
FMR1 Methylation Pattern and Repeat Expansion Screening in a Cohort of Boys with Autism Spectrum Disorders: Correlation of Genetic Findings with Clinical Presentations

 , ,
, ,  , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Recruitment and Sample Collection
2.2. Genetic Investigations
2.2.1. Methylation-Specific Multiplex Ligation-Dependent Probe Amplification (MS-MLPA)
2.2.2. Triplet-Primed PCR (TP-PCR) and Melt Curve Analysis (MCA)
3. Results
3.1. Genetic Results
3.1.1. MS-MLPA
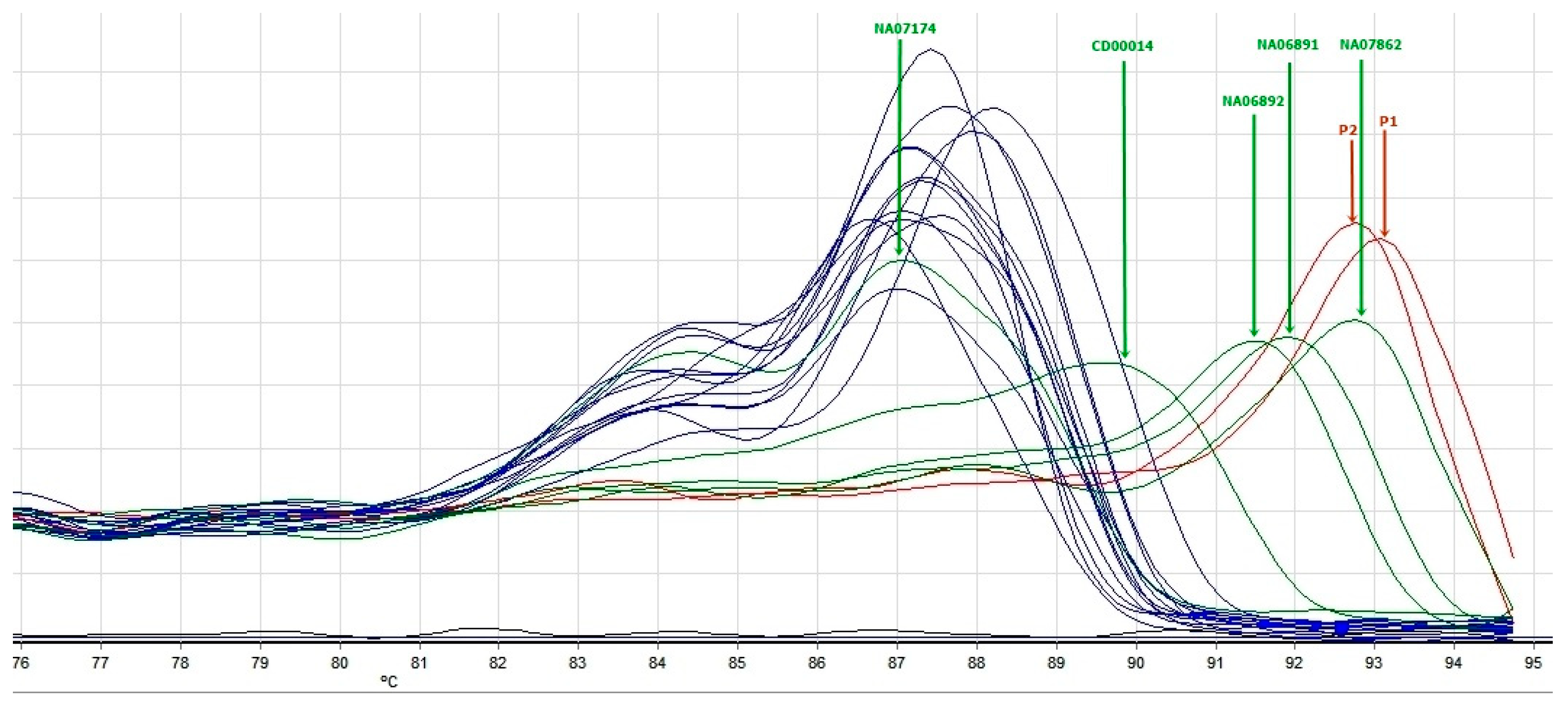
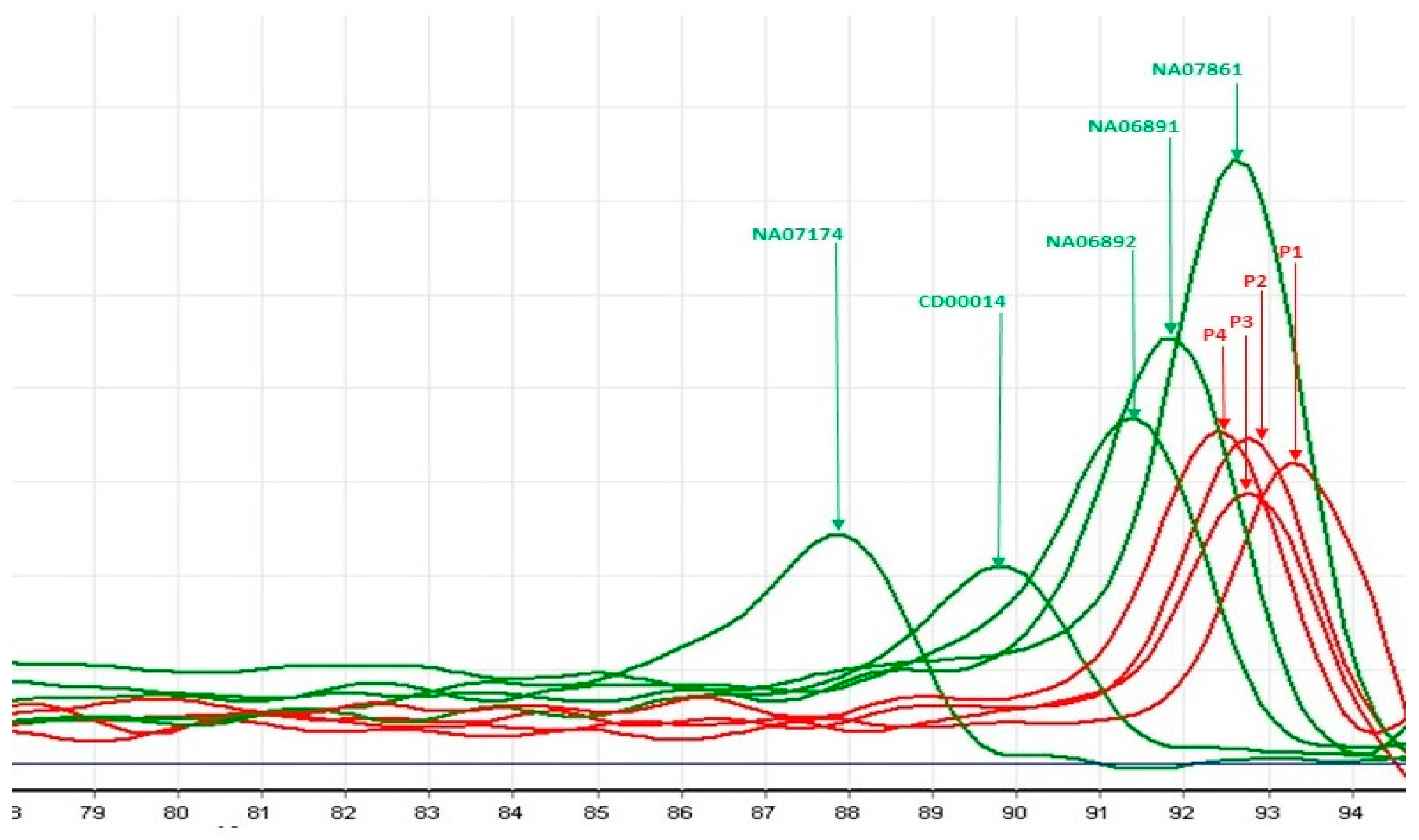
3.1.2. Triplet-Primed PCR
3.2. Clinical Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yakoucheva, L.M.; Muotri, A.R.; Sebat, J. Getting to the Cores of Autism. Cell 2019, 178, 1287–1298. [Google Scholar] [CrossRef]
- Rylaarsdam, L.; Guemez-Gamboa, A. Genetic Causes and Modifiers of Autism Spectrum Disorder. Front. Cell. Neurosci. 2019, 13, 385. [Google Scholar] [CrossRef]
- Crawford, H.; Scerif, G.; Wilde, L.; Beggs, A.; Stockton, J.; Sandhu, P.; Shelley, L.; Oliver, C.; McCleery, J. Genetic modifiers in rare disorders: The case of fragile X syndrome. Eur. J. Hum. Genet. 2021, 29, 173–183. [Google Scholar] [CrossRef]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Arlington, VA, USA, 2013. [Google Scholar] [CrossRef]
- Lord, C.; Brugha, T.S.; Charman, T.; Cusack, J.; Dumas, G.; Frazier, T.; Jones, E.J.H.; Jones, R.M.; Pickles, A.; State, M.W.; et al. Autism spectrum disorder. Nat. Rev. Dis. Primers 2020, 6, 5. [Google Scholar] [CrossRef]
- Online Mendelian Inherintance in Man. Available online: https://omim.org/ (accessed on 11 March 2025).
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.-H.; Kuhl, D.P.A.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Gallagher, A.; Hallahan, B. Fragile X-associated disorders: A clinical overview. J. Neurol. 2012, 259, 401–413. [Google Scholar] [CrossRef]
- Sherman, S.; Pletcher, B.A.; Driscoll, D.A. Fragile X syndrome: Diagnostic and carrier testing. Genet. Med. 2005, 7, 584–587. [Google Scholar] [CrossRef]
- Hagerman, R.; Berry-Kravis, E.; Hazlett, H.; Bailey, D.B., Jr.; Moine, H.; Kooy, R.F.; Tassone, F.; Gantois, I.; Sonenberg, N.; Mandel, J.L.; et al. Fragile X syndrome. Nat. Rev. Dis. Primers 2017, 3, 17065. [Google Scholar] [CrossRef]
- Rauch, A.; Hoyer, J.; Guth, S.; Zweier, C.; Kraus, C.; Becker, C.; Zenker, M.; Hüffmeier, U.; Thiel, C.; Rüschendorf, F.; et al. Diagnostic yield of various genetic approaches in patients with unexplained developmental delay or mental retardation. Am. J. Med. Genet. A 2006, 140, 2063–2074. [Google Scholar] [CrossRef] [PubMed]
- Devlin, B.; Scherer, S.W. Genetic architecture in autism spectrum disorder. Curr. Opin. Genet. Dev. 2012, 22, 229–237. [Google Scholar] [CrossRef]
- Jacobs, P.A.; Bullman, H.; Macpherson, J.; Youings, S.; Rooney, V.; Watson, A.; Dennis, N.R. Population studies of the fragile X: A molecular approach. J. Med. Genet. 1993, 30, 454–459. [Google Scholar] [CrossRef]
- Crawford, D.C.; Meadows, K.L.; Newman, J.L.; Taft, L.F.; Scott, E.; Leslie, M.; Shubek, L.; Holmgreen, P.; Yeargin-Allsopp, M.; Boyle, C.; et al. Prevalence of the fragile X syndrome in African-Americans. Am. J. Med. Genet. 2002, 110, 226–233. [Google Scholar] [CrossRef]
- Coffee, B.; Keith, K.; Albizua, I.; Malone, T.; Mowrey, J.; Sherman, S.L.; Warren, S.T. Incidence of fragile X syndrome by newborn screening for methylated FMR1 DNA. Am. J. Hum. Genet. 2009, 85, 503–514. [Google Scholar] [CrossRef]
- Malachowski, T.; Chandradoss, K.R.; Boya, R.; Zhou, L.; Cook, A.L.; Su, C.; Pham, K.; Haws, S.A.; Kim, J.H.; Ryu, H.S.; et al. Spatially coordinated heterochromatinization of long synaptic genes in fragile X syndrome. Cell 2023, 186, 5840–5858.e36. [Google Scholar] [CrossRef]
- Savatt, J.M.; Myers, S.M. Genetic Testing in Neurodevelopmental Disorders. Front. Pediatr. 2021, 9, 526779. [Google Scholar] [CrossRef]
- Kraan, C.M.; Godler, D.E.; Amor, D.J. Epigenetics of Fragile X Syndrome and Fragile X-related Disorders. Dev. Med. Child. Neurol. 2019, 61, 121–127. [Google Scholar] [CrossRef]
- Coffee, B.; Ikeda, M.; Budimirovic, D.B.; Hjelm, L.N.; Kaufmann, W.E.; Warren, S.T. Mosaic FMR1 deletion causes fragile X syndrome and can lead to molecular misdiagnosis: A case report and review of the literature. Am. J. Med. Genet. A 2008, 146, 1358–1367. [Google Scholar] [CrossRef] [PubMed]
- Myrick, L.; Nakamoto-Kinoshita, M.; Lindor, N.; Kirmani, S.; Cheng, X.; Warren, S.T. Fragile X syndrome due to a missense mutation. Eur. J. Hum. Genet. 2014, 22, 1185–1189. [Google Scholar] [CrossRef] [PubMed]
- Spector, E.; Behlmann, A.; Kronquist, K.; Rose, N.C.; Lyon, E.; Reddi, H.V. Laboratory testing for fragile X, 2021 revision: A technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021, 23, 799–812. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.J.; Hagerman, P.J.; Fragile, X. Syndrome: Lessons Learned and What New Treatment Avenues Are on the Horizon. Annu. Rev. Pharmacol. Toxicol. 2022, 62, 365–381. [Google Scholar] [CrossRef]
- World Health Organization. ICD-10: International Statistical Classification of Diseases and Related Health Problems 10th Revision. Available online: https://icd.who.int/ (accessed on 20 November 2019).
- Nygren, A.O.; Ameziane, N.; Duarte, H.M.; Vijzelaar, R.N.; Waisfisz, Q.; Hess, C.J.; Schouten, J.P.; Errami, A. Methylation-specific MLPA (MS-MLPA): Simultaneous detection of CpG methylation and copy number changes of up to 40 sequences. Nucleic Acids Res. 2005, 33, e128. [Google Scholar] [CrossRef]
- Nygren, A.O.; Lens, S.I.; Carvalho, R. Methylation-specific multiplex ligation-dependent probe amplification enables a rapid and reliable distinction between male FMR1 premutation and full-mutation alleles. J. Mol. Diagn. 2008, 10, 496–501. [Google Scholar] [CrossRef]
- Rajan-Babu, I.S.; Lian, M.; Tran, A.H.; Dang, T.T.; Le, H.T.-M.; Thanh, M.N.; Lee, C.G.; Chong, S.S. Defining the performance parameters of a rapid screening tool for FMR1 CGG-repeat expansions based on direct triplet-primed PCR and melt curve analysis. J. Mol. Diagn. 2016, 18, 719–730. [Google Scholar] [CrossRef]
- Tan, V.J.; Lian, M.; Faradz, S.M.H.; Winarni, T.I.; Chong, S.S. A Single Common Assay for Robust and Rapid Fragile X Mental Retardation Syndrome Screening From Dried Blood Spots. Front. Genet. 2018, 9, 582. [Google Scholar] [CrossRef]
- Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Pieretti, M.; Sutcliffe, J.S.; Richards, S.; Verkerk, A.J.; Holden, J.J.; Fenwick, R.G., Jr.; Warren, S.T.; et al. Variation of the CGG repeat at the fragile X site results in genetic instability: Resolution of the Sherman paradox. Cell 1991, 67, 1047–1058. [Google Scholar] [CrossRef]
- Ranjan, R.; Jha, S.; Prajjwal, P.; Chaudhary, A.; Dudeja, P.; Vora, N.; Mateen, M.A.; Yousuf, M.A.; Chaudhary, B. Neurological, Psychiatric, and Multisystemic Involvement of Fragile X Syndrome Along With Its Pathophysiology, Methods of Screening, and Current Treatment Modalities. Cureus 2023, 15, e35505. [Google Scholar] [CrossRef]
- Nobile, V.; Pucci, C.; Chiurazzi, P.; Neri, G.; Tabolacci, E. DNA Methylation, Mechanisms of FMR1 Inactivation and Therapeutic Perspectives for Fragile X Syndrome. Biomolecules 2021, 11, 296. [Google Scholar] [CrossRef] [PubMed]
- Tabolacci, E.; Nobile, V.; Pucci, C.; Chiurazzi, P. Mechanisms of the FMR1 Repeat Instability: How Does the CGG Sequence Expand? Int. J. Mol. Sci. 2022, 23, 5425. [Google Scholar] [CrossRef] [PubMed]
- Usdin, K.; Kumari, D. Repeat-mediated epigenetic dysregulation of the FMR1 gene in the fragile X-related disorders. Front. Genet. 2015, 6, 192. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.; Sharma, M.R.; Shi, X.; Agrawal, R.K.; Joseph, S. Fragile X mental retardation protein regulates translation by binding directly to the ribosome. Mol. Cell 2014, 54, 407–417. [Google Scholar] [CrossRef]
- Miyashiro, K.Y.; Beckel-Mitchener, A.; Purk, T.P.; Becker, K.G.; Barret, T.; Liu, L.; Carbonetto, S.; Weiler, I.J.; Greenough, W.T.; Eberwine, J. RNA cargoes associating with FMRP reveal deficits in cellular functioning in Fmr1 null mice. Neuron 2003, 37, 417–431. [Google Scholar] [CrossRef] [PubMed]
- Bagni, C.; Zukin, R.S. A Synaptic Perspective of Fragile X Syndrome and Autism Spectrum Disorders. Neuron 2019, 101, 1070–1088. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dobre, M.; Gaina, G.; Erbescu, A.; Glangher, A.; Linca, F.I.; Ioana, D.; Severin, E.M.; Rad, F.; Iliescu, M.C.; Papuc, S.M.; et al. FMR1 Methylation Pattern and Repeat Expansion Screening in a Cohort of Boys with Autism Spectrum Disorders: Correlation of Genetic Findings with Clinical Presentations. Genes 2025, 16, 903. https://doi.org/10.3390/genes16080903
Dobre M, Gaina G, Erbescu A, Glangher A, Linca FI, Ioana D, Severin EM, Rad F, Iliescu MC, Papuc SM, et al. FMR1 Methylation Pattern and Repeat Expansion Screening in a Cohort of Boys with Autism Spectrum Disorders: Correlation of Genetic Findings with Clinical Presentations. Genes. 2025; 16(8):903. https://doi.org/10.3390/genes16080903
Chicago/Turabian StyleDobre, Maria, Gisela Gaina, Alina Erbescu, Adelina Glangher, Florentina Ionela Linca, Doina Ioana, Emilia Maria Severin, Florina Rad, Mihaela Catrinel Iliescu, Sorina Mihaela Papuc, and et al. 2025. "FMR1 Methylation Pattern and Repeat Expansion Screening in a Cohort of Boys with Autism Spectrum Disorders: Correlation of Genetic Findings with Clinical Presentations" Genes 16, no. 8: 903. https://doi.org/10.3390/genes16080903
APA StyleDobre, M., Gaina, G., Erbescu, A., Glangher, A., Linca, F. I., Ioana, D., Severin, E. M., Rad, F., Iliescu, M. C., Papuc, S. M., Hinescu, M. E., Arghir, A., & Budișteanu, M. (2025). FMR1 Methylation Pattern and Repeat Expansion Screening in a Cohort of Boys with Autism Spectrum Disorders: Correlation of Genetic Findings with Clinical Presentations. Genes, 16(8), 903. https://doi.org/10.3390/genes16080903