Complete Genome Sequence of the Soybean Symbiont Bradyrhizobium japonicum Strain USDA6T

Abstract
: The complete nucleotide sequence of the genome of the soybean symbiont Bradyrhizobium japonicum strain USDA6T was determined. The genome of USDA6T is a single circular chromosome of 9,207,384 bp. The genome size is similar to that of the genome of another soybean symbiont, B. japonicum USDA110 (9,105,828 bp). Comparison of the whole-genome sequences of USDA6T and USDA110 showed colinearity of major regions in the two genomes, although a large inversion exists between them. A significantly high level of sequence conservation was detected in three regions on each genome. The gene constitution and nucleotide sequence features in these three regions indicate that they may have been derived from a symbiosis island. An ancestral, large symbiosis island, approximately 860 kb in total size, appears to have been split into these three regions by unknown large-scale genome rearrangements. The two integration events responsible for this appear to have taken place independently, but through comparable mechanisms, in both genomes.1. Introduction
The process of symbiotic nitrogen fixation is of both agricultural and ecological significance. It is a notable component of agricultural production. The establishment of symbiosis between legume plants and rhizobia allows for cooperative functioning between these two organisms, and is vital for agriculture. Soybeans are nodulated by slow-growing bacteria belonging to various species within the genus Bradyrhizobium [1]. In general, only some Bradyrhizobium japonicum strains are being used in the production of commercial inoculants for soybeans, whereas various other Bradyrhizobium spp. span wide regions of the world and exhibit great genetic diversity. Based on geographical and climatic differences in their habitats, the genus Bradyrhizobium has maintained a high degree of genetic diversity [2]. For the classification of B. japonicum, the strain USDA6T has been used as the type strain for this bacterial species. USDA6T was originally isolated from nodules of a soybean planted in Japan, and has been used for research since it was entered in a culture collection in 1929 [3]. USDA6T has effective symbiotic nitrogen fixation capacity [4].
Since the nucleotide sequence of the genome of B. japonicum strain USDA110 was determined in 2002 [5], it has been used for genomic research directed towards investigating soybean symbionts. USDA110 is a strain isolated from the nodule of a soybean grown in Florida, USA in 1957 [4]. Largely because of the superior characteristics of this organism with respect to symbiotic nitrogen fixation, it has been widely used in research in molecular genetics and physiology. The genome of USDA110 is a single circular chromosome, 9.1 Mb in length, and carries 14 genomic islands as DNA segments inserted into tRNA genes [5]. In addition to these genomic islands, a notable large genomic island, known as the symbiosis island, is located downstream of a valine-tRNA gene (trnV).
The symbiosis island region carries genes required for the establishment and maintenance of nitrogen-fixing symbiosis with the host plant. The symbiosis island was first identified as an integrated island in the trnF gene of Mesorhizobium loti ICMP3153 [6]. In addition, the integration of other symbiosis islands into the chromosomes of other rhizobial strains has been reported. It is remarkable that symbiosis islands have only been identified in some rhizobial strains, because, in general, fast-growing rhizobia have the symbiotic plasmid as a cassette for symbiotic nitrogen fixation genes [7]. Complete nucleotide sequences for the islands have been determined for four strains of rhizobia, M. loti MAFF303099, M. loti R7A, Azorhizobium caulinodans ORS571, and B. japonicum USDA110 [5,8-10].
The systematic analysis of the gene expression and the structures of these genomes were conducted using DNA array technology methods based on the USDA110 genome sequence [11-15]. Comparative genomic hybridization (CGH), using such DNA array technology, has been applied for the determination of global genome variations among closely related bacteria [16]. CGH analyses of nine strains of B. japonicum were performed to clarify their genomic variations [15]. The profiles indicated that the genomes of the nine strains could be classified into three broad genome types (USDA110, USDA122, and USDA6 groups) based on the lower signal intensity from regions located in the putative genomic islands of USDA110.
CGH analysis of B. japonicum USDA6T showed genomic variations between USDA6T and USDA110 [15]. Lower intensities were detected in the regions corresponding to all 14 small genomic islands identified on the USDA110 genome, which is a common feature of other members of genomes belonging to the USDA6 group. This observation indicates that the insertions of genomic islands cause obvious variation between USDA110 and the diverse genomes categorized in the USDA6 group. Phylogenetic analyses based on internal transcribed spacer region of ribosomal DNA (ITS) sequence data have also shown that USDA6T belongs to a clade distant from that of USDA110 [15]. These analyses indicate that USDA110 and USDA6T are more divergent than previously thought.
The CGH profile is an effective tool for the categorization of closely related bacterial genomes, but the analysis is restricted to comparisons of regions existing in the reference genome. In order to investigate the genomic features of the type strain of B. japonicum and conduct detailed comparative analysis between the two B. japonicum strains, we carried out genome sequencing of USDA6T, and compared the genomic features between USDA6T and USDA110.
2. Results and Discussion
2.1. Genome Sequencing
In order to assess the genome size of USDA6T and to confirm its divergence from USDA110 before sequencing, the composition of fragments of genomic DNA digested with two restriction endonucleases, PmeI and SwaI, was analyzed using pulsed-field gel electrophoresis (PFGE). The sizes of the fragments were estimated by relative mobility analysis and by comparing them to the size markers. The double digestion of USDA6T DNA yielded eight fragments ranging from 95 to 2,200 kb (Table 1). The sum of the fragment sizes was approximately 9.3 Mb. The restriction pattern of USDA6T DNA was different from the predicted pattern based on the USDA110 genome sequence, in terms of fragment number (fifteen fragments) and fragment sizes (from 1 kb to 2,152 kb). This suggests that USDA6T is phylogenetically divergent from USDA110.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total size (kb) | PmeI-SwaI fragments sizes (kb) | |
|---|---|---|
| USDA6T (PFGE-gel) | 9,295 | 2,200, 1,850, 1,800, 1,500, 740, 650, 460, 95 |
| USDA6T (computed) | 9,207 | 2,322, 1,702, 1,694, 1,407, 788, 682, 508, 103 |
| USDA110 (computed) | 9,106 | 2,152, 1,911, 1,481, 954, 784, 445, 413, 333, 162, 126, 124, 112, 106, 2, 1 |
The draft nucleotide sequence of the entire USDA6T genome was deduced initially by assembling a total of 651,020 files containing single-read sequences from the 454/Roche GS-FLX system. The sum of these sequence reads corresponds to 28.7 genome equivalents. The initial assembly produced 151 contigs with lengths of 500 bp or more. The total size of the contigs was 9,195,176 bp.
Two libraries were prepared: a bacterial artificial chromosome (BAC) library (Bj006b) contained inserts of an average length of 82 kb, and a cosmid library (Bj006c) contained inserts of an average length of 30 kb. In total, 2,382 clones from the Bj006b BAC library and 1,929 clones from the Bj006c cosmid library were used to scaffold 137 contigs. The clones were end-sequenced, and these sequences were used to map the contigs, based on output data from BLASTN searches. As a result, these contigs were arranged into a single large scaffold based on the end-sequence information from the Bj006c and Bj006b clones. In order to obtain the nucleotide sequence with accuracy sufficient for subsequent comparative genome analyses, the draft sequences were visually edited and additional sequencing was performed to close the gaps. Integrity assessment of the completed genome sequence was performed by comparing the insert lengths of each Bj006c and Bj006b clone with the computed distance between the corresponding end sequences in the whole-genome sequence.
2.2. Structural Features of the USDA6T Genome
2.2.1. Sequence Features
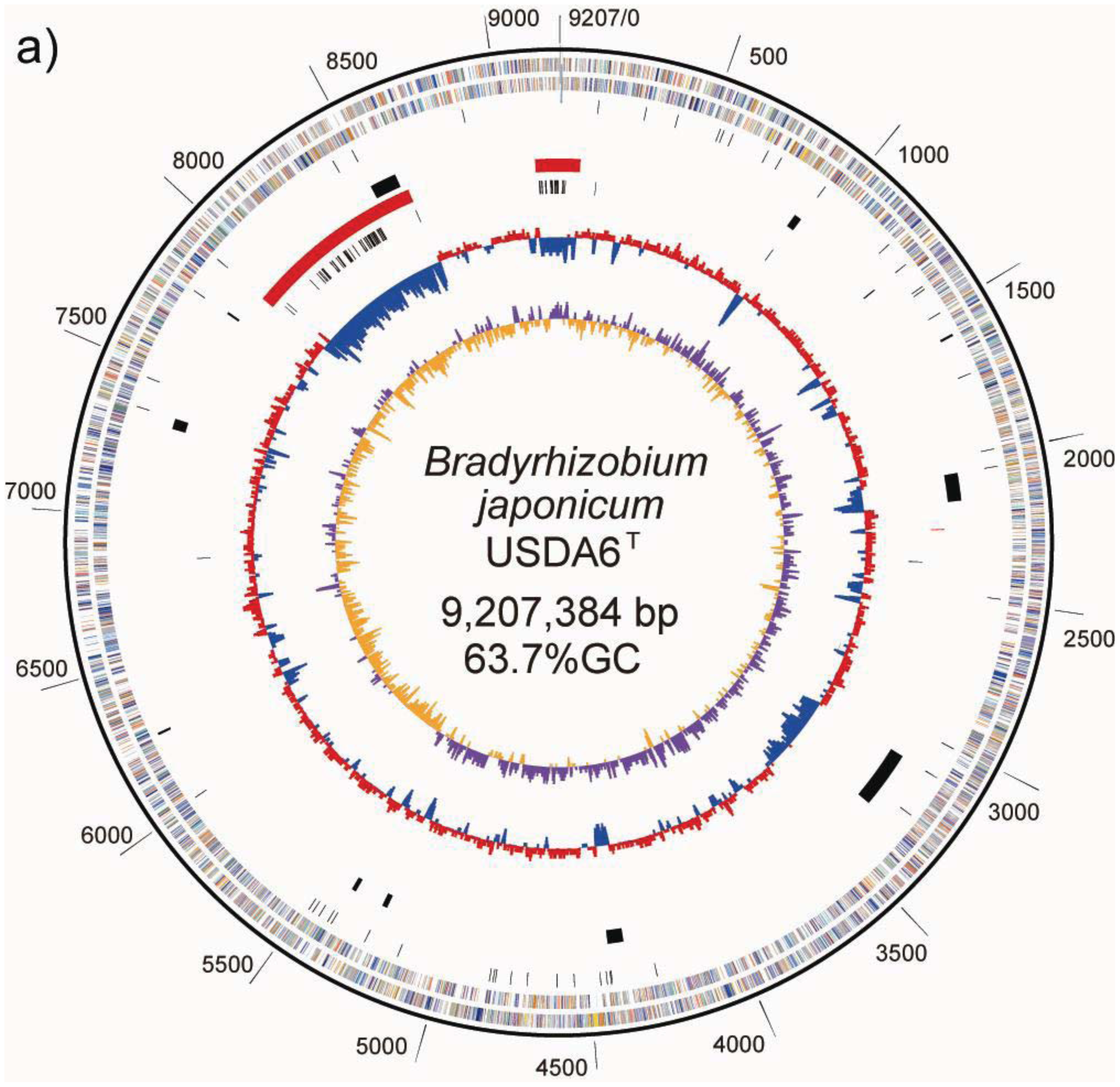
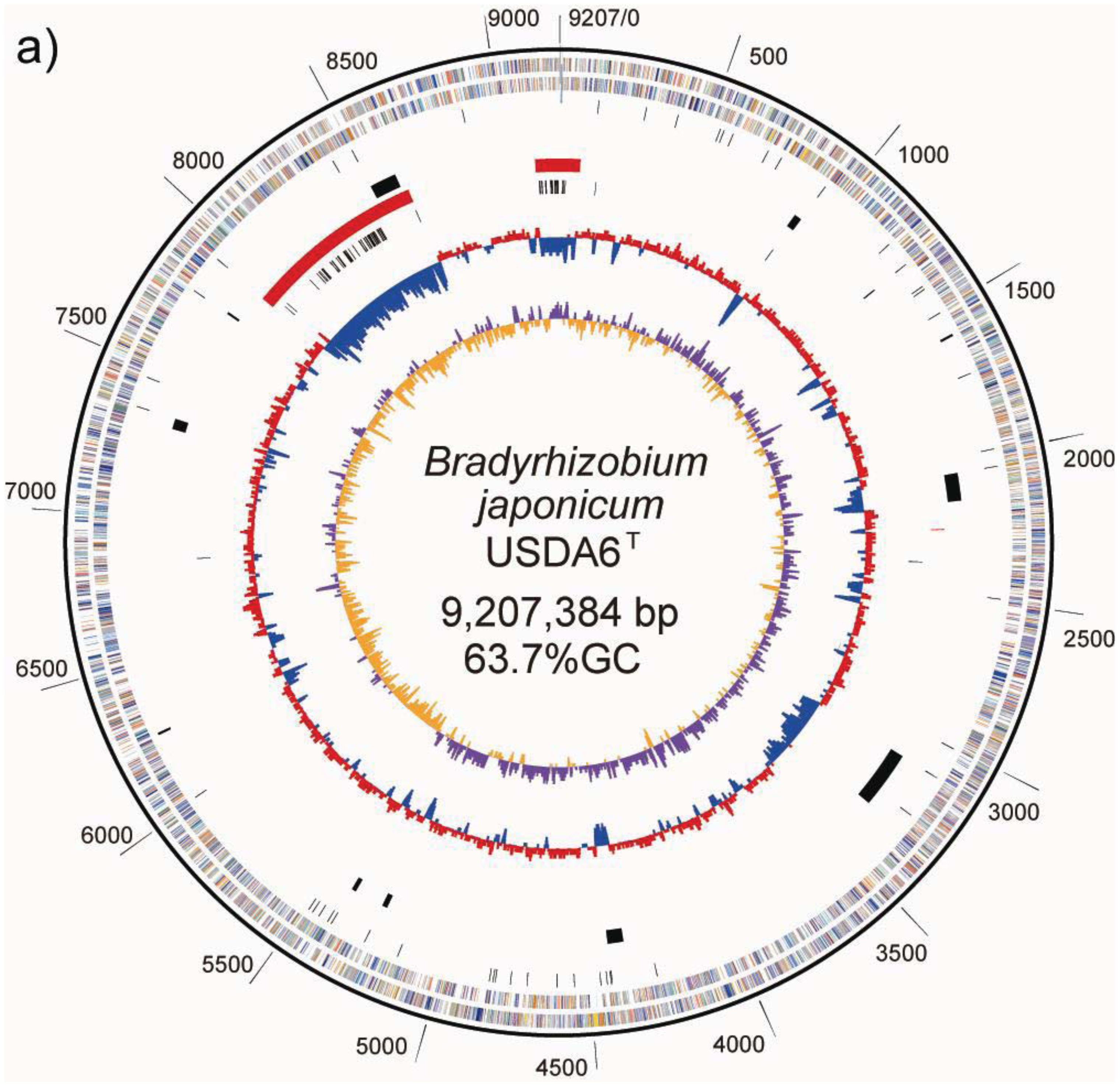
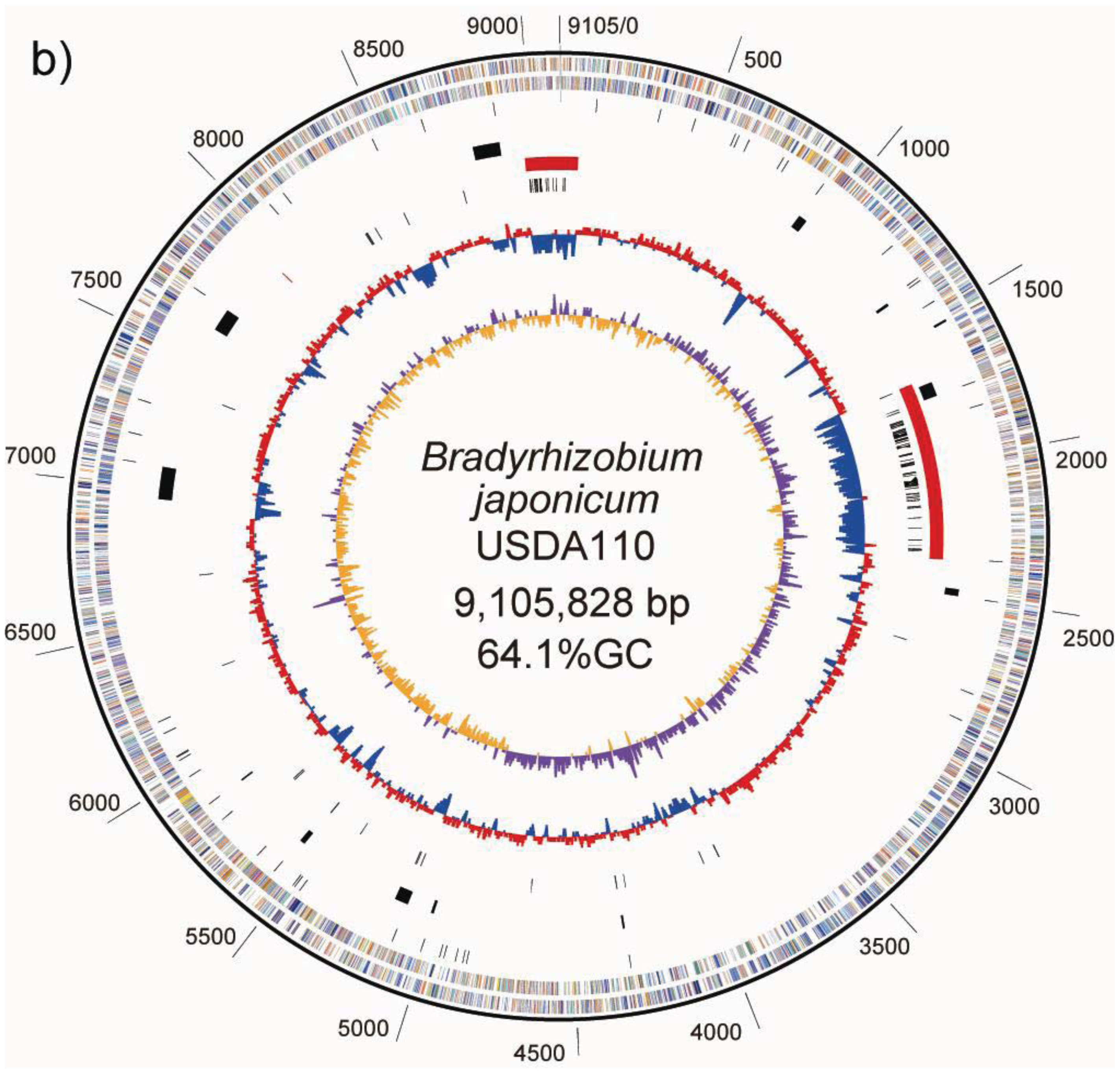
The chromosome of B. japonicum USDA6T is a circular molecule of 9,207,384 bp with an average GC content of 63.67% (Table 2, Figure 1a). No plasmid was detected during the course of this study, including in PFGE patterns. Nucleotide position was assigned as corresponding to the nucleotide sequence initiation site of the B. japonicum USDA110 chromosome [5]. Briefly, numbering began at one of the recognition sites of the restriction enzyme Pac I (TTAATTAA). Hypothetical PmeI and SwaI restriction patterns were calculated for the genome sequence of USDA6T, whose size was fixed at 9,207 kb. The restriction patterns are shown in Table 1. The resulting eight fragments, from 103 kb to 2,322 kb, roughly corresponded to the physical restriction patterns of the total DNA seen in PFGE analysis. This supports the structural integrity of the complete genome sequence.
The USDA6T genome sequence is available in public DNA databases (DDBJ/GenBank/EMBL) under the accession number AP012206.
| USDA6T | USDA 110 | |
|---|---|---|
| Size (bp) | 9,207,384 | 9,105,828 |
| G + C content (%) | 63.67 | 64.06 |
| tRNA coding genes | 51 | 50 |
| rRNA gene clusters | 2 | 1 |
| Protein-encoding genes | 8,829 | 8,317 |
| Gene density (bp) | 1,043 | 1,095 |
| Genes assigned to COG | 5,859 | 5,834 |
| Not in COGs | 2,970 | 2,483 |
| Genomic islands | 15 | 14 |
| Insertion sequences | 69 | 104 |
2.2.2. rRNA- and tRNA-Encoding Genes
Two copies of the rRNA gene cluster (rrn) were identified on the USDA6T chromosome in the order 16S-trnI-trnA-23S-5S. One of the rrns (rrn1) is located at coordinates 1,584,400–1,589,790 bp. The second one (rrn2) was found at coordinates 6,251,101–6,256,492 bp, as an exact copy of the first one.
In the case of USDA110, a single copy of rrn was identified on the chromosome (Table 2). In the 5′ flanking region of the rrn cluster, bll1407 (unknown function) was found, and in the 3′ region of the cluster, bll1408 (unknown function) was found. This gene arrangement (bll1407-rrn-bll1408) is conserved in the rrn1 of USDA6T (BJ6T15130-rrn1-BJ6T15190), whereas the organization around rrn2 is completely different. In the region including rrn2, at coordinates 6,233,372-6,260,564, 15 protein-encoding genes, BJ6T61710-BJ6T61810 and BJ6T61870-BJ6T61900, were found, but no orthologs of these genes were found in the USDA110 genome. Briefly, this makes it possible to deduce that the region containing rrn2 was deleted in the course of evolution of the USDA110 genome.
A total of 51 tRNA genes, including two copies each of trnI-GAT and trnA-TGC in the rrns, which represent 46 tRNA species, are located in the USDA6T genome by prediction analysis using the tRNA scan-SE program. Most of the tRNA genes are dispersed on the genome and are likely to be transcribed as single units, except for those in the rRNA gene cluster. There are four copies of trnI-CAU composed of identical sequences (three on USDA110). One copy (BJ6T75390) of the trnI-CAU gene is located at 7,790,768–7,790,843 bp and is considered to have originated from the genomic backbone. The other trnI-CAU (BJ6T31990) is located within one of the nested genomic islands (BJ6TGI06: described in section “2.2.7. Genomic islands and symbiosis island”) inserted in trnK-CUU (BJ6T30200). Two trnI-CAUs, BJ6T81540 and BJ6T82220, are encoded by sequences within the symbiosis island, as already shown for the USDA110 genome. The trnI gene copies are located within genomic islands, suggesting that they were acquired by lateral gene transfer from an unknown donor.
The nucleotide sequences of the majority of the tRNA genes of USDA6T are identical to or highly similar to (97% identity or more) each of their counterparts in the USDA110 genome. However, two exceptions, trnS-GCU (BJ6T46380) and trnC-GCA (BJ6T53250), showed lower identities at 84% and 85%, respectively, with their corresponding genes in the USDA110 genome, whereas they showed 97% and 100%, identities, respectively, to their counterparts in Rhodopseudomonas palustris CGA009, which belongs to a different clade within the Bradyrhizobiaceae [17]. These results show that the USDA6T and USDA110 genomes include intraspecific variations at the molecular level, even in tRNA gene constitution.
2.2.3. Protein-Encoding Genes
Potential protein-encoding regions were predicted using the MetaGeneAnnotator program [18] and a similarity search, as described in section “3.4. Gene assignment, annotation, and information analyses”. The total number of putative protein-encoding genes assigned to the genome was 8,829 (Table 2). The average gene density was estimated to be one gene in every 1,043 bp. Each of the putative protein-encoding genes began with one of three codons: ATG (7,183 genes), GTG (1,084 genes), or TTG (562 genes).
Clusters of Orthologous Groups (COG) assignment for predicted gene products was performed [19]. A total of 5,859 putative gene products encoded by USDA6T were assigned to COG identifications classified into 18 COG categories. For comparison, 5,834 of 8,317 genes in USDA110 were classified into 18 COG categories using the same parameters (Table 2). As shown by comparing the numbers of genes in each category between USDA6T and USDA110 in Table 3, there is a marked underrepresentation of genes classified in category L; “Replication, recombination, and repair” in USDA6T. This is ascribed to the presence of a smaller number of transposase genes in USDA6T than in USDA110 [described in 2.2.6. “Insertion sequences (ISs)”].
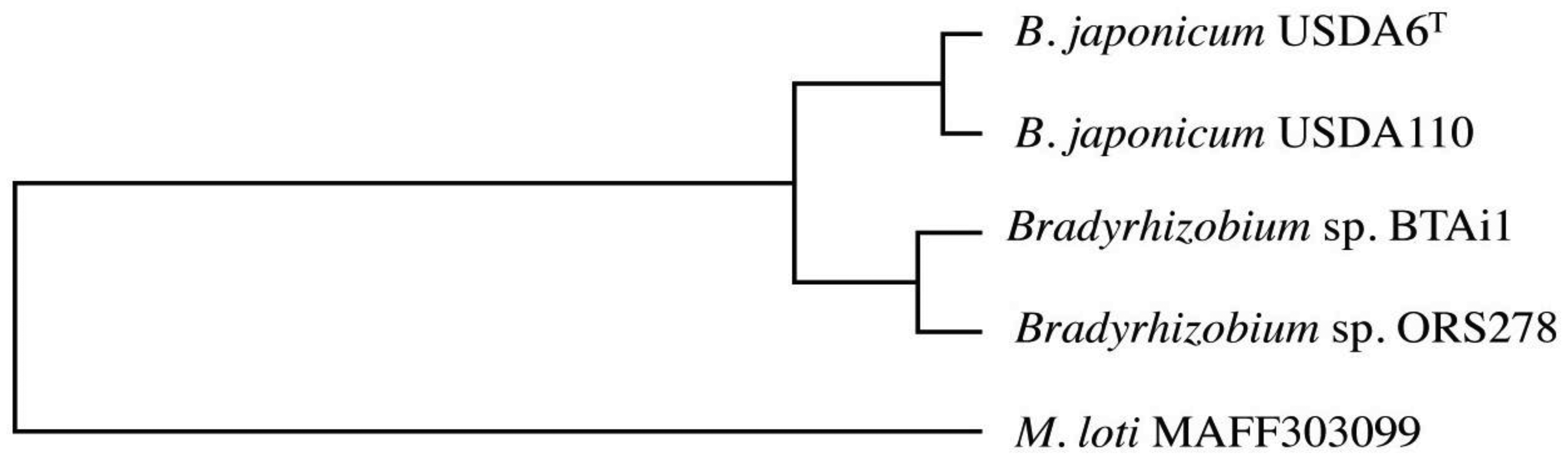
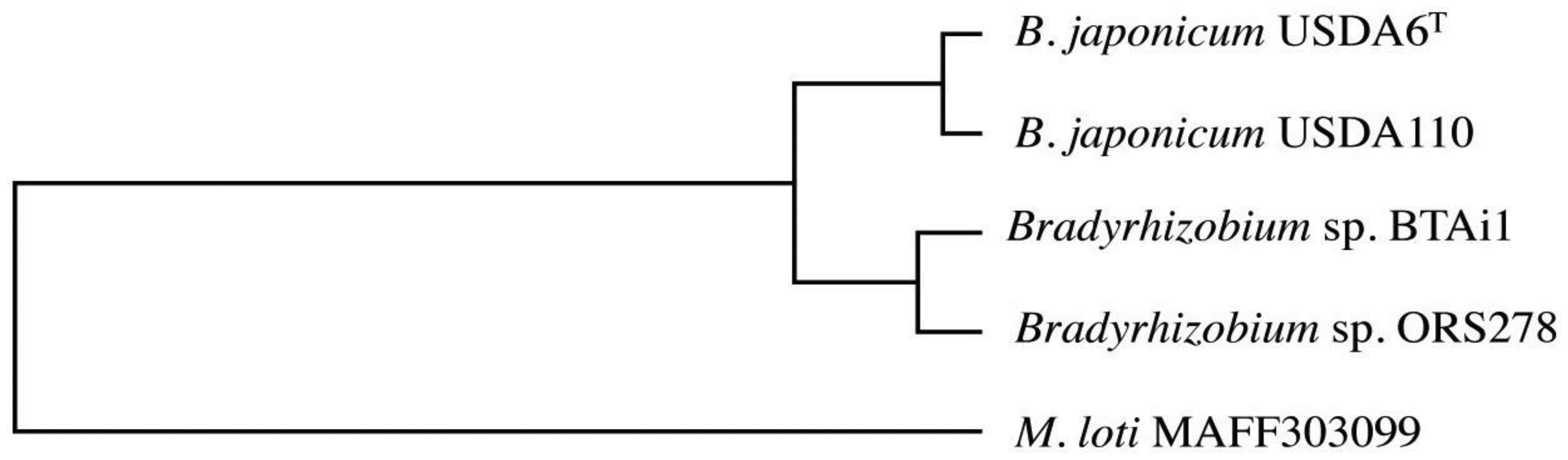
In order to evaluate conservation of the genes in the genome of USDA6T, we carried out ortholog cluster analysis using information on genes predicted in USDA6T and USDA110 as well as in Bradyrhizobium sp. BTAi1, Bradyrhizobium sp. ORS278, and Mesorhizobium loti MAFF303099 [8,20]. The data from this analysis were assessed by focusing on orthologous clusters showing single linkages among the five bacterial strains. As a result, 705 groups were extracted as acceptable orthologous clusters from the grouping data, and 682 of these exhibited patterns corresponding to the phylogenetic tree generated using 16S rRNA genes or ITS sequences from Bradyrhizobiaceae members (Figure 2) [15]. This result shows that many of the protein-encoding genes that maintained a single orthologous relationship within Bradyrhizobiaceae members had evolved at a similar pace.
| COG code | USDA110 entire genome | USDA6T | |
|---|---|---|---|
| entire genome | GIs a | ||
| Information storage and processing | |||
| J: Translation, ribosomal structure, and biogenesis | 197 (2.4%) | 203 (2.3%) | 1 (0.2%) |
| K: Transcription | 468 (5.6%) | 487 (5.5%) | 10 (1.7%) |
| L: Replication, recombination, and repair | 333 (4.0%) | 290 (3.3%) | 30 (5.1%) |
| Cellular processes and signaling | |||
| D: Cell cycle control, cell division, and chromosome partitioning | 33 (0.4%) | 30 (0.3%) | 0 (0.0%) |
| T: Signal transduction mechanisms | 330 (4.0%) | 346 (3.9%) | 30 (5.1%) |
| M: Cell wall/membrane/envelope biogenesis | 263 (3.2%) | 245 (2.8%) | 9 (1.5%) |
| N: Cell motility | 243 (2.9%) | 233 (2.6%) | 9 (1.5%) |
| O: Posttranslational modification, protein turnover, and chaperones | 213 (2.6%) | 216 (2.4%) | 16 (2.7%) |
| Metabolism | |||
| C: Energy production and conversion | 419 (5.0%) | 411 (4.7%) | 4 (0.7%) |
| G: Carbohydrate transport and metabolism | 389 (4.7%) | 409 (4.6%) | 4 (0.7%) |
| E: Amino acid transport and metabolism | 717 (8.6%) | 710 (8.0%) | 14 (2.4%) |
| F: Nucleotide transport and metabolism | 93 (1.1%) | 89 (1.0%) | 0 (0.0%) |
| H: Coenzyme transport and metabolism | 183 (2.2%) | 179 (2.0%) | 0 (0.0%) |
| I: Lipid transport and metabolism | 289 (3.5%) | 271 (3.1%) | 2 (0.3%) |
| P: Inorganic ion transport and metabolism | 281 (3.4%) | 296 (3.4%) | 5 (0.8%) |
| Q: Secondary metabolites biosynthesis, transport and catabolism | 356 (4.3%) | 356 (4.0%) | 11 (1.9%) |
| Poorly characterized | |||
| R: General function prediction only | 583 (7.0%) | 599 (6.8%) | 26 (4.4%) |
| S: Function unknown | 444 (5.3%) | 489 (5.5%) | 19 (3.2%) |
| not in COGs | 2,483 (29.9%) | 2,970 (33.6%) | 402 (67.9%) |
| Total | 8,317 | 8,829 | 592 |
The percentage of assigned genes out of the total number of genes is shown in parentheses.aThe number of predicted genes assigned inside fifteen GIs of USDA6T is shown.
2.2.4. Putative Replication Origin
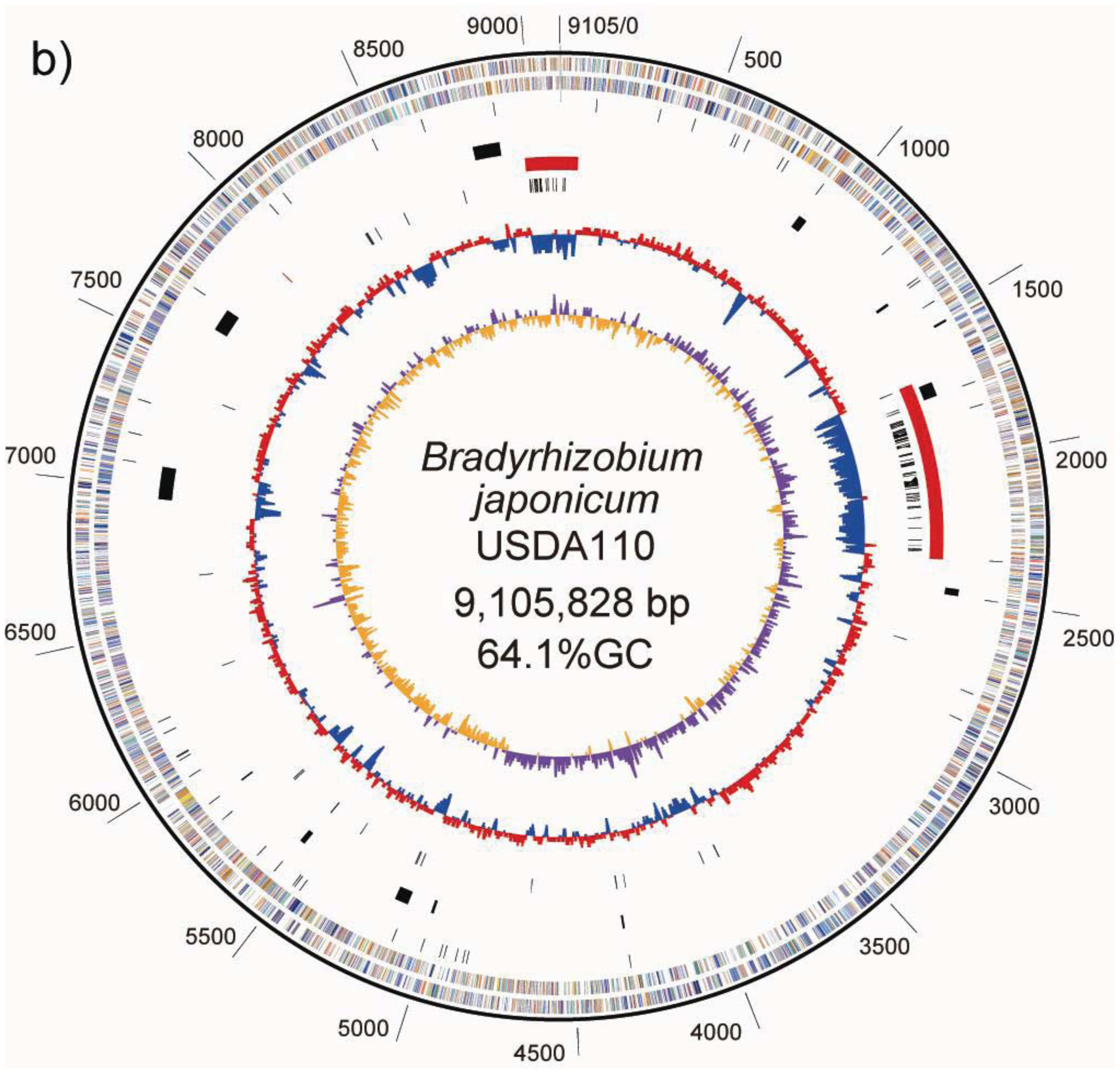
A GC-skew analysis was performed to predict the locations of the replication origin and terminus on the chromosome [21]. We observed that shifts in GC-skew values occur in two loci on the USDA6T chromosome, at coordinates 650 and 5,430 kb, as shown in Figure 1a. Shifts of the same sort were found at 705 kb and 4,900 kb in USDA110 (Figure 1b) [5].
The conserved sequence pattern required to convert a dimer chromosome to monomers after aberrant DNA duplication led to the designation of dif, a 28-mer sequence [22]. The genome sequence of USDA6T revealed that dif is located at coordinates 5,304,527–5,304,554, near one side of the GC-skew shift. It is likely that DNA replication terminates in this region.
Another shift in GC-skew was detected at the diagonal position of dif. This seems to indicate the presence of a putative replication origin in the USDA6T chromosome. The occurrence of a gene cluster around this putative origin of replication coincides with other typical origins from the Bradyrhizobiaceae [10]. In the case of USDA110, the putative origin is positioned between bll0636 (conserved hypothetical protein) and blr0637 (conserved hypothetical protein). In USDA6T, BJ6T06040 and BJ6T06050 were found to be orthologs of these two genes, respectively. This intergenic region showed a nucleotide sequence alignment with 82% identity over 391 bp at coordinates 629,186–629,573 with the corresponding region of the USDA110 sequence. This suggests that the sequence corresponding to the origin of replication is conserved in this region.
2.2.5. Structural Features of the USDA6T Genome
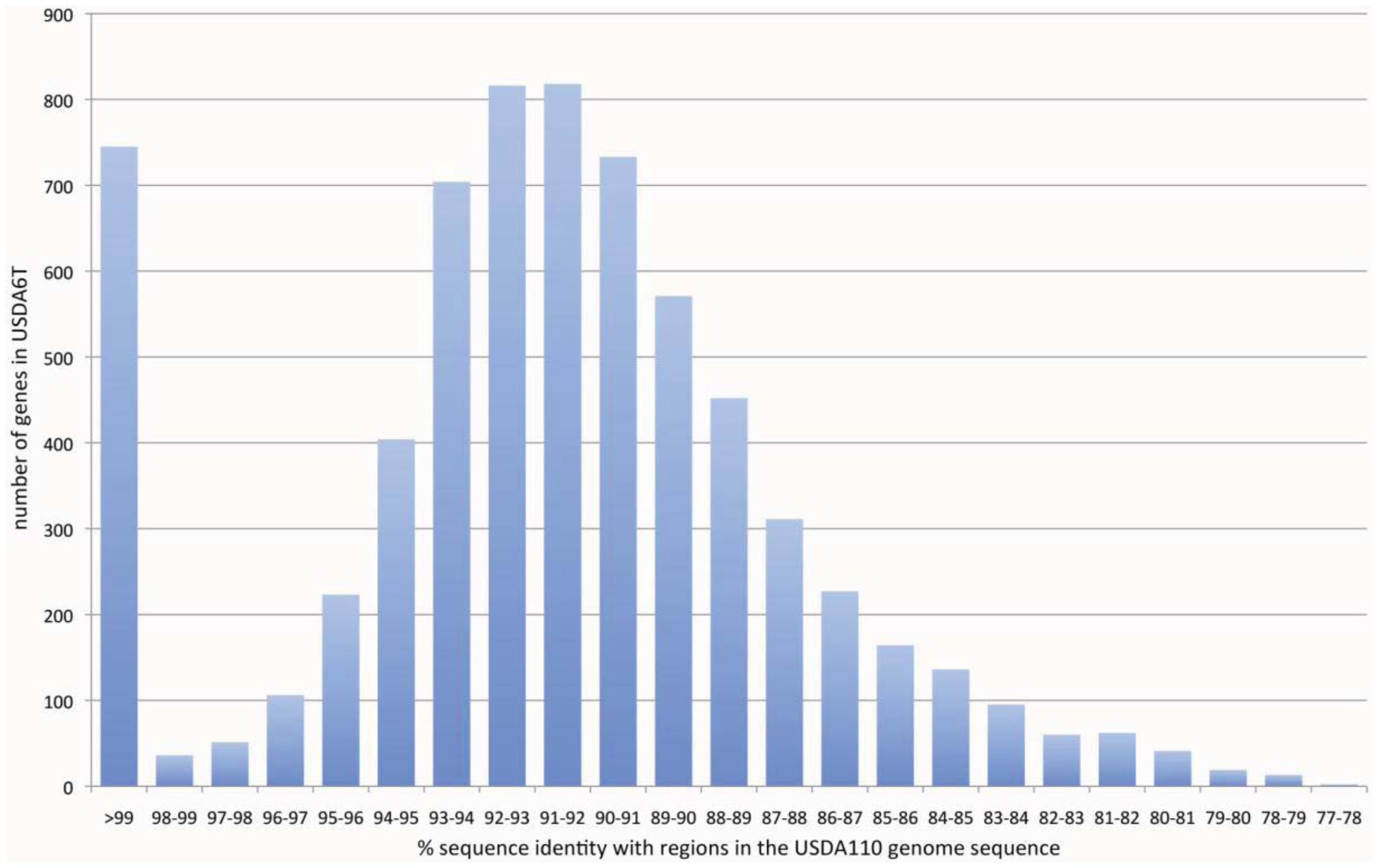
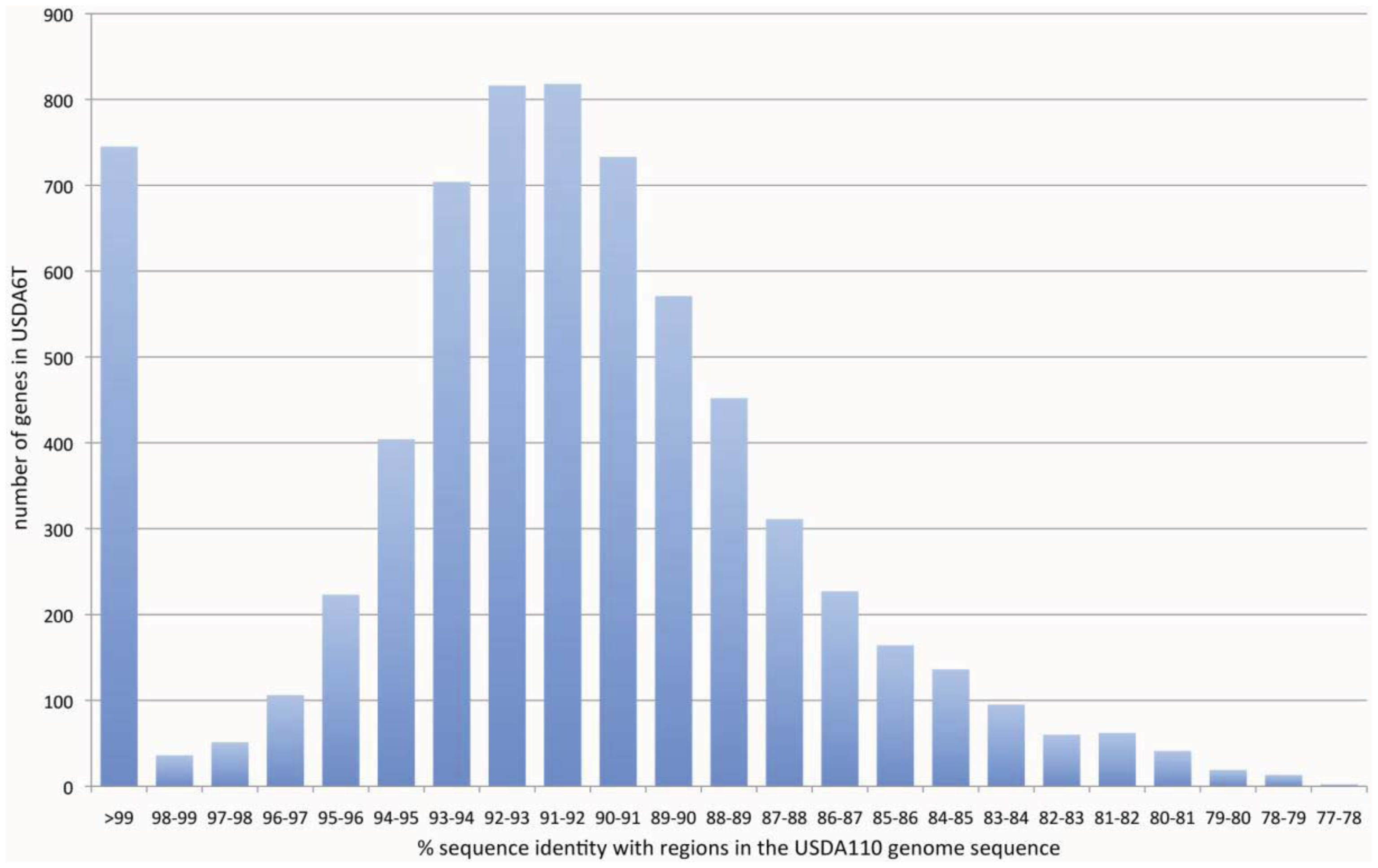
As described in “2.1. Genome Sequencing”, the results of PFGE pattern analysis suggested different genomic proportions, in terms of restriction digest patterns, between USDA6T and USDA110 (Table 1), while the whole-genome sequence analysis focused on sequence conservation between the gene set from USDA6T and the gene set from USDA110. For this purpose, the predicted protein-coding regions of USDA6T were compared with those of USDA110 at the nucleotide sequence level by BLASTN best-hit analysis. The resulting sequence alignments were evaluated based on both the blast bit score of each alignment and the % identity. On this basis, all protein genes were classified into three groups: (1) distinct identical gene, score 100 or more, with over 99% identity; (2) significant similarity, score 100 or more, with less than 99% identity; (3) no similarity, score of less than 100.
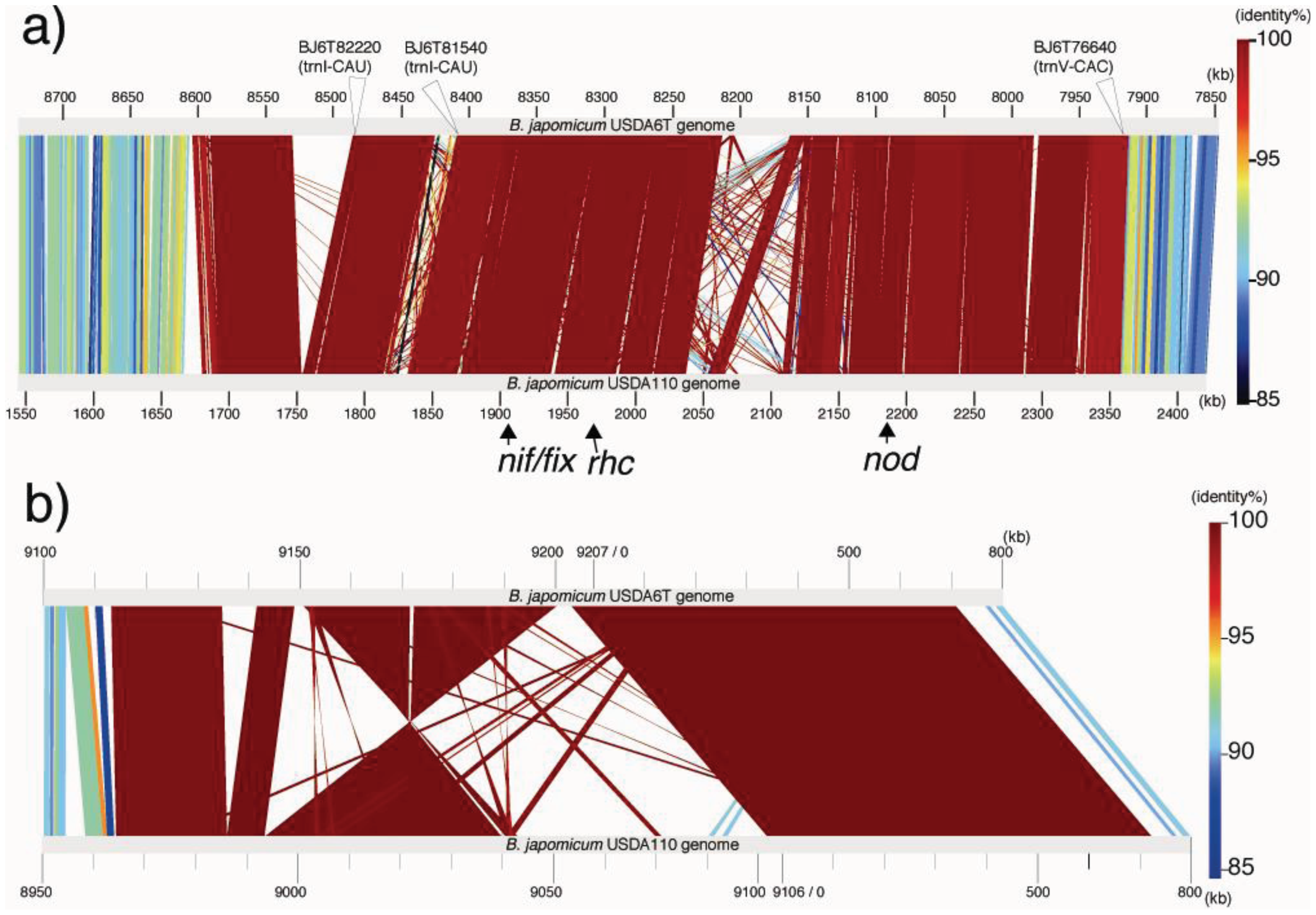
As a result of this classification, the number (and percentage) of “distinct identical genes” was 744 (8.5%); the number of genes with “significant similarity” was 6,045 (68.5%); and the number of genes with “no similarity” was 2,039 (23%). Among the USDA6T genes categorized as having “significant similarity”, 3,071 genes have identity percentages within the range of 90% to 94% (Figure 3), and are distributed evenly throughout the genome (data not shown). However, 732 of the genes classified as “distinct identical gene” were found to be limited to three loci with the following coordinates: 2,248,280–2,252,640 (Locus B), 7,920,139–8,614,416 (Locus A), and 9,113,996-0-70,356 (Locus C). The distribution breakdown for the distinct identical genes is: Locus B, seven genes; Locus A, 541 genes; and Locus C, 184 genes. The gene list is shown in Supplemental table 1. These three loci correspond to three parts of the putative symbiosis island (Figure 1a: described in the section “2.2.7. Genomic islands and symbiosis island”).
A dot plot of a BLASTN comparison of the whole-genome sequences of strains USDA6T and USDA110 is presented in Supplemental Figure 2. BLASTN was performed with default parameters, except that the threshold for the alignment was set to a cutoff E-value of 10−20. The results from this comparison showed the following genomic characteristics:
- (1)
Although the dot plot depicts the colinearity of major regions of the two chromosomes, alignment of a region including the replication origin between 8.7 Mb and 1.5 Mb of the USDA6T chromosome harbors a large inversion, compared with USDA110. The two replication origins were positioned similarly; at 0.69 Mb of USDA110 and 0.63 Mb of USDA6T;
- (2)
Many intensively dotted areas in both the 8.0–8.5 Mb and the 9.1–9.2 Mb regions of the USDA6T genome shows the existence of highly copied insertion sequences inside the putative symbiosis island (Table 4: described in section “2.2.6. Insertion sequences”);
- (3)
A lack of lines indicates regions of unique DNA sequence in each genome. Major deflections of the segments along either axis indicate insertions of genomic islands;
- (4)
Red lines corresponding to 7.9–8.6 Mb and 9.1–0.7 Mb on the vertical axis indicate the presence of homologous blocks of sequence. The high level of matching sequence alignment suggests that the two regions share a recent common ancestor that includes the insertion of a symbiosis island. These regions match in Locus A and Locus C, described above.
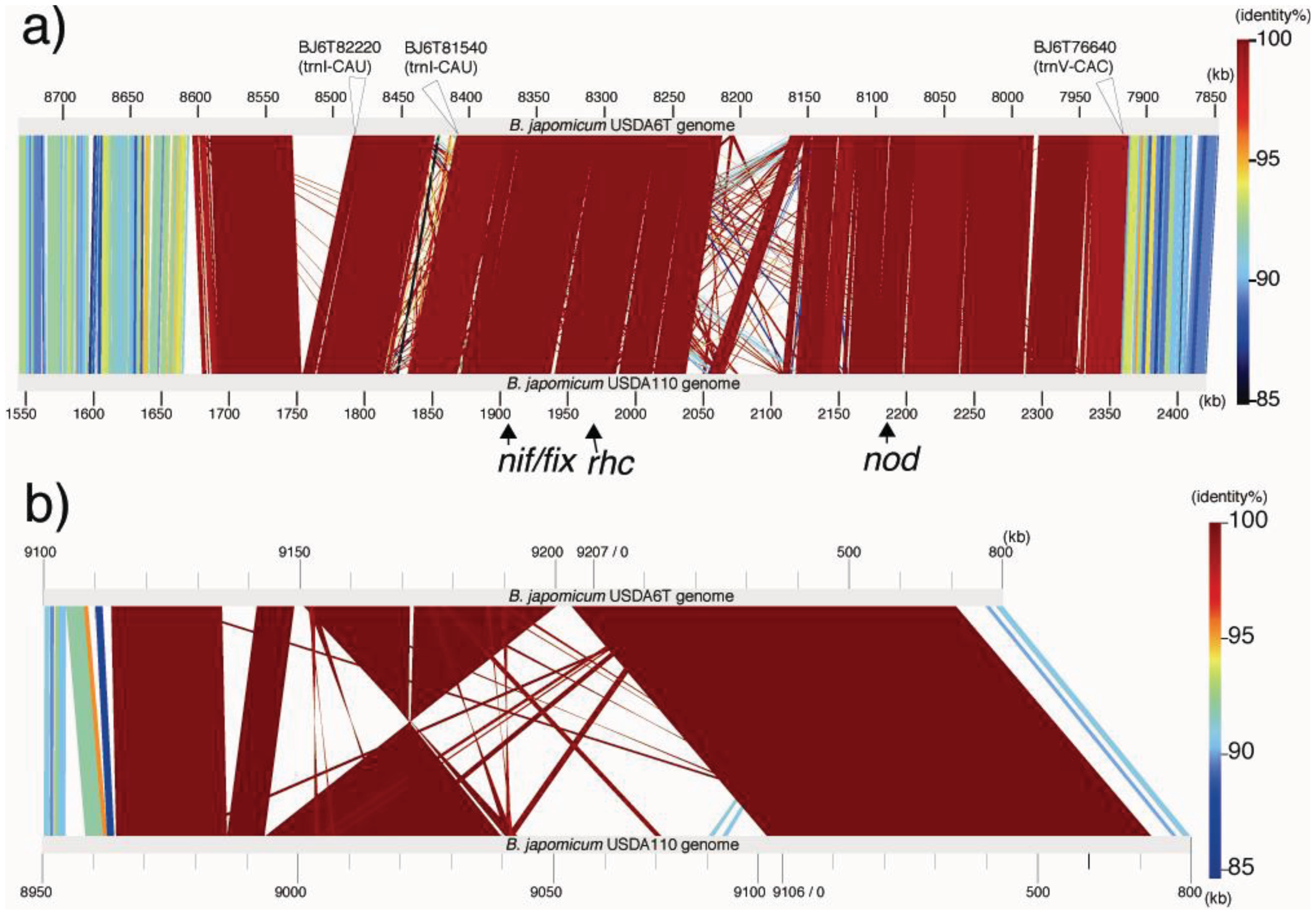
Comparisons of linear representations were performed by matching the USDA110 genome with Locus A and Locus C. The approximately 890 kb regions including Locus A are depicted in Figure 4a. The sequence of USDA110 corresponding to Locus A is located at coordinates 1,680,702–2,362,044 of the whole-genome sequence. Locus C is equivalent to a USDA110 region at coordinates 8,974,971-0-70,365 (Figure 4b). The degree of almost all similarities within each long alignment part is represented by red color, denoting their relationships with distinct identical sequences. The fact that these nucleotid sequences in the alignments are included in both coding and intergenic regions shows that there is little intergenic variation in these loci (Figure 4). The variations in these regions depend on the insertion of transposable elements or deletions. Strain-specific islets more than 10 kb in size were found in three parts in USDA6T and four parts in USDA110. Therefore, such events are attributed to the significant variations between these loci.
CGH using a DNA array of USDA110 has been applied to some B. japonicum strains, including the USDA6T, by Itakura et al. [15]. The signal ratio for each probe was plotted along the genome of USDA110. The CGH profile showed that almost all genomic islands of USDA110 were absent in USDA6T. However, the signals within the symbiosis island (1.68–2.36 Mb and 8.98-0-0.08 Mb of USDA110 genome) were detected at higher intensity than the ones in the genomic backbone region. Comparative genome analysis shows that the results of the CGH profile analysis indicated the presence of a highly conserved region between the two different strains.
2.2.6. Insertion Sequences (ISs)
ISs are simple mobile DNA elements capable of moving through the genome by transposition. Transposase, an enzyme encoded in ISs, is involved in site-specific DNA recombination required for transposition in bacteria. A number of different ISs found in the bacterial genome sequence have been classified into various families based on structural features of transposases encoded in the ISs [23]. With respect to ISs found in the USDA110 genome, 104 copies have been classified into 20 groups, each comprising 1 to 15 members on the basis of the type of transposase [5]. As shown on the USDA110 map in Figure 1b and Table 4, 62 out of 104 ISs (60%) were located in presumptive symbiosis island A, and 19 (18%) were found in presumptive symbiosis island C (described in the section “2.2.7. Genomic islands and symbiosis island”).
| IS name | USDA110 | USDA6T | ||||
|---|---|---|---|---|---|---|
| entire genome | symbiosis island A | symbiosis island C | entire genome | symbiosis island A | symbiosis island C | |
| RSalpha | 15 | 8 | 2 | 9 | 7 | 2 |
| RSbeta | 12 | 7 | 4 | 7 | 3 | 4 |
| FK1 | 6 | 5 | 1 | 5 | 4 | 1 |
| IS1632 | 2 | 2 | 0 | 2 | 2 | 0 |
| ISB20 | 3 | 2 | 1 | 2 | 1 | 1 |
| ISB27 | 3 | 2 | 1 | 3 | 2 | 1 |
| ISBj2 | 10 | 9 | 0 | 10 | 8 | 2 |
| ISBj3 | 4 | 2 | 0 | 1 | 1 | 0 |
| ISBj4 | 6 | 0 | 1 | 1 | 0 | 1 |
| ISBj5 | 5 | 1 | 2 | 5 | 1 | 2 |
| ISBj6 | 1 | 0 | 1 | 0 | 0 | 0 |
| ISBj7 | 11 | 9 | 2 | 10 | 8 | 2 |
| ISBj8 | 4 | 2 | 2 | 5 | 4 | 1 |
| ISBj9 | 2 | 0 | 1 | 0 | 0 | 0 |
| ISBj10 | 1 | 1 | 0 | 2 | 0 | 2 |
| ISBj11 | 5 | 3 | 0 | 1 | 1 | 0 |
| ISBj12 | 1 | 1 | 0 | 2 | 0 | 2 |
| ISBj13 | 3 | 3 | 0 | 1 | 1 | 0 |
| ISBj14 | 7 | 3 | 0 | 1 | 0 | 1 |
| ISBj15 | 3 | 2 | 1 | 2 | 2 | 0 |
| Total | 104 | 62 | 19 | 69 | 45 | 22 |
To examine the distribution of ISs on the USDA6T genome, similarity searches using the BLASTN program were conducted using the ISs assigned on the USDA110 genome as a query against the entire USDA6T genome sequence. The regions harboring alignments longer than 100 bp, which showed cutoff E-values of up to 10−60, were then evaluated. The sequences of these regions were classified according to the corresponding IS groups reported in USDA110. As a result, a total of 69 copies of ISs were assigned identities in the USDA6T genome (Table 4). For all ISs in the USDA110 genome, except ISBj6 and ISBj9, corresponding copies were identified on the USDA6T genome. However, a clear difference in the total copy number of ISs between USDA6T and USDA110 was revealed. This is concordant with the results of the COG analysis, in which USDA6T showed a lower density of transposase genes in the “L” category: Replication, recombination and repair (Table 3).
As shown on the USDA6T map in Figure 1a and Table 4, 45 out of 69 ISs (65%) are located in presumptive symbiosis island A, and 22 (32%) are found in island C (described in the section “2.2.7. Genomic islands and symbiosis island”). Compared with USDA110, there is a marked tendency for ISs in USDA6T to be concentrated in discrete parts of the genome equivalent to the symbiosis island rather than to be distributed evenly throughout the genome (Figure 4a). This observation suggests that there is a comparatively lower degree of plasticity in the USDA6T genome than in the USDA110 genome, due to the smaller number of mobile repetitive elements.
2.2.7. Genomic Islands and Symbiosis Island
A genomic island (GI) is a region that forms syntenic groups of multiple accessory genes encoding diverse functions and characteristics [24]. Acquisition of such a genetic element confers advantages on the bacterium in comparison to other bacteria that colonize the same environment under the same ambient conditions. The majority of GIs occur as traits of integrative elements inserted into the sequenced genome through horizontal transfer. Their sequences are often inserted into various tRNA genes in various bacterial genomes, with the entire tRNA gene flanking the inserted sequence at one end, and the duplicated portion of tRNA gene flanking the inserted sequence at the other end [24].
Duplication of portions of tRNA genes were found at ten loci in the USDA6T genome (Figure 1a). In seven out of ten loci, GIs were identified as a single pair of tRNA genes and the portion. However, multiple duplications of such portion of the tRNA genes were observed in the remaining three loci; four copies in trnK-CTT (BJ6T30200), two in trnQ-TTG (BJ6T71420), and two in trnI-CAT (BJ6T82220) (Table 5). This suggests that discrete GIs were independently integrated into the same tRNA gene, and that this frequently occurred in the course of recent bacterial evolution. Multiple insertions of GIs into trnK-CTT and trnI-CAT have also been reported in the USDA110 genome [5]. These two tRNA genes may be hotspots of GI integration in B. japonicum.
As a result, fifteen varieties of typical GIs with duplicated portions of tRNA genes at both ends were found in the USDA6T genome. The GC content in all islands is lower than the average GC content (63.67%) in the genome (Table 5). The sizes of these GIs are diverse. GI segments ranging from 2.9 to 100 kb separate these duplications, and besides, no apparent nucleotide sequence similarity was detected in any of the fifteen GIs, nor was any such similarity detected in the fourteen GIs from USDA110, except for one pair: BJ6TGI15 in USDA6T and trnI1-element in USDA110, which are distinct identical sequences inserted in each symbiosis island.
| GI name | tRNA gene (gene_ID) | GI-left end | GI-right end | GI-length (bp) | GC% | duplication length (bp) | gene_ID of integrase |
|---|---|---|---|---|---|---|---|
| BJ6TGI01 | trnR-CCG (BJ6T08940) | 911,924 | 937,940 | 26,017 | 55.7 | 50 | BJ6T08690 |
| BJ6TGI02 | trnR-ACG (BJ6T12260) | 1,307,833 | 1,310,731 | 2,899 | 62.7 | 18 | - |
| BJ6TGI03 | trnE-CTC (BJ6T19770) | 2,046,607 | 2,146,757 | 100,151 | 59.7 | 48 | BJ6T20830 |
| BJ6TGI04 | trnK-CTT (BJ6T30200) | 3,118,227 | 3,192,248 | 74,022 | 59.5 | 46 | BJ6T31070 |
| BJ6TGI05 | trnK-CTT (BJ6T30200) | 3,192,295 | 3,236,376 | 44,082 | 60.9 | 46 | BJ6T31620 |
| BJ6TGI06 | trnK-CTT (BJ6T30200) | 3,236,423 | 3,322,375 | 85,953 | 59.0 | 46 | - |
| BJ6TGI07 | trnK-CTT (BJ6T30200) | 3,322,421 | 3,326,439 | 4,019 | 58.6 | 45 | BJ6T32590 |
| BJ6TGI08 | trnT-TGT (BJ6T43300) | 4,371,641 | 4,428,687 | 57,047 | 58.8 | 47 | BJ6T42570 |
| BJ6TGI09 | trnS-TGA (BJ6T51900) | 5,251,688 | 5,269,566 | 17,879 | 58.4 | 14 | BJ6T51650 |
| BJ6TGI10 | trnN-GTT (BJ6T53120) | 5,380,012 | 5,394,916 | 14,905 | 59.3 | 53 | BJ6T52900 |
| BJ6TGI11 | trnQ-TTG (BJ6T71420) | 7,326,115 | 7,332,736 | 6,622 | 62.7 | 47 | - |
| BJ6TGI12 | trnQ-TTG (BJ6T71420) | 7,332,777 | 7,363,200 | 30,424 | 62.1 | 39 | - |
| BJ6TGI13 | trnI-CAT (BJ6T75390) | 7,790,844 | 7,797,321 | 6,478 | 61.1 | 48 | - |
| BJ6TGI14 | trnI-CAT (BJ6T82220) | 8,494,412 | 8,539,486 | 45,075 | 58.5 | 49 | BJ6T82530 |
| BJ6TGI15 | trnI-CAT (BJ6T82220) | 8,539,534 | 8,591,258 | 51,725 | 59.5 | 47 | BJ6T82650 |
A total of 592 genes were predicted in the fifteen genomic islands, and 190 of these were assigned into 18 COG categories along with the other genes in the USDA6T genome as described in the section “2.2.3. Protein-encoding genes” (Table 3). This result shows that there is marked overrepresentation of COG categories L (DNA replication, recombination and repair) and T (Signal transduction mechanisms) inside the GIs, due to the presence of integrase and two-component regulator system proteins, respectively. Each of the ten GIs contains a putative integrase gene (Table 5), which implies a function for the GI in integrative events. Although the effects stemming from the addition of two-component regulator system-related genes to the USDA6T genome remain unclear, these genes may confer novel abilities that permit environmental adaptation by USDA6T.
There are other potential areas for presumptive GIs within flanking tRNA genes. These regions have no duplications in either end of their segments; however, their sequences show lower GC content. We found five such candidate GIs, with coordinates at 262–288 kb (59.5% GC) in trnS-GGA (BJ6T02870), 1,473–1,503 kb (59.6% GC) in trnL-CAA (BJ6T14350), 2,488–2,544 kb (61.1% GC) in trnM-CAT (BJ6T24130), 8,831–8,882 kb (62.7% GC) in trnM-CAT (BJ6T85680) and 7,920–8,614 kb (59.0% GC) in trnV-CAC (BJ6T76640). Although each of the first four regions listed above represents a specific sequence in USDA6T compared to USDA110, the putative GI flanking trnV is consistent with a region described as Locus A in “2.2.5. Structural features of the USDA6T genome” (Figure 4a), and shows the significant similarity to “symbiosis island A” from USDA110 (Figure 1b) [5].
A symbiosis island carries genes required for nitrogen-fixing symbiosis within the host plant. In the case of USDA110, a 680-kb DNA segment was found to correspond to a symbiosis island [5]. A trnV-CAC was found at one end of this region, but there is no trace of any partial terminal duplication of the gene sequence at the other end of this low GC region. However, a 45-bp segment of the 3′ terminal portion of the trnV gene flanking 6 kb of segments with lower GC contents was found 3,528 kb away from the trnV flanking the symbiosis island (Figure 1b). Another important characteristic of ISs is that they tend to be concentrated in the symbiotic region of the rhizobial genome [7]. The 680-kb segment tagged as a symbiosis island contains 60% (62) of the identified ISs in the genome of USDA110 (Table 4). The other remarkable region containing a high number (19) of ISs and showing low GC content is located at coordinates 8,975-0-70 kb (Figure 1b). Because this 201-kb DNA segment is similar to a symbiosis island in terms of the above features, it is supposed that this segment originated from a common, ancestral, laterally transferred DNA.
A fairly similar situation to USDA110 was found in the USDA6T genome. For USDA6T, the presence of a 695-kb DNA region with low GC content at coordinates 7,920–8,614 kb (59.0%) was revealed, with remarkable features reminiscent of a symbiosis island (Figure 1a). This region included most of the nod, nif, and fix genes, and trnV-CAC (BJ6T76640) was found at one end of this region at coordinates 7,919,767–7,919,841, but no trace of terminal duplication of trnV was detected at the other end of this region. However, a 45-bp segment of the 3′ terminal portion of the trnV-CAC gene was found at coordinates 2,248,279–2,248,223, which is 3,536-kb away from the intact trnV-CAC gene (Figure 1a), and a putative integrase gene (BJ6T21770) was found next to this tRNA partial segment. The GC content of the 4-kb region (coordinates 2,248–2,252 kb) adjacent to this gene is lower than the average GC content of the entire genome (60.1% vs. 63.67%). Among the 69 ISs identified in the USDA6T genome, 45 (65%) are located in the 695-kb region, and 22 (32%) of the remaining ISs are located in the third 164-kb region at coordinates 9,114-0-70 kb, which also has a lower than average GC content (59.85%) (Figure 1a, Table 5). The nucleotide sequences of these three regions correspond to the regions described as Locus A, Locus B, and Locus C in “2.2.5. Structural features of the USDA6T genome”, except for the intervening strain-specific insertions and small inversions (Figure 4b). This suggests that an ancient larger symbiosis island may have been split into three smaller ones: the 695-kb DNA region (putative symbiosis island A at coordinates 7,920–8,614 kb), the 4-kb region (putative symbiosis island B at coordinates 2,248–2,252 kb), and the 164-kb region (putative symbiosis island C at coordinates 9,114-0-70 kb). This is postulated to have taken place by unknown large-scale genome rearrangement through comparable mechanisms after independent integration events into both chromosomes.
2.3. Comparative Aspects of Genetic Organization between USDA6T and USDA110
A comparison between the putative gene products of USDA6T and USDA110 was carried out by BLASTP analysis with the aim to determining the evolutionary diversity of B. japonicum strains. This analysis revealed that 7,651 (86.7%) putative protein genes in the USDA6T are conserved in the USDA110 genome. Among these conserved genes, 739 genes (8.4% of total genes) show significant high similarity to those in the USDA110 genome (more than 99% identity). Most of the conserved genes with high similarity are located in the symbiosis island. This result is consistent with the nucleotide sequence level comparison as described in “2.2.5. Structural features of the USDA6T genome”.
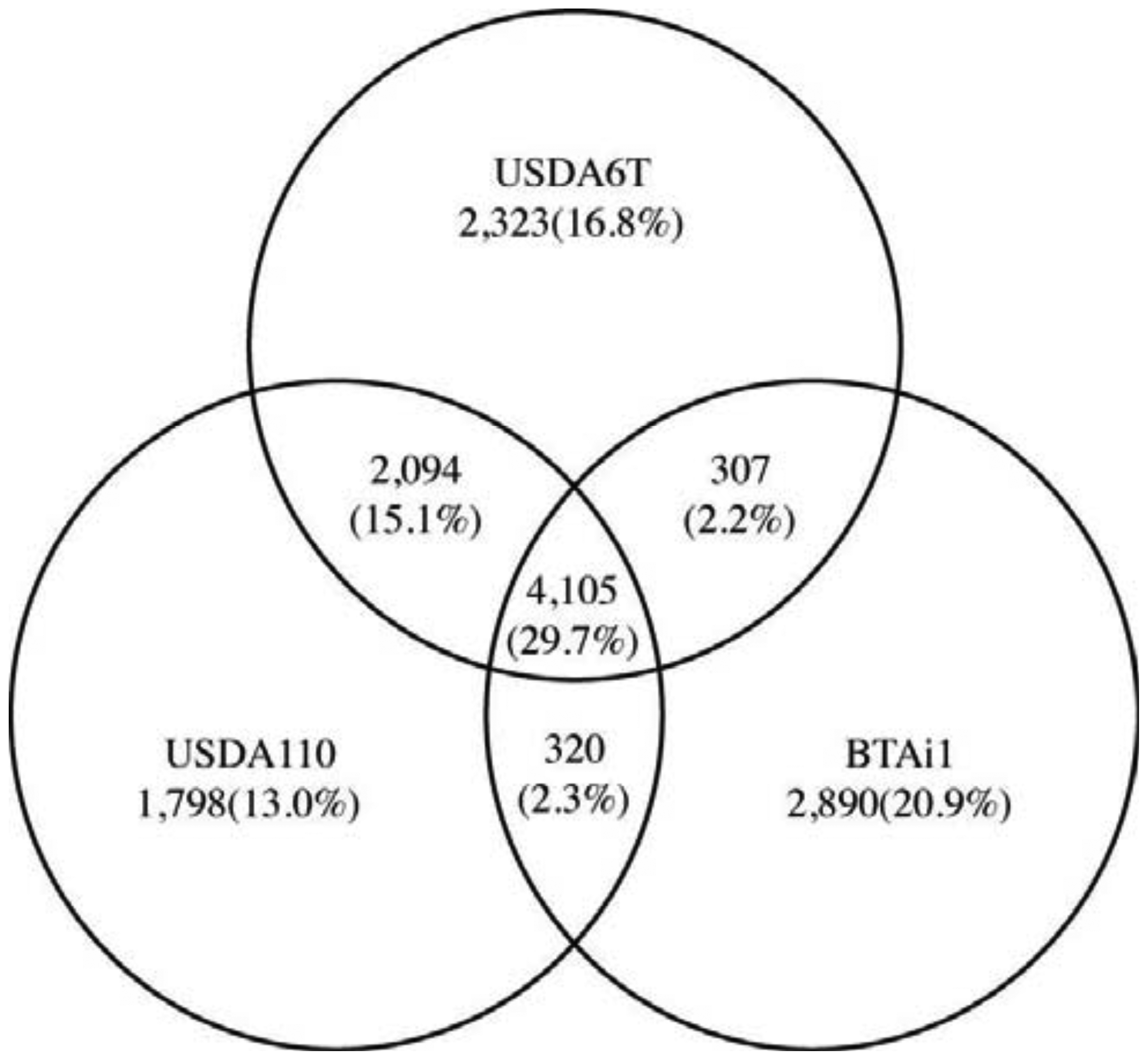
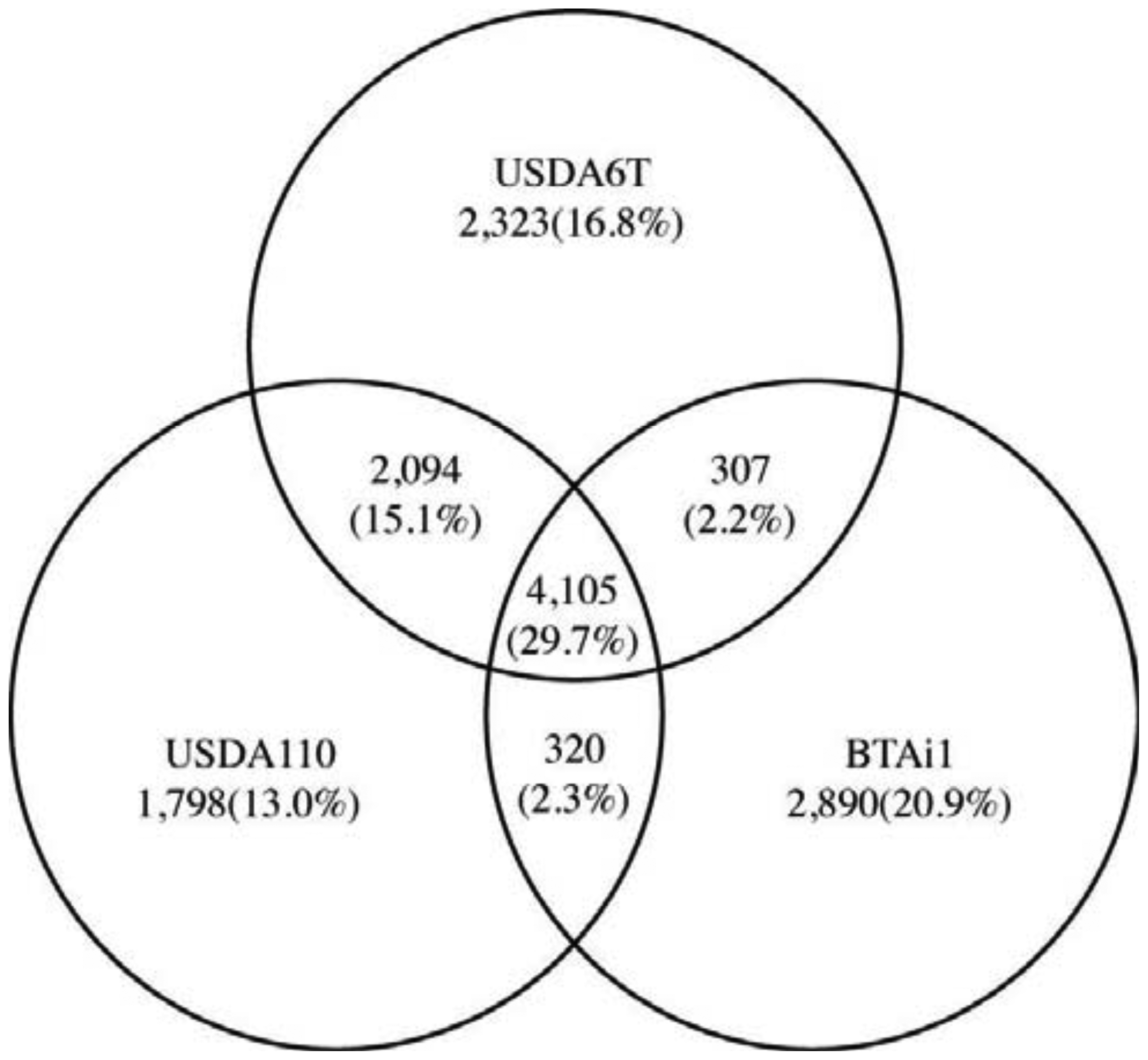
We subsequently performed comparative analysis by comparing genetic data from Bradyrhizobium sp. BTAi1 with that from the two B. japonicum strains in order to identify the genetic characteristics that are commonly shared by two B. japonicum strains. Bradyrhizobium sp. BTAi1 possesses the ability to form nodules on stems of Aeschynomene spp., and to perform nitrogen fixation in nodules, but BTAi1 has neither symbiosis islands nor symbiotic plasmids [25]. A comparison of the gene constitutions of these strains revealed 4,105 genes commonly conserved in all three strains. This is 29.7% of the combined non-redundant set of genes (13,837) (Figure 5). This group is anticipated to contain the essential genes that are indispensable to support bacterial cellular life, in addition to genes involved in nitrogen fixation and interactions with the host. The number of genes conserved between USDA6T and USDA110 is 2,094 (15.1%). This is significantly larger than 307 (2.2%) between USDA6T and BTAi1 and 320 (2.3%) between USDA110 and BTAi1. These may indicate that many of genes conserved between USDA6T and USDA110 can become the significant candidates in charge of functions involved in soybean symbiosis.
Conserved genes related to nitrogen fixation are present in all three strains. The nif/fix gene clusters, nifDKENX-nifST-nifB-nifZ-nifHQW-fixBCX and fixR-nifA-fixA, are positioned at two loci in the symbiosis islands of both USDA110 (blr1743-bsr1775 and blr2036-blr2038) and USDA6T (BJ6T80440-80760 and BJ6T78160-78180), with identical gene arrangements. Although the BTAi1 genome possesses no symbiosis island, similar arrangements of the nif/fix gene clusters (BBta_5869-5923 and BBta_5933-5935) are conserved in the genome, except for fixA (BBta_5872) flanking fixB (BBta_5871) in the first cluster, which is separated from the second cluster which included fixR (BBta_5935) and nifA (BBta_5933). Another gene cluster of fix genes (fixK2, fixLJ, fixNOPQ, fixGHIS), which is essential for microoxic respiration, is positioned outside of the symbiosis island in both USDA110 and USDA6T. This cluster is also conserved among all three strains, USDA110 (bll2757-bsr2770), USDA6T (BJ6T70170-70040), and BTAi1 (BBta_2786-2798).
Approximately half of the genes assigned to the symbiosis island were classified into the orthologous group, whose members are conserved between USDA6T and USDA110 (data not shown). The nod genes, involved in nodulation, and the rhc genes, which encode the type III secretion system, were classified into this group. The nod genes nolZY-nolA-nodD2-nodD1YABCSUIJ-nolMNO-nodZ are conserved as a single cluster in each strain (bsl2015-blr2035 in USDA110 and BJ6T78190-BJ6T78390 in USDA6T), and the rhc genes (ttsI-rhcC2, nopB-rhcJ-nolUV-rhcNQRSTU, rhcC1-nopLC, rhcV; bll1800-bll1843 in USDA110 and BJ6T80240-BJ6T79820 in USDA6T) were also positioned at a single locus within the symbiosis island in each strain (Figure 4a) [26].
B. japonicum possesses the capacity to infect plant cells, suggesting that the carbohydrate configuration of the cell surface is important for association with the host during infection [27]. The B. japonicum cell is studded with cyclic beta-glucan, lipopolysaccharide (LPS), and exopolysaccharide (EPS) as major extracellular polysaccharides. Some of the genes responsible for synthesis of these polysaccharides and for their secretion have been identified in B. japonicum USDA110. The genes for synthesis of cyclic beta-glucan and LPS are conserved in all three of the strains, including Bradyrhizobium sp. BTAi1, and are found in a similar arrangement in each gene cluster. However, most of the genes involved in EPS synthesis were classified into the group of genes that are conserved between USDA6T and USDA110 but are not found in Bradyrhizobium sp. BTAi1.
Cyclic beta-glucans are localized within the periplasmic compartment, or are excreted into the extracellular medium of the cultures. The cell surface cyclic beta-glucans are recognized to provide functions related to the B. japonicum infection process [28]. Three genes, ndvB (blr4614), ndvD (blr4613), and ndvC (blr4612), have been identified as functional genes for biosynthesis of cyclic beta-glucans in USDA110. These genes show significant similarity to the predicted genes, ndvB (BJ6T50730), ndvD (BJ6T50740), and ndvC (BJ6T50750), suggesting a general metabolic pathway for cyclic beta-glucans in USDA6T that is assumed to be similar to that in USDA110.
Lipopolysaccharides (LPSs) are glycolipid molecules, which universally constitute the major component of the outer membrane of proteobacteria. Two LPS biosynthesis-related genes, lspL homolog (bll5920) and galE (blr5931), were identified in one locus on the USDA110 genome [29]. At this locus, there is a gene cluster that included thirteen genes (bll5919-blr5931) with predicted functions in sugar modification. The gene arrangement in the cluster is completely conserved in USDA6T (BJ6T37870-BJ6T37740).
Exopolysaccharides (EPSs) are bacterial homopolymers composed of a single type of sugar or heteropolysaccharides containing different types of sugars. Rhizobial EPSs are synthesized through a series of sequential steps. Two loci including a cluster of genes involved in EPS synthesis were found in USDA110 [29]. The first cluster (bll7571-blr7578) was identified as exoUM-metA-exoPTB. In the USDA6T genome, BJ6T17210-BJ6T17140 at coordinates 1,796,656–1,804,352 contain the counterparts to this exo gene cluster. The second cluster in the USDA110 genome consists of ten genes (blr2374-blr2383), including lspL (UDP-glucuronic acid epimerase) and ugdH (UDP-glucose 6-dehydrogenase). In the USDA6T genome, the region harboring BJ6T74640 to BJ6T74510 corresponds to this gene cluster, except for two genes (BJ6T74530 and BJ6T74540), which are inserted between lspL (BJ6T74520) and ugdH (BJ6T74550). BJ6T74530 and BJ6T74540 are homologs of the nodulation genes nolK (GDP-fucose synthetase) and noeL (GDP-mannose 4,6-dehydratase), respectively [30,31]. It is not clear at the moment why the noeL-nolK genes are inserted within this cluster in the USDA6T genome, and how these gene products contribute to EPS biosynthesis. Further analysis of these points within the specific portion of the gene cluster remains to be performed.
Two reaction systems, denitrification in free-living cells and the uptake of hydrogen produced during nitrogen fixation, have been investigated in B. japonicum [32,33]. The genes encoding key functional enzymes in each system form clusters with other, including functionally related genes, nosRZDFYLX (bll0314-blr0320) for denitrification and hupNCUVSLCDFGHIJK, hypABFCDE, and hoxXA (bll6949-bll6927) for the uptake hydrogenase have been identified on the USDA110 genome. However, some B. japonicum strains, including USDA6T, do not possess genes involved in these functions [15]. The availability of genomic information for USDA6T allows us to compare it with the USDA110 genome and characterize its gene constitution, thus providing a glimpse into the source of differences in gene distribution between the two strains. Comparative analysis of these genes showed that nosRZDFYLX (bll0314-blr0320) was a unique gene set in USDA110. No functional component of the nos gene cluster was found in USDA6T, and thus USDA6T would be expected to lack nitrous oxidase activity. Denitrification in B. japonicum also depends on the nitrate reductase (napEDABC: bsr7036-blr7040), nitrite reductase (nirK: blr7089) and nitric oxide reductase (norCBQD: blr3214-blr3217) [34], however these USDA6T orthologs were found (BJ6T23740-BJ6T23700, BJ6T23260 and BJ6T65370-BJ6T65340, respectively). Notably, the coding region for the nos gene cluster (bll0314-blr0320) in USDA110 flanks the trnS-GGA gene at coordinates 333,518–333,607, while the flanking region of trnS-GGA (BJ6T02870) in USDA6T suggested a genomic island at 262–288 kb (59.5% GC) as described in “2.2.7 Genomic islands and symbiosis island”. This suggests that the nos gene cluster is part of an unknown GI in USDA110 and that it was possibly inserted in the USDA110 genome during the course of evolution.
A complete set of hup-hyp-hox cluster genes, hupNCUVSLCDFGHIJK, hypABFCDE and hoxXA (bll6949-bll6927), was found at coordinates 7,624,531–7,649,197 in the USDA110 genome [5]. This indicates that this strain has the functional role of hydrogenase uptake [35]. Since this gene cluster is found inside the genomic island trnM1-element at coordinates 7,619,541–7,701,499, it is possible that high nitrogen fixation by USDA110 might have been acquired through the insertion of a GI into its genome. On the other hand, the hup-hyp-hox cluster is missing from USDA6T. In USDA6T, the trnM-CAT gene (BJ6T24130), corresponding to the one targeted by the trnM1-element of USDA110, was detected. However, the tRNA gene (BJ6T24130) is flanked by a region of lower GC content (61.1% GC) at coordinates 2,488–2,544 kb whose sequence is not coincident with ones of the trnM1-element. The existence of such tRNA gene (BJ6T24130) in USDA6T may show that it has the potential to gain the ability to take up the hydrogen and to improve efficiency of nitrogen fixation.
USDA110 possess a trb gene cluster, trbBCDEJLFGI (bll8279-bll8288). The trb genes are related to conjugal transfer of mobilizable genetic elements, and this cluster bore a marked similarity to the virB gene cluster, which comprises the Type IV secretion system (T4SS) [36]. Notably, the trbLFGI genes showed similarity to virB6-B10 genes, which encode proteins that comprise the channel for the Type IV secretion system [37]. On the other hand, trbCDE genes have similarity to virB2-B4 genes as well. virB2 encodes pilus components, VirB3 is an assembly factor for T pilus formation, and virB4 encodes an ATPase [37]. In the USDA110 genome, trbBCDEJLFGI (bll8279-bll8288) was found at coordinates 9,068,090–9,077,553. This locus is partly comprised of a region conserved between USDA110 and USDA6T (Figure 4b). This region corresponds to the “putative symbiosis island C” described in section “2.2.7 Genomic islands and symbiosis island”. However, this gene cluster could not be identified in the USDA6T genome. Presumably, it would have been deleted from the corresponding region in the USDA6T genome. The effect of a lack of trb genes is uncertain. If the trb genes are functionally related to T4SS, their deletion in USDA6T might cause differences in host range between USDA6T and USDA110.
3. Experimental Section
3.1. Bacterial Strain and Genomic Libraries
Bradyrhizobium japonicum strain USDA6T and its cultivation have been described in Itakura et al. [15]. The entire genome of USDA6T was sequenced using Sanger-ABI3730 and 454/Roche GS-FLX machines. Total cellular DNA for 454 sequence analysis was purified according to the instruction manual for QIAGEN Genomic-tip (QIAGEN, Hilden, Germany). After the DNA was sheared by nebulization, a genomic library containing inserts of 0.55 kb in average length was constructed to perform pyrosequencing. For genome sequencing using the Sanger method, two random genomic libraries were constructed from the total cellular DNA of USDA6T. Megabase genomic DNA embedded in agarose plugs was used for construction of BAC and cosmid libraries as previously mentioned [38,39]. The BAC library (Bj006b) contained inserts of 82 kb in average length that were cloned into a BAC vector, pCC1BAC (Epicentre Bio., Madison, WI, USA), and the cosmid library (Bj006c) contained inserts of 30 kb in average length that were cloned into the cosmid vector pKS800 [39].
3.2. Pulsed-Field Gel Electrophoresis
The DNA fragments were separated by pulsed-field gel electrophoresis (PFGE). Preparation of intact genomic DNA and restriction enzyme digestion of DNA were performed based on the protocol reported previously [40]. In brief, the DNA was digested with two restriction endonucleases, PmeI and SwaI (NEB, Ipswich, MA, USA). The resulting fragments were resolved using the CHEF Mapper gel electrophoresis system (BioRad, Richmond, CA, USA) with 0.5× TBE buffer and 1% Pulsed field agarose (BioRad) at 14 °C in a temperature controlled cooling unit. To facilitate separation of fragments of 50–1,000 kb in size, contour-clamped homogeneous electric field (CHEF) electrophoresis was conducted using a pulse time of 40–60 seconds, field strength 6 V/cm, and run time of 24 h. Two stepwise CHEF conditions were applied to resolve the larger fragments as follows. The first step utilized a pulse time of 1,200–1,469 seconds, field strength of 2 V/cm, and runtime of 72 h. The second step utilized a pulse time of 79–146 seconds, field strength of 6 V/cm, and runtime of 11 h. Saccharomyces cerevisiae chromosomes, Hansenula wingei chromosomes (BioRad), and MidRange I PFG Marker (NEB) were used as size standards for the PFGE analyses.
3.3. DNA Sequencing
The whole-genome shotgun method was applied to genome sequencing using sequence data obtained by the use of a 454/Roche GS-FLX machine, in combination with BAC end-sequencing. Pyrosequencing for the production of the shotgun single reads was performed using one-half of the picotiter plate in the 454/Roche GS-FLX system, resulting in 28.7-fold (651,020 reads) sequence coverage of the genome. The sequences at both ends of the clones from the Bj006b and Bj006c libraries were analyzed using a Dye-terminator Cycle Sequencing Kit and a 3730XL Sequencer (Applied Biosystems, Foster City, CA, USA). The end-sequence data from the BAC clones facilitated the gap-closure process and provided the scaffolding for reconstruction of the sequence of the entire genome [5].
The accumulated sequence files were assembled in two steps. A total of 651,020 sequence files were processed using a MIRA3 assembler [41]. The contigs resulting from the initial assembly were read out as consensus sequences in FASTA format. The contig sequences were assembled together with the end-sequences from 3,072 BAC and 1,968 cosmid clones using Phred/Phrap software [42]. GSFLX-derived data and Sanger-derived reads for USDA6T were imported into Consed [42], edited, and verified. We filled the remaining gaps in the sequence by primer walking, using the cosmids or the BAC clones as templates. The integrity of the reconstructed genome sequence was assessed by chromosome walking using the end sequences of the BAC clones.
3.4. Gene Assignment, Annotation, and Information Analyses
Regions encoding rRNA genes (16S, 23S, and 5S) were identified by similarity searches against data deposited into an in-house database, RhizoBase [43]. tRNA genes were predicted using the tRNA scan-SE 1.23 program [44].
Prediction of protein-coding regions was carried out using a combination of the MetaGeneAnnotator program [18] and similarity searches against predicted genes from the USDA110 genome. All protein-coding regions 120 bp or longer were translated into amino acid sequences. The putative protein-encoding genes used either ATG, GTG, or TTG as their start codons. All of the predicted genes for structural RNAs and proteins were denoted following their ordering on the genome by a serial number with the prefix “BJ6T”, representing the strain USDA6T. The putative protein-encoding genes were subjected to subsequent similarity searches against the protein databases. In order to assign functions to the protein-encoding genes, they were annotated based on the best hit information from sequence similarity searches against a gene set derived from the USDA110 genome database.
Comparison between two genomic nucleotide sequences, USDA6T and USDA110, was performed using GenomeMatcher v.1.6 [45]. GC-skew analysis was performed as described by Lobry [46]. To examine the similarity of the nucleotide sequences of the USDA6T genes or genome to those of the USDA110 strain, similarity searches was conducted using the BLASTN program [47].
In order to characterize the protein gene products encoded by USDA6T, similarity searches using the BLASTP program were performed against databases including all protein gene products from the USDA110 genome or the Bradyrhizobium sp. BTAi1 genome, and against the NCBI non-redundant protein sequences (nr) database [48]. A cutoff E-value of 10−4 was considered significant. Predicted protein-encoding genes from USDA6T and USDA110 were classified into the categories of COGs according to the results of BLASTP searches against the COG reference data set [19]. Functional motifs of the predicted gene products in the genomes of USDA6T and USDA110 were assigned using the HMMSCAN program with domain searches against the Pfam 25.0 database [49,50].
Orthologous genes among the three strains, USDA6T, USDA110, and Bradyrhizobium sp. BTAi1, were predicted by reciprocal BLAST best hit with E-value cutoff of 10−4. Orthologous relationships were depicted in a Venn diagram. The genes of five bacterial strains investigated, B. japonicum USDA6T, USDA110, Bradyrhizobium sp. BTAi1, Bradyrhizobium sp. ORS278, and M. loti MAFF303099, were classified into clusters by single linkage clustering using the BLASTCLUST program using as criteria, 60% or more amino acid identity and the alignment extended over at least 0.7 times the length of the query sequence. Phylogenetic patterns of the orthologous genes were inferred for the amino acid sequences using the neighbor-joining algorithm and the CLUSTALW program [51]. Phylogenetic trees were constructed with MEGA 5 software [52].
4. Conclusions
The genome of B. japonicum USDA6T was sequenced and the genomic features of two strains, USDA6T and USDA110 were compared. The analysis revealed typical genomic features of the type strain of B. japonicum, and integration of a symbiosis island similar to one of USDA110 into the USDA6T genome. The present results suggest that a larger symbiosis island, approximately 860 kb in size, was retained in an ancestral genome. The progenitor symbiosis island was split into three regions by unknown large-scale genome rearrangements through comparable mechanisms after independently integrating into both genomes. The genome information for USDA6T, along with that for USDA110, provides new insights into the genetic diversity of the genus Bradyrhizobium, which could be used to further study the evolutionary relationships among soybean symbionts as well as their individual characteristics.
Acknowledgments
This work was supported by Kyoto Sangyo University Research Grants, by the Kazusa DNA Research Institute foundation, and by Grants-in-Aid for Scientific Research (B) 21310130 and for Challenging Exploratory Research 23658057 from the Ministry of Education, Science, Sports, and Culture of Japan.
Supplementary Files
References
- van Berkum, P.; Eardly, B.D. Molecular Evolutionary Systematics of the Rhizobiaceae. In The Rhizobiaceae; Spaink, H.P., Kondorosi, A., Hooykaas, P.J.J., Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1998; pp. 1–24. [Google Scholar]
- Kuykendall, L.D. Genus I. Bradyrhizobium Jordan 1982, 137vp. In Bergey's Manual of Systematic Bacteriology, 2nd ed.; Garrity, G.M., Brenner, D.J., Krieg, N.R., Staley, J.T., Eds.; Springer: New York, NY, USA, 2005; Volume 2, pp. 438–443. [Google Scholar]
- USDA ARS National Rhizobium Germplasm Resource Center home page. Available online: http://bldg6.arsusda.gov/pberkum/Public/page5/cc5a.html#japonicum/ (accessed on 10 October 2011).
- Keyser, H.H.; Griffin, R.F. Beltsville Rhizobium Culture Collection Catalog; USDA-ARS: Beltsville, MD, USA, 1987. [Google Scholar]
- Kaneko, T.; Nakamura, Y.; Sato, S.; Minamisawa, K.; Uchiumi, T.; Sasamoto, S.; Watanabe, A.; Idesawa, K.; Iriguchi, M.; Kawashima, K.; et al. Complete genomic sequence of nitrogen-fixing symbiotic bacterium Bradyrhizobium japonicum USDA110. DNA Res. 2002, 9, 189–197. [Google Scholar]
- Sullivan, J.T.; Ronson, C.W. Evolution of rhizobia by acquisition of a 500-kb symbiosis island that integrates into a phe-tRNA gene. Proc. Natl. Acad. Sci. USA 1998, 95, 5145–5149. [Google Scholar]
- MacLean, A.M.; Finan, T.M.; Sadowsky, M.J. Genomes of the symbiotic nitrogen-fixing bacteria of legumes. Plant Physiol. 2007, 144, 615–622. [Google Scholar]
- Kaneko, T.; Nakamura, Y.; Sato, S.; Asamizu, E.; Kato, T.; Sasamoto, S.; Watanabe, A.; Idesawa, K.; Ishikawa, A.; Kawashima, K.; et al. Complete genome structure of the nitrogen-fixing symbiotic bacterium Mesorhizobium loti. DNA Res. 2000, 7, 331–338. [Google Scholar]
- Sullivan, J.T.; Trzebiatowski, J.R.; Cruickshank, R.W.; Gouzy, J.; Brown, S.D.; Elliot, R.M.; Fleetwood, D.J.; McCallum, N.G.; Rossbach, U.; Stuart, G.S.; et al. Comparative sequence analysis of the symbiosis island of Mesorhizobium loti strain R7A. J. Bacteriol. 2002, 184, 3086–3095. [Google Scholar]
- Lee, K.B.; de Backer, P.; Aono, T.; Liu, C.T.; Suzuki, S.; Suzuki, T.; Kaneko, T.; Yamada, M.; Tabata, S.; Kupfer, D.M.; et al. The genome of the versatile nitrogen fixer Azorhizobium caulinodans ORS571. BMC Genomics 2008, 9, 271. [Google Scholar]
- Wei, M.; Takeshima, K.; Yokoyama, T.; Minamisawa, K.; Mitsui, H.; Itakura, M.; Kaneko, T.; Tabata, S.; Saeki, K.; Omori, H.; et al. Temperature-dependent expression of type III secretion system genes and its regulation in Bradyrhizobium japonicum. Mol. Plant Microbe Interact. 2010, 23, 628–637. [Google Scholar]
- Wei, M.; Yokoyama, T.; Minamisawa, K.; Mitsui, H.; Itakura, M.; Kaneko, T.; Tabata, S.; Saeki, K.; Omori, H.; Tajima, S.; et al. Soybean seed extracts preferentially express genomic loci of Bradyrhizobium japonicum in the initial interaction with soybean, Glycine max (L.). Merr DNA Res. 2008, 15, 201–214. [Google Scholar]
- Chang, W.S.; Franck, W.L.; Cytryn, E.; Jeong, S.; Joshi, T.; Emerich, D.W.; Sadowsky, M.J.; Xu, D.; Stacey, G. An oligonucleotide microarray resource for transcriptional profiling of Bradyrhizobium japonicum. Mol. Plant Microbe Interact. 2007, 20, 1298–1307. [Google Scholar]
- Hauser, F.; Lindemann, A.; Vuilleumier, S.; Patrignani, A.; Schlapbach, R.; Fischer, H.M.; Hennecke, H. Design and validation of a partial-genome microarray for transcriptional profiling of the Bradyrhizobium japonicum symbiotic gene region. Mol. Genet. Genomics 2006, 275, 55–67. [Google Scholar]
- Itakura, M.; Saeki, K.; Omori, H.; Yokoyama, T.; Kaneko, T.; Tabata, S.; Ohwada, T.; Tajima, S.; Uchiumi, T.; Honnma, K.; et al. Genomic comparison of Bradyrhizobium japonicum strains with different symbiotic nitrogen-fixing capabilities and other Bradyrhizobiaceae members. ISME J. 2009, 3, 326–339. [Google Scholar]
- Huyghe, A.; Francois, P.; Schrenzel, J. Characterization of microbial pathogens by DNA microarrays. Infect. Genet. Evol. 2009, 9, 987–995. [Google Scholar]
- Larimer, F.W.; Chain, P.; Hauser, L.; Lamerdin, J.; Malfatti, S.; Do, L.; Land, M.L.; Pelletier, D.A.; Beatty, J.T.; Lang, A.S.; et al. Complete genome sequence of the metabolically versatile photosynthetic bacterium Rhodopseudomonas palustris. Nat. Biotechnol. 2004, 22, 55–61. [Google Scholar]
- Noguchi, H.; Taniguchi, T.; Itoh, T. MetaGeneAnnotator: Detecting species-specific patterns of ribosomal binding site for precise gene prediction in anonymous prokaryotic and phage genomes. DNA Res. 2008, 15, 387–396. [Google Scholar]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG database: An updated version includes eukaryotes. BMC Bioinformatics 2003, 4, 41. [Google Scholar]
- Molouba, F.; Lorquin, J.; Willems, A.; Hoste, B.; Giraud, E.; Dreyfus, B.; Gillis, M.; de Lajudie, P.; Masson-Boivin, C. Photosynthetic bradyrhizobia from Aeschynomene spp. are specific to stem-nodulated species and form a separate 16S ribosomal DNA restriction fragment length polymorphism group. Appl. Environ. Microbiol. 1999, 65, 3084–3094. [Google Scholar]
- Lobry, J.R. Asymmetric substitution patterns in the two DNA strands of bacteria. Mol. Biol. Evol. 1996, 13, 660–665. [Google Scholar]
- Blakely, G.W.; Davidson, A.O.; Sherratt, D.J. Sequential strand exchange by XerC and XerD during site-specific recombination at dif. J. Biol. Chem. 2000, 275, 9930–9936. [Google Scholar]
- Chandler, M.; Mahillon, J. Insertion sequences revisited. In Mobile DNA II; Craig, N.L., Craigie, R., Gellert, M., Lambowitz, A., Eds.; American Society for Microbiology: Washington, DC, USA, 2002; pp. 305–366. [Google Scholar]
- Juhas, M.; van der Meer, J.R.; Gaillard, M.; Harding, R.M.; Hood, D.W.; Crook, D.W. Genomic islands: Tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol. Rev. 2009, 33, 376–393. [Google Scholar]
- Giraud, E.; Moulin, L.; Vallenet, D.; Barbe, V.; Cytryn, E.; Avarre, J.C.; Jaubert, M.; Simon, D.; Cartieaux, F.; Prin, Y.; et al. Legumes symbioses: Absence of Nod genes in photosynthetic bradyrhizobia. Science 2007, 316, 1307–1312. [Google Scholar]
- Göttfert, M.; Röthlisberger, S.; Kündig, C.; Beck, C.; Marty, R.; Hennecke, H. Potential symbiosis specific genes uncovered by sequencing a 410-kilobase DNA region of the Bradyrhizobium japonicum chromosome. J. Bacteriol. 2001, 183, 1405–1412. [Google Scholar]
- Puvanesarajah, V.; Schell, F.M.; Gerhold, D.; Stacey, G. Cell surface polysaccharides from Bradyrhizobium japonicum and a nonnodulating mutant. J. Bacteriol. 1987, 169, 137–141. [Google Scholar]
- Bhagwat, A.A.; Mithöfer, A.; Pfeffer, P.E.; Kraus, C.; Spickers, N.; Hotchkiss, A.; Ebel, J.; Keister, D.L. Further studies of the role of cyclic beta-glucans in symbiosis. An NdvC mutant of Bradyrhizobium japonicum synthesizes cyclodecakis-(1->3)-beta-glucosyl. Plant Physiol. 1999, 119, 1057–1064. [Google Scholar]
- Quelas, J.I.; Mongiardini, E.J.; Casabuono, A.; López-García, S.L.; Althabegoiti, M.J.; Covelli, J.M.; Pérez-Giménez, J.; Couto, A.; Lodeiro, A.R. Lack of galactose or galacturonic acid in Bradyrhizobium japonicum USDA 110 exopolysaccharide leads to different symbiotic responses in soybean. Mol. Plant Microbe Interact. 2010, 23, 1592–1604. [Google Scholar]
- Mergaert, P.; van Montagu, M.; Holsters, M. The nodulation gene nolK of Azorhizobium caulinodans is involved in the formation of GDP-fucose from GDP-mannose. FEBS Lett. 1997, 409, 312–316. [Google Scholar]
- Lamrabet, Y.; Bellogín, R.A.; Cubo, T.; Espuny, R.; Gil, A.; Krishnan, H.B.; Megias, M.; Ollero, F.J.; Pueppke, S.G.; Ruiz-Sainz, J.E.; et al. Mutation in GDP-fucose synthesis genes of Sinorhizobium fredii alters Nod factors and significantly decreases competitiveness to nodulate soybeans. Mol. Plant Microbe Interact. 1999, 12, 207–217. [Google Scholar]
- Velasco, L.; Mesa, S.; Xu, C.A.; Delgado, M.J.; Bedmar, E.J. Molecular characterization of nosRZDFYLX genes coding for denitrifying nitrous oxide reductase of Bradyrhizobium japonicum. Antonie Van Leeuwenhoek 2004, 85, 229–235. [Google Scholar]
- Baginsky, C.; Brito, B.; Imperial, J.; Palacios, J.M.; Ruiz-Argüeso, T. Diversity and evolution of hydrogenase systems in rhizobia. Appl. Environ. Microbiol. 2002, 68, 4915–4924. [Google Scholar]
- Bedmar, E.J.; Robles, E.F.; Delgado, M.J. The complete denitrification pathway of the symbiotic, nitrogen-fixing bacterium Bradyrhizobium japonicum. Biochem. Soc. Trans. 2005, 33, 141–144. [Google Scholar]
- Minamisawa, K. Comparison of extracellular polysaccharide composition, rhizobitoxine production, and hydrogenase phenotype among various strains of Bradyrhizobium japonicum. Plant Cell Physiol. 1989, 30, 877–884. [Google Scholar]
- Ding, H.; Hynes, M.F. Plasmid transfer systems in the rhizobia. Can. J. Microbiol. 2009, 55, 917–927. [Google Scholar]
- Cascales, E.; Christie, P.J. The versatile bacterial type IV secretion systems. Nat. Rev. Microbiol. 2003, 1, 137–149. [Google Scholar]
- Sato, S.; Nakamura, Y.; Kaneko, T.; Asamizu, E.; Kato, T.; Nakao, M.; Sasamoto, S.; Watanabe, A.; Ono, A.; Kawashima, K.; et al. Genome structure of the legume, Lotus japonicus. DNA Res. 2008, 15, 227–239. [Google Scholar]
- Hattori, Y.; Omori, H.; Hanyu, M.; Kaseda, N.; Mishima, E.; Kaneko, T.; Tabata, S.; Saeki, K. Ordered cosmid library of the Mesorhizobium loti MAFF303099 genome for systematic gene disruption and complementation analysis. Plant Cell Physiol. 2002, 43, 1542–1557. [Google Scholar]
- Birren, B.; Lai, E. Pulsed Field Gel Electrophoresis: A Practical Guide; Academic Press: San diego, CA, USA, 1993; pp. 25–106. [Google Scholar]
- Chevreux, B.; Wetter, T.; Suhai, S. Genome sequence assembly using trace signals and additional sequence information. Computer Science and Biology: Proceedings of the German Conference on Bioinformatics, GCB'99, Hannover, Germany, 4–6 October 1999; pp. 45–56.
- Philip Green, Department of Genome Sciences, University of Washington, Seattle, USA: Phred, Phrap, Consed. Available online http://www.phrap.org/phredphrapconsed.html/ (accessed on 10 October 2011).
- RhizoBase: The genome database for rhizobia. Available online: http://genome.kazusa.or.jp/rhizobase/ (accessed on 10 October 2011).
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar]
- Ohtsubo, Y.; Ikeda-Ohtsubo, W.; Nagata, Y.; Tsuda, M. GenomeMatcher: A graphical user interface for DNA sequence comparison. BMC Bioinformatics 2008, 9, 376. [Google Scholar]
- Lobry, J.R. Asymmetric substitution patterns in the two DNA strands of bacteria. Mol. Biol. Evol. 1996, 13, 660–665. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI reference sequences (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007, 35, D61–D65. [Google Scholar]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar]
- Finn, R.D.; Tate, J.; Mistry, J.; Coggill, P.C.; Sammut, S.J.; Hotz, H.R.; Ceric, G.; Forslund, K.; Eddy, S.R.; Sonnhammer, E.L.; et al. The Pfam protein families database. Nucleic Acids Res. 2008, 36, D281–D288. [Google Scholar]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011. [Google Scholar] [CrossRef]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kaneko, T.; Maita, H.; Hirakawa, H.; Uchiike, N.; Minamisawa, K.; Watanabe, A.; Sato, S. Complete Genome Sequence of the Soybean Symbiont Bradyrhizobium japonicum Strain USDA6T. Genes 2011, 2, 763-787. https://doi.org/10.3390/genes2040763
Kaneko T, Maita H, Hirakawa H, Uchiike N, Minamisawa K, Watanabe A, Sato S. Complete Genome Sequence of the Soybean Symbiont Bradyrhizobium japonicum Strain USDA6T. Genes. 2011; 2(4):763-787. https://doi.org/10.3390/genes2040763
Chicago/Turabian StyleKaneko, Takakazu, Hiroko Maita, Hideki Hirakawa, Nobukazu Uchiike, Kiwamu Minamisawa, Akiko Watanabe, and Shusei Sato. 2011. "Complete Genome Sequence of the Soybean Symbiont Bradyrhizobium japonicum Strain USDA6T" Genes 2, no. 4: 763-787. https://doi.org/10.3390/genes2040763
APA StyleKaneko, T., Maita, H., Hirakawa, H., Uchiike, N., Minamisawa, K., Watanabe, A., & Sato, S. (2011). Complete Genome Sequence of the Soybean Symbiont Bradyrhizobium japonicum Strain USDA6T. Genes, 2(4), 763-787. https://doi.org/10.3390/genes2040763