. [Google Scholar]
- Karboul, A.; Mazza, A.; Gey van Pittius, N.C.; Ho, J.L.; Brousseau, R.; Mardassi, H. Frequent homologous recombination events in Mycobacterium tuberculosis PE/PPE multigene families: Potential role in antigenic variability. J. Bacteriol. 2008, 190, 7838–7846. [Google Scholar]
- Ochman, H.; Lerat, E.; Daubin, V. Examining bacterial species under the specter of gene transfer and exchange. Proc. Natl. Acad. Sci. USA 2005, 102, 6595–6599. [Google Scholar]
- Shapiro, B.J.; David, L.A.; Friedman, J.; Alm, E.J. Looking for Darwin's footprints in the microbial world. Trends Microbiol. 2009, 17, 196–204. [Google Scholar]
- Fraser, C.; Hanage, W.P.; Spratt, B.G. Recombination and the nature of bacterial speciation. Science 2007, 315, 476–480. [Google Scholar]
- Didelot, X.; Bowden, R.; Street, T.; Golubchik, T.; Spencer, C.; McVean, G.; Sangal, V.; Anjum, M.F.; Achtman, M.; Falush, D.; et al. Recombination and Population Structure in Salmonella enterica. PLoS Genet. 2011, 7. [Google Scholar] [CrossRef]
- Didelot, X.; Achtman, M.; Parkhill, J.; Thomson, N.R.; Falush, D. A bimodal pattern of relatedness between the Salmonella Paratyphi A and Typhi genomes: Convergence or divergence by homologous recombination? Genome Res. 2007, 17, 61–68. [Google Scholar]
- Stokes, H.W.; Hall, R.M. A novel family of potentially mobile DNA elements encoding site-specific gene-integration functions: Integrons. Mol. Microbiol. 1989, 3, 1669–1683. [Google Scholar]
- Hall, R.M.; Collis, C.M. Mobile gene cassettes and integrons: Capture and spread of genes by site-specific recombination. Mol. Microbiol. 1995, 15, 593–600. [Google Scholar]
- Rowe-Magnus, D.A.; Mazel, D. Integrons: Natural tools for bacterial genome evolution. Curr. Opin. Microbiol. 2001, 4, 565–569. [Google Scholar]
- Rowe-Magnus, D.A.; Mazel, D. The role of integrons in antibiotic resistance gene capture. Int. J. Med. Microbiol. 2002, 292, 115–125. [Google Scholar]
- Matic, I.; Rayssiguier, C.; Radman, M. Interspecies gene exchange in bacteria: The role of SOS and mismatch repair systems in evolution of species. Cell 1995, 80, 507–515. [Google Scholar]
- Matic, I.; Taddei, F.; Radman, M. Genetic barriers among bacteria. Trends Microbiol. 1996, 4, 69–72. [Google Scholar]
- Matic, I.; Taddei, F.; Radman, M. No genetic barriers between Salmonella enterica serovar typhimurium and Escherichia coli in SOS-induced mismatch repair-deficient cells. J. Bacteriol. 2000, 182, 5922–5924. [Google Scholar]
- Denamur, E.; Lecointre, G.; Darlu, P.; Tenaillon, O.; Acquaviva, C.; Sayada, C.; Sunjevaric, I.; Rothstein, R.; Elion, J.; Taddei, F.; et al. Evolutionary implications of the frequent horizontal transfer of mismatch repair genes. Cell 2000, 103, 711–721. [Google Scholar]
- Martinez, J.L.; Sanchez, M.B.; Martinez-Solano, L.; Hernandez, A.; Garmendia, L.; Fajardo, A.; Alvarez-Ortega, C. Functional role of bacterial multidrug efflux pumps in microbial natural ecosystems. FEMS Microbiol. Rev. 2009, 33, 430–449. [Google Scholar]
- Baquero, F.; Martinez, J.L.; Canton, R. Antibiotics and antibiotic resistance in water environments. Curr. Opin. Biotechnol. 2008, 19, 260–265. [Google Scholar]
- Martinez, J.L.; Baquero, F.; Andersson, D.I. Predicting antibiotic resistance. Nat. Rev. Microbiol. 2007, 5, 958–965. [Google Scholar]
- Toft, C.; Andersson, S.G. Evolutionary microbial genomics: Insights into bacterial host adaptation. Nat. Rev. Genet. 2010, 11, 465–475. [Google Scholar]
- Salverda, M.L.; de Visser, J.A.; Barlow, M. Natural evolution of TEM-1 beta-lactamase: Experimental reconstruction and clinical relevance. FEMS Microbiol. Rev. 2010, 34, 1015–1036. [Google Scholar]
- Moran, N.A.; McLaughlin, H.J.; Sorek, R. The dynamics and time scale of ongoing genomic erosion in symbiotic bacteria. Science 2009, 323, 379–382. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Boto, L.; Martínez, J.L. Ecological and Temporal Constraints in the Evolution of Bacterial Genomes. Genes 2011, 2, 804-828. https://doi.org/10.3390/genes2040804
Boto L, Martínez JL. Ecological and Temporal Constraints in the Evolution of Bacterial Genomes. Genes. 2011; 2(4):804-828. https://doi.org/10.3390/genes2040804
Chicago/Turabian StyleBoto, Luis, and Jose Luis Martínez. 2011. "Ecological and Temporal Constraints in the Evolution of Bacterial Genomes" Genes 2, no. 4: 804-828. https://doi.org/10.3390/genes2040804
APA StyleBoto, L., & Martínez, J. L. (2011). Ecological and Temporal Constraints in the Evolution of Bacterial Genomes. Genes, 2(4), 804-828. https://doi.org/10.3390/genes2040804