
Genotypic and Phenotypic Characterization of Axonal Charcot–Marie–Tooth Disease in Childhood: Identification of One Novel and Four Known Mutations

 , , , and
, , , and
Abstract
1. Introduction
2. Methods
2.1. Study Design, Setting, and Ethical Approval
2.2. Patient Characterization
- Neurological findings—muscle strength by Medical Research Council (MRC) scale, deep-tendon reflexes (DTRs), sensory deficits, presence of camptodactyly or hyperlaxity.
- Orthopedic features—type of foot deformity (pes cavus, rocker-bottom, pes calcaneovalgus), genu recurvatum, pectus excavatum, scoliosis.
- Systemic/laboratory data—serum creatine kinase (CK), liver enzymes, lactate, and abdominal ultrasonography or echocardiography when clinically indicated.
2.3. Neuroimaging
2.4. Electrophysiological Evaluation
2.5. Genetic Testing and Bioinformatic Analysis
- Whole-Exome Sequencing (WES)—applied to three sporadic cases. Library preparation employed the Twist Human Core Exome Kit (Twist Bioscience, South San Francisco, CA, USA); paired-end (2 × 150 bp) sequencing was performed on an Illumina NovaSeq 6000 platform (Illumina, San Diego, CA, USA), achieving a mean on-target depth > 100×. Reads were aligned to GRCh38 with BWA-MEM; variant calling used the GATK v4 pipeline.
- Targeted CMT Gene Panel—applied to the two siblings (Cases 4a/4b). A 72-gene hereditary neuropathy panel (TruSight™ Inherited Neuropathy, Illumina) was sequenced on an Illumina MiSeq.
3. Results
3.1. Case 1
3.2. Case 2
3.3. Case 3
3.4. Case 4a
3.5. Case 4b
4. Discussion
Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shy, M.E. Peripheral neuropathies. In Merrit’s Neurology, 12th ed.; Rowland, L.P., Ed.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2005; pp. 733–747. [Google Scholar]
- Dyck, P.J. Neuronal atrophy and degeneration predominantly affecting peripheral sensory and autonomic neurons. In Peripheral Neuropathy; Dyck, P.J., Thomas, P.K., Lambert, E.H., Bunge, R., Eds.; WB Saunders: Philadelphia, PA, USA, 1984; Volume 2. [Google Scholar]
- Pareyson, D.; Saveri, P.; Pisciotta, C. New developments in Charcot-Marie-Tooth neuropathy and related diseases. Curr. Opin. Neurol. 2017, 30, 471–480. [Google Scholar] [CrossRef] [PubMed]
- VarSome. VarSome The Human Genomics Community. Available online: https://varsome.com/variant/hg38/NEFH%20c.2434_2436del?annotation-mode=germline (accessed on 27 December 2022).
- Laurà, M.; Milani, M.; Morbin, M.; Moggio, M.; Ripolone, M.; Jann, S.; Scaioli, V.; Taroni, F.; Pareyson, D. Rapid progression of late-onset axonal Charcot-Marie-Tooth disease associated with a novel MPZ mutation in the extracellular domain. J. Neurol. Neurosurg. Psychiatry 2007, 78, 1263–1266. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.K.; Choi, B.-O. Analyzing clinical and genetic aspects of axonal Charcot–Marie-Tooth disease. J. Genet. Med. 2021, 18, 83–93. [Google Scholar] [CrossRef]
- Ikenberg, E.; Reilich, P.; Abicht, A.; Heller, C.; Schoser, B.; Walter, M.C. Charcot-Marie-Tooth disease type 2CC due to a frameshift mutation of the neurofilament heavy polypeptide gene in an Austrian family. Neuromuscul. Disord. 2019, 29, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Rebelo, A.P.; Abrams, A.J.; Cottenie, E.; Horga, A.; Gonzalez, M.; Bis, D.M.; Sanchez, M.A.; Pinto, M.; Buglo, E.; Markel, K.; et al. Cryptic amyloidogenic elements in the 3-prime UTRs of neurofilament genes trigger axonal neuropathy. Am. J. Hum. Genet. 2016, 98, 597–614. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. A Study of ACTX-401 for the Treatment of IGHMBP2-Related Disorders. Available online: https://clinicaltrials.gov/study/NCT05152823 (accessed on 1 March 2025).
- Mandarakas, M.R.; Menezes, M.P.; Rose, K.J.; Shy, R.; Eichinger, K.; Foscan, M.; Estilow, T.; Kennedy, R.; Herbert, K.; Bray, P.; et al. Development and validation of the Charcot-Marie-Tooth Disease Infant Scale. Brain 2018, 141, 3319–3330. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.D.; Huang, L.; Tetreault, M.; Majewski, J.; Boycott, K.M.; Bulman, D.E. Autosomal recessive axonal polyneuropathy in a sibling pair due to a novel homozygous mutation in IGHMBP2. Neuromuscul. Disord. 2015, 25, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, X.; Hu, Z.; Mao, X.; Zi, X.; Xia, K.; Tang, B.; Zhang, R. IGHMBP2-related clinical and genetic features in a cohort of Chinese Charcot-Marie Tooth disease type 2 patients. Neuromuscul. Disord. 2017, 27, 193–199. [Google Scholar] [CrossRef] [PubMed]
- OMIM. Carcot-Marie-Tooth Disease Caused Due to DYNCH1 Mutation Can Have Variable Phenotype. Available online: https://www.omim.org/entry/614228 (accessed on 27 December 2022).
- Tekin, H.G.; Edem, P.; Özyılmaz, B. Spinal muscular atrophy with predominant lower extremity (SMA-LED) with no signs other than pure motor symptoms at the intersection of multiple overlap syndrome. Brain Dev. 2022, 44, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.H.S.; van Alfen, N.; Thuestad, I.J.; Ip, J.; Chan, A.O.; Mak, C.; Chung, B.H.; Verrips, A.; Kamsteeg, E.J. A recurrent de novo DYNC1H1 tail domain mutation causes spinal muscular atrophy with lower extremity predominance, learning difficulties and mild brain abnormality. Neuromuscul. Disord. 2018, 28, 750–756. [Google Scholar] [CrossRef] [PubMed]
- Amabile, S.; Jeffries, L.; McGrath, J.M.; Ji, W.; Spencer-Manzon, M.; Zhang, H.; Lakhani, S.A. DYNC1H1-related disorders: A description of four new unrelated patients and a comprehensive review of previously reported variants. Am. J. Med. Genet. Part A 2020, 182, 2049–2057. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.J.; Lyu, G.Z.; Zhang, V.W.; Jin, H. Missense mutation in DYNC1H1 gene caused psychomotor developmental delay and muscle weakness: A case report. World J. Clin. Cases 2021, 9, 9302–9309. [Google Scholar] [CrossRef] [PubMed]
- Baumann, M.; Schreiber, H.; Schlotter-Weigel, B.; Loscher, W.; Stucka, R.; Karall, D.; Strom, T.M.; Bauer, P.; Krabichler, B.; Fauth, C.; et al. MPV17 mutations in juvenile- and adult-onset axonal sensorimotor polyneuropathy. Clin. Genet. 2019, 95, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Spinazzola, A.; Viscomi, C.; Fernandez-Vizarra, E.; Carrara, F.; D’Adamo, P.; Calvo, S.; Marsano, R.M.; Donnini, C.; Weiher, H.; Strisciuglio, P.; et al. MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nat. Genet. 2006, 38, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Karadimas, C.L.; Vu, T.H.; Holve, S.A. Navajo neurohepatopathy is caused by a mutation in the MPV17 gene. Am. J. Hum. Genet. 2006, 79, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Blakely, E.L.; Butterworth, A.; Hadden, R.D.M.; Bodi, I.; He, L.; McFarland, R.; Taylor, R.W. MPV17 mutation causes neuropathy and leukoencephalopathy with multiple mtDNA deletions in muscle. Neuromusc. Disord. 2012, 22, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.R.; Hong, Y.B.; Jung, S.C.; Lee, J.H.; Kim, Y.J.; Park, H.J.; Lee, J.; Koo, H.; Lee, J.S.; Jwa, D.H.; et al. A novel homozygous MPV17 mutation in two families with axonal sensorimotor polyneuropathy. BMC Neurol. 2015, 15, 179. [Google Scholar] [CrossRef] [PubMed]
- OMIM. Carcot-Marie-Tooth Disease Caused Due to MPV17 Mutation Can Have Variable Phenotype. Available online: https://www.omim.org/entry/618400 (accessed on 27 December 2022).
- National Center for Biotechnology Information. ClinVar; Variation ID: 214660. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/214660/ (accessed on 16 July 2025).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | 1 | 2 | 3 | 4a | 4b |
|---|---|---|---|---|---|
| Nerve conduction study | Sensorimotor polyneuropathy consistent with axonal degeneration | Severe sensorimotor polyneuropathy with axonal degeneration and demyelination | Normal | Severe sensorimotor polyneuropathy with axonal degeneration and demyelination | Severe sensorimotor polyneuropathy with axonal degeneration and demyelination |
| MRI | Brain: Normal Spine: Normal | Brain: Normal Spine: Normal | Brain: Abnormal Spine: Normal | Brain: Abnormal Spine: Normal (Figure 4a) | Brain: Abnormal Spine: Normal (Figure 4b) |
| Genetic test | NEFH (NM_021076.4) | IGHMBP2 (NM_002180.3) | DYNC1H1 (NM_001376.5) | MPV17 (NM_002437.5) | |
| Clinical Phenotype | CMT2CC | CMT2S | CMT2O | CMT2EE | |
| Protein change | p.K812del | p.G857Afs*27 | p.K671E | p.R41W | |
| ACMG Classification | VUS (PM4, PM2) | LP (PVS1, PM2) | VUS (PM2, PP2) | LP (PM2, PM5, PP3, PP2, PP5) | |
| HGMD Variant | c.2434_2436del; p.(Lys812del) | c.2568_2569del; p.(G857Afs*27) | c.2011A > G; p.(K671E) | c.121C > T; p.(R41W) | |
| Zygosity | Heterozygous | Homozygous | Heterozygous | Homozygous | |
| Case | 1 | 2 | 3 | 4a | 4b |
|---|---|---|---|---|---|
| Age at diagnosis (years) | 159/12 | 99/12 | 14 | 1011/12 | 88/12 |
| Onset age | At birth | 12 months old | At birth | 7 years old | 8 years 6 months old |
| Gender | Female | Female | Male | Male | Female |
| Parental consanguinity | First-degree cousins | First-degree cousins | Third-degree cousins | First-degree cousins | First-degree cousins |
| Cognitive function | Normal | Normal | Normal | Moderate intellectual disability | Mild intellectual disability |
| High-arched palate | (+) | (−) | (−) | (−) | (−) |
| Muscle strength | Upper extremity distal: 4/5 (mild) Lower extremity distal: 4/5 (mild) | Upper extremity distal: 4/5 (mild) Lower extremity distal: 4/5 (mild) | Upper extremity distal: 4/5 (mild) Lower extremity distal: 4/5 (mild) | Upper extremity distal: 4/5 Lower extremity distal: 3–4/5 (mild-moderate) | Upper extremity distal: 4/5 (mild) Lower extremity distal: 4/5 (mild) |
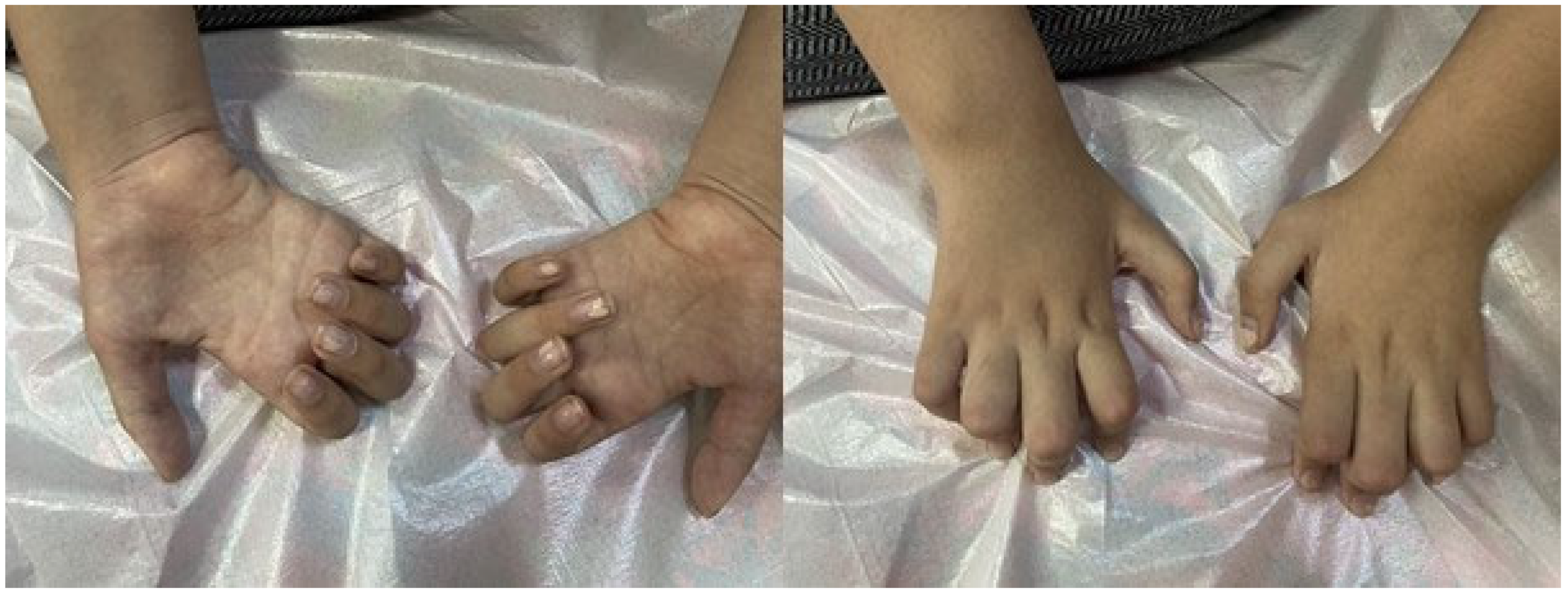
| Muscle atrophy | Lower extremity distal, upper distal, and thenar and hypothenar (Figure 1) | Lower extremity distal, thenar, and hypothenar | Lower extremity distal | Lower extremity distal, thenar, and hypothenar atrophy | Lower extremity distal |
| DTR | Absent | Absent | Absent | Absent | Absent |
| Hyperlaxity | (+) | (+) | (−) | (+) | (−) |
| Camptodactyly | (+) | (+) | (−) | (+) | (−) |
| Foot deformity | Pes cavus, rocker bottom feet | Pes cavus deformity, rocker bottom feet (Figure 2) | Pes calcaneovalgus deformity, rocker bottom feet (Figure 3) | Pes cavus deformity, contractures in the ankle | Pes cavus deformity, contractures in the ankle |
| Others | Hypoesthesia Pectus excavatum and scoliosis | Small hands and feet, weak grip Genu recurvatum prominent (Figure 2) | Bifid uvula (Figure 3) | Moderate liver enzyme elevation Nasal speech | Moderate liver enzyme elevation Exhibiting fluctuating |
| ALT: 66 (5–40 U/L) AST: 46 (5–40 U/L) NT-ProBNP: 13 pg/mL Lactate: 3.2 (0.7–2 mmol/L) Urine ketone: +2 Slightly coarse granular appearance in the liver parenchyma on abdominal USG ECHO: Normal ECG: Normal | ALT: 53 (5–40 U/L) AST: 79 (5–40 U/L) NT-ProBNP: 15 pg/mL Lactate: NA Urine ketone: +3 ECHO: Bicuspid aorta, ascending aortic dilatation ECG: Normal | ||||
| CK (29–200 U/L) | 315 U/L | 568 U/L | 632 U/L | 54 U/L | 33 U/L |
| R Sural Nerve | R Tibial Nerve | R Peroneal Nerve | ||||
|---|---|---|---|---|---|---|
| SNAP (µV) (Reference ≥ 4) | SCV (m/s) (Reference ≥ 39) | CMAP (mV) (Reference ≥ 3) | MCV (m/s) (Reference ≥ 39) | CMAP (mV) (Reference ≥ 2.5) | MCV (m/s) (Reference ≥ 39) | |
| Case 1 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Case 2 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Case 3 | 30.7 | 40.3 | 4.3 | 42.9 | 3.8 | 43.8 |
| Case 4a | 2.2 | 36.3 | 0.0 | 0.0 | 0.0 | 0.0 |
| Case 4b | 1.8 | 34.8 | 0.0 | 0.0 | 0.0 | 0.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
İpek, R.; Çavdartepe, B.E.; Bozdoğan, S.T.; Altunışık, E.; Akalın, A.; Yaman, M.; Akın, A.; Kumandaş, S. Genotypic and Phenotypic Characterization of Axonal Charcot–Marie–Tooth Disease in Childhood: Identification of One Novel and Four Known Mutations. Genes 2025, 16, 917. https://doi.org/10.3390/genes16080917
İpek R, Çavdartepe BE, Bozdoğan ST, Altunışık E, Akalın A, Yaman M, Akın A, Kumandaş S. Genotypic and Phenotypic Characterization of Axonal Charcot–Marie–Tooth Disease in Childhood: Identification of One Novel and Four Known Mutations. Genes. 2025; 16(8):917. https://doi.org/10.3390/genes16080917
Chicago/Turabian Styleİpek, Rojan, Büşra Eser Çavdartepe, Sevcan Tuğ Bozdoğan, Erman Altunışık, Akçahan Akalın, Mahmut Yaman, Alper Akın, and Sefer Kumandaş. 2025. "Genotypic and Phenotypic Characterization of Axonal Charcot–Marie–Tooth Disease in Childhood: Identification of One Novel and Four Known Mutations" Genes 16, no. 8: 917. https://doi.org/10.3390/genes16080917
APA Styleİpek, R., Çavdartepe, B. E., Bozdoğan, S. T., Altunışık, E., Akalın, A., Yaman, M., Akın, A., & Kumandaş, S. (2025). Genotypic and Phenotypic Characterization of Axonal Charcot–Marie–Tooth Disease in Childhood: Identification of One Novel and Four Known Mutations. Genes, 16(8), 917. https://doi.org/10.3390/genes16080917