
GPR143-Associated Ocular Albinism in a Hispanic Family and Review of the Literature

 , , , , , , ,
, , , , , , ,
Abstract
1. Introduction
2. Materials and Methods
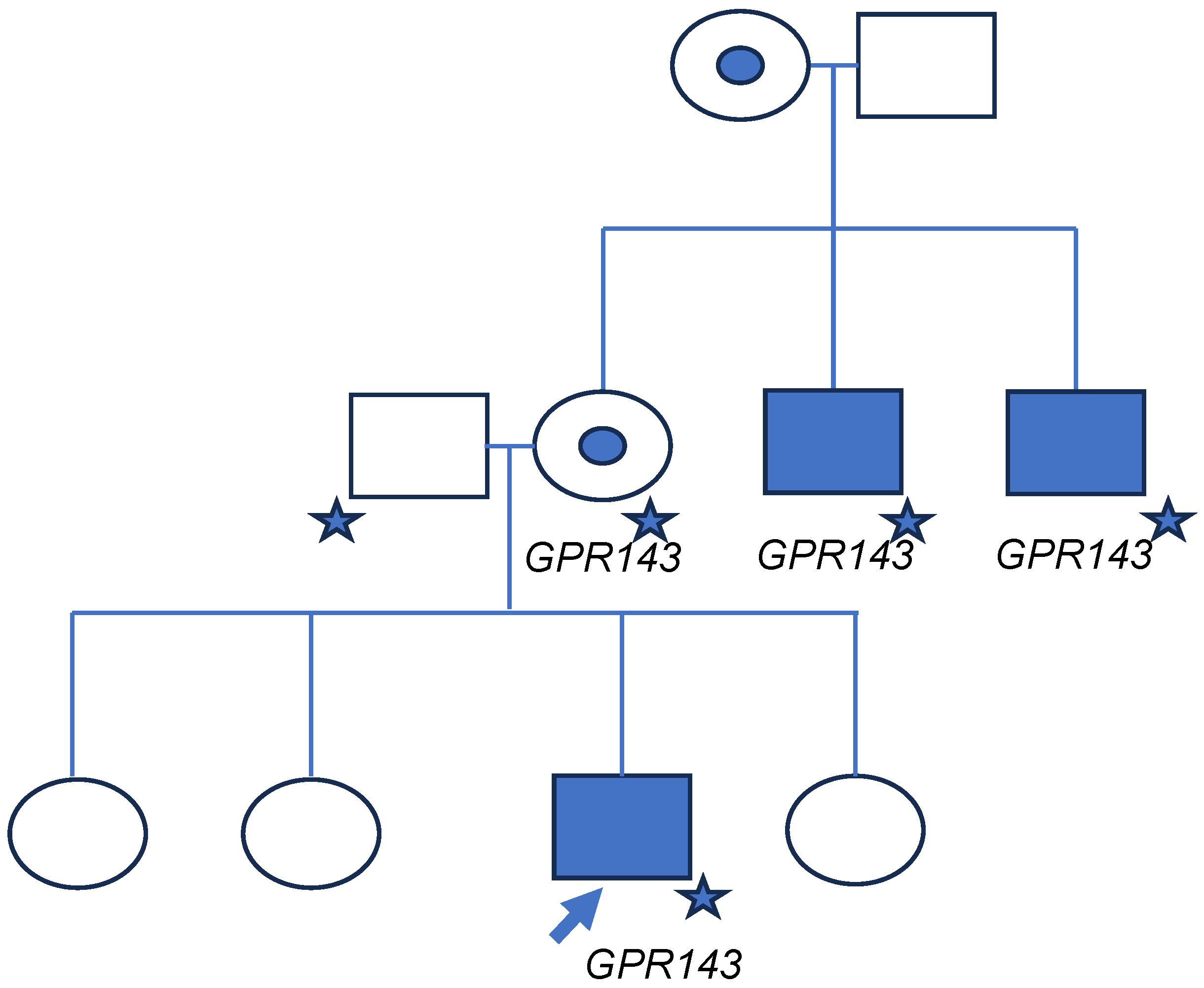
3. Results
Literature Review
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| OA | Ocular Albinism |
| OD | Right eye |
| OS | Left eye |
| WES | Whole exome sequencing |
| WGS | Whole genome sequencing |
References
- Tsang, S.H.; Sharma, T. X-linked Ocular Albinism. Adv. Exp. Med. Biol. 2018, 1085, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Trebušak Podkrajšek, K.; Stirn Kranjc, B.; Hovnik, T.; Kovač, J.; Battelino, T. GPR143 gene mutation analysis in pediatric patients with albinism. Ophthalmic Genet. 2012, 33, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Galli, J.; Loi, E.; Morandi, A.; Scaglioni, V.; Rossi, A.; Molinaro, A.; Pasini, N.; Semeraro, F.; Ruberto, G.; Fazzi, E. Neurodevelopmental Profile in Children Affected by Ocular Albinism. Neuropediatrics 2022, 53, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; You, B.; Xu, K.; Zhang, X.; Xie, Y.; Li, Y. GPR143 genotypic and ocular phenotypic characterisation in a Chinese cohort with ocular albinism. Ophthalmic Genet. 2021, 42, 717–724. [Google Scholar] [CrossRef] [PubMed]
- De Silva, S.R.; Arno, G.; Robson, A.G.; Fakin, A.; Pontikos, N.; Mohamed, M.D.; Bird, A.C.; Moore, A.T.; Michaelides, M.; Webster, A.R.; et al. The X-linked retinopathies: Physiological insights, pathogenic mechanisms, phenotypic features and novel therapies. Prog. Retin. Eye Res. 2021, 82, 100898. [Google Scholar] [CrossRef] [PubMed]
- Nanda, T.; Coombs, A.; Shi, A.; Baumal, C.R. Macular Findings in Carriers of Ocular Albinism With a Novel GPR143 Mutation. Ophthalmic Surg. Lasers Imaging Retin. 2022, 53, 460–463. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Walker, L.C.; Hoya, M.; Wiggins, G.A.R.; Lindy, A.; Vincent, L.M.; Parsons, M.T.; Canson, D.M.; Bis-Brewer, D.; Cass, A.; Tchourbanov, A.; et al. Using the ACMG/AMP framework to capture evidence related to predicted and observed impact on splicing: Recommendations from the ClinGen SVI Splicing Subgroup. Am. J. Hum. Genet. 2023, 110, 1046–1067. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pan, Q.; Yi, C.; Xu, T.; Liu, J.; Jing, X.; Hu, B.; Wang, Y. A novel mutation, c.494C>A (p.Ala165Asp), in the GPR143 gene causes a mild phenotype in a Chinese X-linked ocular albinism patient. Acta Ophthalmol. 2016, 94, 417–418. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.S.; Bohnsack, B.L.; Ing, A.; Drackley, A.; Castelluccio, V.; Zhang, K.X.; Ralay-Ranaivo, H.; Rossen, J.L. Diagnostic Yield of Genetic Testing for Ocular and Oculocutaneous Albinism in a Diverse United States Pediatric Population. Genes 2023, 14, 135. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xiao, X.; Zhang, Q. Iris hyperpigmentation in a Chinese family with ocular albinism and the GPR143 mutation. Am. J. Med. Genet. A 2009, 149, 1786–1788. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Liao, X.; Cai, S.P.; Lan, C.; Wang, Y.; Zhou, X.; Yin, Y.; Yu, W.; Liu, X. A novel nonsense mutation of the GPR143 gene identified in a Chinese pedigree with ocular albinism. PLoS ONE 2012, 7, e43177. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mao, X.; Chen, M.; Yu, Y.; Liu, Q.; Yuan, S.; Fan, W. Identification of a novel GPR143 mutation in a large Chinese family with isolated foveal hypoplasia. BMC Ophthalmol. 2021, 21, 156. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cai, X.B.; Wu, K.C.; Zhang, X.; Lv, J.N.; Jin, G.H.; Xiang, L.; Chen, J.; Huang, X.F.; Pan, D.; Lu, B.; et al. Whole-exome sequencing identified ARL2 as a novel candidate gene for MRCS (microcornea, rod-cone dystrophy, cataract, and posterior staphyloma) syndrome. Clin. Genet. 2019, 96, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.W.; Schiff, E.R.; Tailor, V.K.; Malka, S.; Neveu, M.M.; Theodorou, M.; Moosajee, M. Prospective Study of the Phenotypic and Mutational Spectrum of Ocular Albinism and Oculocutaneous Albinism. Genes 2021, 12, 508. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, J.; Jia, Y.; Wang, L.; Bu, J. A previously unidentified deletion in G protein-coupled receptor 143 causing X-linked congenital nystagmus in a Chinese family. Indian J. Ophthalmol. 2016, 64, 813–817. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, J.Y.; Ren, X.; Yang, X.; Guo, T.; Yao, Q.; Li, L.; Dai, X.; Zhang, M.; Wang, L.; Liu, M.; et al. Identification of a novel GPR143 mutation in a large Chinese family with congenital nystagmus as the most prominent and consistent manifestation. J. Hum. Genet. 2007, 52, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Micale, L.; Augello, B.; Fusco, C.; Turturo, M.G.; Granatiero, M.; Piemontese, M.R.; Zelante, L.; Cecconi, A.; Merla, G. GPR143 mutational analysis in two Italian families with X-linked ocular albinism. Genet. Test. Mol. Biomarkers 2009, 13, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Wang, Z.; Zhang, J.; Hu, L.; Kong, X. Identification of a novel GPR143 deletion in a Chinese family with X-linked congenital nystagmus. Mol. Vis. 2008, 14, 1015–1019. [Google Scholar] [PubMed] [PubMed Central]
- Jia, X.; Yuan, J.; Jia, X.; Ling, S.; Li, S.; Guo, X. GPR143 mutations in Chinese patients with ocular albinism type 1. Mol. Med. Rep. 2017, 15, 3069–3075. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gao, X.; Liu, T.; Cheng, X.; Dai, A.; Liu, W.; Li, R.; Zhang, M. A novel GPR143 mutation in a Chinese family with X-linked ocular albinism type 1. Mol. Med. Rep. 2020, 21, 240–248. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fang, S.; Guo, X.; Jia, X.; Xiao, X.; Li, S.; Zhang, Q. Novel GPR143 mutations and clinical characteristics in six Chinese families with X-linked ocular albinism. Mol. Vis. 2008, 14, 1974–1982. [Google Scholar] [PubMed] [PubMed Central]
- Peng, Y.; Meng, Y.; Wang, Z.; Qin, M.; Li, X.; Dian, Y.; Huang, S. A novel GPR143 duplication mutation in a Chinese family with X-linked congenital nystagmus. Mol. Vis. 2009, 15, 810–814. [Google Scholar] [PubMed] [PubMed Central]
- Sepúlveda-Vázquez, H.E.; Villanueva-Mendoza, C.; Zenteno, J.C.; Villegas-Ruiz, V.; Pelcastre-Luna, E.; García-Aguirre, G. Macular optical coherence tomography findings and GPR143 mutations in patients with ocular albinism. Int. Ophthalmol. 2014, 34, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Naruto, T.; Okamoto, N.; Masuda, K.; Endo, T.; Hatsukawa, Y.; Kohmoto, T.; Imoto, I. Deep intronic GPR143 mutation in a Japanese family with ocular albinism. Sci. Rep. 2015, 5, 11334. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Han, R.; Wang, X.; Wang, D.; Wang, L.; Yuan, Z.; Ying, M.; Li, N. GPR143 Gene Mutations in Five Chinese Families with X-linked Congenital Nystagmus. Sci. Rep. 2015, 5, 12031. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zou, X.; Li, H.; Yang, L.; Sun, Z.; Yuan, Z.; Li, H.; Sui, R. Molecular genetic and clinical evaluation of three Chinese families with X-linked ocular albinism. Sci. Rep. 2017, 7, 33713. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, Y.; Guo, X.; Wei, A.; Zhu, W.; Li, W.; Lian, S. Identification of a novel mutation in a Chinese family with X-linked ocular albinism. Eur. J. Ophthalmol. 2009, 19, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Liang, D.; Xue, J.; Liu, J.; Wu, L. A novel GPR143 splicing mutation in a Chinese family with X-linked congenital nystagmus. Mol. Vis. 2011, 17, 715–722. [Google Scholar] [PubMed] [PubMed Central]
- Schiaffino, M.V.; Bassi, M.T.; Galli, L.; Renieri, A.; Bruttini, M.; De Nigris, F.; Bergen, A.A.; Charles, S.J.; Yates, J.R.; Meindl, A.; et al. Analysis of the OA1 gene reveals mutations in only one-third of patients with X-linked ocular albinism. Hum. Mol. Genet. 1995, 4, 2319–2325. [Google Scholar] [CrossRef] [PubMed]
- Camand, O.; Boutboul, S.; Arbogast, L.; Roche, O.; Sternberg, C.; Sutherland, J.; Levin, A.; Héon, E.; Menasche, M.; Dufier, J.; et al. Mutational analysis of the OA1 gene in ocular albinism. Ophthalmic Genet. 2003, 24, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Schnur, R.E.; Gao, M.; Wick, P.A.; Keller, M.; Benke, P.J.; Edwards, M.J.; Grix, A.W.; Hockey, A.; Jung, J.H.; Kidd, K.K.; et al. OA1 mutations and deletions in X-linked ocular albinism. Am. J. Hum. Genet. 1998, 62, 800–809. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- d’Addio, M.; Pizzigoni, A.; Bassi, M.T.; Baschirotto, C.; Valetti, C.; Incerti, B.; Clementi, M.; De Luca, M.; Ballabio, A.; Schiaffino, M.V. Defective intracellular transport and processing of OA1 is a major cause of ocular albinism type 1. Hum. Mol. Genet. 2000, 9, 3011–3018. [Google Scholar] [CrossRef] [PubMed]
- Oetting, W.S. New insights into ocular albinism type 1 (OA1): Mutations and polymorphisms of the OA1 gene. Hum. Mutat. 2002, 19, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Yang, L.; Li, H.; Huang, L.; Li, N. Evaluation of the iris thickness changes for the Chinese families with GPR143 gene mutations. Exp. Eye Res. 2019, 189, 107819. [Google Scholar] [CrossRef] [PubMed]
- Kim, U.S.; Cho, E.; Kim, H.J. A novel nonsense mutation of GPR143 gene in a Korean kindred with X-linked congenital nystagmus. Int. J. Ophthalmol. 2016, 9, 1367–1370. [Google Scholar] [CrossRef]
- Rim, J.H.; Lee, S.T.; Gee, H.Y.; Lee, B.J.; Choi, J.R.; Park, H.W.; Han, S.H.; Han, J. Accuracy of Next-Generation Sequencing for Molecular Diagnosis in Patients With Infantile Nystagmus Syndrome. JAMA Ophthalmol. 2017, 135, 1376–1385. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ohtsubo, M.; Sato, M.; Hikoya, A.; Hosono, K.; Minoshima, S.; Hotta, Y. Case of Japanese patient with x-linked ocular albinism associated with GPR143 gene mutation. Jpn. J. Ophthalmol. 2010, 54, 624–626. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.H.; Oh, E.H.; Shin, J.H.; Kim, H.S.; Choi, S.Y.; Choi, K.D.; Lee, C.; Choi, J.H. Identification of a novel GPR143 mutation in X-linked ocular albinism with marked intrafamilial phenotypic variability. J. Genet. 2018, 97, 1479–1484. [Google Scholar] [CrossRef] [PubMed]
- Bassi, M.T.; Bergen, A.A.; Bitoun, P.; Charles, S.J.; Clementi, M.; Gosselin, R.; Hurst, J.; Lewis, R.A.; Lorenz, B.; Meitinger, T.; et al. Diverse prevalence of large deletions within the OA1 gene in ocular albinism type 1 patients from Europe and North America. Hum. Genet. 2001, 108, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, T.; Schwartz, M. X-linked ocular albinism: Prevalence and mutations--a national study. Eur. J. Hum. Genet. 1998, 6, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, G.; Meindl, A.; Bechmann, M.; Schworm, H.D.; Achatz, H.; Boergen, K.P.; Kampik, A.; Berninger, T.; Meitinger, T. X-linked ocular albinism (Nettleship-Falls): A novel 29-bp deletion in exon 1. Carrier detection by ophthalmic examination and DNA analysis. Graefes Arch. Clin. Exp. Ophthalmol. 2001, 239, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Burns, W.N.; Schiaffino, M.V.; Lewis, R.A. Repeated transmission of X-linked ocular albinism type 1 by a carrier oocyte donor. Fertil. Steril. 1998, 70, 1169–1172. [Google Scholar] [CrossRef] [PubMed]
- Tijmes, N.T.; Bergen, A.B.; De Jong, P.T. Paucity of signs in X linked ocular albinism with a 700 kb deletion spanning the OA1 gene. Br. J. Ophthalmol. 1998, 82, 457–458. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lam, B.L.; Fingert, J.H.; Shutt, B.C.; Singleton, E.M.; Merin, L.M.; Brown, H.H.; Sheffield, V.C.; Stone, E.M. Clinical and molecular characterization of a family affected with X-linked ocular albinism (OA1). Ophthalmic Genet. 1997, 18, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, L.; Ferrero, G.B.; Grillo, A.; Bassi, M.T.; Roth, E.J.; Wapenaar, M.C.; van Ommen, G.J.; Mohandas, T.K.; Rocchi, M.; Zoghbi, H.Y.; et al. A high resolution deletion map of human chromosome Xp22. Nat. Genet. 1993, 4, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Montoya Delgado, M.J.; Astiazarán, M.C.; Casanova Imken, F.; Ramírez Estudillo, A.; Hernández Vázquez, Á. Ocular albinism with mutation in GPR143: Findings in wide-field autofluorescence and optical coherence tomography. Arch. Soc. Esp. Oftalmol. 2019, 94, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e524. [Google Scholar] [CrossRef] [PubMed]
- Vetrini, F.; Tammaro, R.; Bondanza, S.; Surace, E.M.; Auricchio, A.; De Luca, M.; Ballabio, A.; Marigo, V. Aberrant splicing in the ocular albinism type 1 gene (OA1/GPR143) is corrected in vitro by morpholino antisense oligonucleotides. Hum. Mutat. 2006, 27, 420–426. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Clinical Finding | Patients with Clinical Finding | Percentage of Patients with Clinical Finding |
|---|---|---|
| Reduced Visual Acuity | 149/149 | 100% |
| Visual Acuity (N = 95 patients) | 0.71 ± 0.23 [IQR 0.6, 0.9]. | |
| Nystagmus | 165/167 | 99% |
| Foveal Hypoplasia | 113/117 | 97% |
| Fundus Hypopigmentation | 91/115 | 79% |
| Iris Hypopigmentation | 92/130 | 71% |
| Variant | # of Literature Reports |
|---|---|
| c.251G>A, p.Gly84Asp | 3 |
| c.353G>A, p.Gly118Glu | 4 |
| c.455+3A>G | 3 |
| c.703G>A, p.Glu235Lys | 4 |
| c.733C>T, p.Arg245 * | 7 |
| Type of Variant | # of Variants | % of Total Variants (N = 127) |
|---|---|---|
| Missense | 39 | 31% |
| Large Deletion | 33 | 26% |
| Frameshift | 25 | 20% |
| Splicing | 20 | 16% |
| Nonsense | 9 | 7% |
| Synonymous | 1 | 1% |
| Missense | Large Deletion | Frameshift | Splicing | Nonsense | p-Value | |
|---|---|---|---|---|---|---|
| Reduced Visual Acuity | 100% (47 of 47 patients) | 100% (27 of 27 patients) | 100% (24 of 24 patients) | 100% (31 of 31 patients) | 100% (27 of 27 patients) | >0.9999 |
| Nystagmus | 97% (43 of 44 patients) | 100% (40 of 40 patients) | 100% (27 of 27 patients) | 100% (36 of 36 patients) | 100% (19 of 19 patients) | >0.9999 |
| Iris Hypopigmentation | 82% (29 of 35 patients) | 83% (19 of 23 patients) | 50% (11 of 22 patients) | 76% (25 of 33 patients) | 44% (8 of 18 patients) | 0.0063 |
| Fundus Hypopigmentation | 82% (19 of 23 patients) | 88% (15 of 17 patients) | 64% (14 of 22 patients) | 84% (27 of 32 patients) | 84% (21 of 25 patients) | 0.3228 |
| Foveal Hypoplasia | 95% (22 of 23 patients) | 100% (19 of 19 patients) | 95% (18 of 19 patients) | 100% (31 of 31 patients) | 96% (23 of 24 patients) | 0.5837 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aneja, A.; Bohnsack, B.L.; Allegretti, V.; Goetsch Weisman, A.; Drackley, A.; Ing, A.; McMullen, P.; Skol, A.; Ralay Ranaivo, H.; Yap, K.L.; et al. GPR143-Associated Ocular Albinism in a Hispanic Family and Review of the Literature. Genes 2025, 16, 911. https://doi.org/10.3390/genes16080911
Aneja A, Bohnsack BL, Allegretti V, Goetsch Weisman A, Drackley A, Ing A, McMullen P, Skol A, Ralay Ranaivo H, Yap KL, et al. GPR143-Associated Ocular Albinism in a Hispanic Family and Review of the Literature. Genes. 2025; 16(8):911. https://doi.org/10.3390/genes16080911
Chicago/Turabian StyleAneja, Anushree, Brenda L. Bohnsack, Valerie Allegretti, Allison Goetsch Weisman, Andy Drackley, Alexander Ing, Patrick McMullen, Andrew Skol, Hantamalala Ralay Ranaivo, Kai Lee Yap, and et al. 2025. "GPR143-Associated Ocular Albinism in a Hispanic Family and Review of the Literature" Genes 16, no. 8: 911. https://doi.org/10.3390/genes16080911
APA StyleAneja, A., Bohnsack, B. L., Allegretti, V., Goetsch Weisman, A., Drackley, A., Ing, A., McMullen, P., Skol, A., Ralay Ranaivo, H., Yap, K. L., Rathbun, P., Gordon, A., & Rossen, J. L. (2025). GPR143-Associated Ocular Albinism in a Hispanic Family and Review of the Literature. Genes, 16(8), 911. https://doi.org/10.3390/genes16080911